
Abstract
The low-density lipoprotein receptor-related protein 1 (LRP-1) is a multiligand endocytic receptor, which plays a pivotal role in controlling cytoskeleton dynamics during cancer cell migration. Its rapid endocytosis further allows efficient clearance of extracellular ligands. Concanavalin-A (ConA) is a lectin used to trigger in vitro physiological cellular processes, including cytokines secretion, nitric oxide production, and T-lymphocytes activation. Given that ConA exerts part of its effects through cytoskeleton remodeling, we questioned whether it affected LRP-1 expression, intracellular trafficking, and cell surface function in grade IV U87 glioblastoma cells. Using flow cytometry and confocal microscopy, we found that loss of the cell surface 600-kDa mature form of LRP-1 occurs upon ConA treatment. Consequently, internalization of the physiological α 2-macroglobulin and the synthetic angiopep-2 ligands of LRP-1 was also decreased. Silencing of known mediators of ConA, such as the membrane type-1 matrix metalloproteinase, and the Toll-like receptors (TLR)-2 and TLR-6 was unable to rescue ConA-mediated LRP-1 expression decrease, implying that the loss of LRP-1 was independent of cell surface relayed signaling. The ConA-mediated reduction in LRP-1 expression was emulated by the actin cytoskeleton-disrupting agent cytochalasin-D, but not by the microtubule inhibitor nocodazole, and required both lysosomal- and ubiquitin-proteasome system-mediated degradation. Our study implies that actin cytoskeleton integrity is required for proper LRP-1 cell surface functions and that impaired trafficking leads to specialized compartmentation and degradation. Our data also strengthen the biomarker role of cell surface LRP-1 functions in the vectorized transport of therapeutic angiopep bioconjugates into brain cancer cells.
Introduction
Low-density lipoprotein receptor-related protein 1 (LRP-1) is a member of the LDL receptor family of endocytic receptors formed of one extracellular 515 kDa subunit and one cytoplasmic 85 kDa subunit; 1 the mature receptor having been generated by the cleavage of a 600 kDa propeptide by furin. 2 The LRP family contains several functionally dynamic receptors involved in the cellular internalization of more than 40 circulating physiological ligands, including apolipoprotein E, 3 α2-macroglobulin, 4 factor VIII, 5 lipoproteins, 6 and amyloid-β. 7 Once the ligand is bound, the LRP-1/ligand complex undergoes clathrin-mediated endocytosis in order that the ligand is further targeted within specialized intracellular compartments. 8 Disruption of the LRP-1 gene in mice was found to be embryonically lethal, presumably because LRP-1 transduces intracellular signaling and is involved in many essential functions. 9
Over the past few decades, it has emerged that LRP-1 plays a significant role in the malignant phenotype of brain cancer cells, where it is tethered to the actin network and focal adhesion sites. 10 Through LRP1-dependent actin network remodeling, the activating ERK and inhibiting JNK signaling pathways contribute to the adhesive states of cancer cells, which favor invasion. 11 Intriguingly, both positive and negative effects of the LRP-1 status and function have been reported for cancer cells. LRP-1 displayed protective properties in the CNS malignant phenotype, due to its ability to clear the extracellular compartment of proteases. 12 The status of LRP-1 expression was also assessed in human glioma cell lines, 13 in in vivo glioblastomas, 14 and was found to be particularly elevated in U87 glioblastoma cells 15 as well as CD133+ pediatric brain tumor cells. 4 Several studies have also demonstrated that LRP-1 blockade reduced the invasive phenotype in numerous cancer cell models. 16 In glioblastoma cells, LRP-1 silencing reduced cell invasion and migration abilities, despite elevated levels of matrix metalloproteinase-2 (MMP-2) in the extracellular compartment. 16 Furthermore, its cell surface interactions with the CD44 protein implicated LRP-1 in both internalization and recycling, with LRP-1/CD44 complexes being found at the migratory front of carcinoma cells. 17 This association of LRP-1 compartmentation at the leading edge of migrating/invading cancer cells is relevant to its role in brain tumor development, and understanding of its cell surface expression will be crucial for the development of future therapeutic strategies. Interestingly, both LRP-1 and CD44 are cleaved by membrane type-1 matrix metalloproteinase (MT1-MMP),18,19 a transmembrane matrix metalloproteinase that plays a fundamental role in cell motility. 20 Regulation of the invasive phenotype of glioma cells involving a MT1-MMP/CD44/caveolin-1 interaction has been described21,22 through, in part, its rapid tracking/recycling to the plasma membrane from trans-Golgi network/endosome storage compartments. 23
Recently, the ligand internalization functions and recycling of LRP-1 to the cell surface have been exploited for the vectorized transport of synthetic cargo peptides, termed angiopep, through the blood–brain barrier and to the brain.24,25 This successful strategy led to the design of receptor-mediated internalization strategies through high brain permeable anticancer drugs such as paclitaxel-angiopep bioconjugates to gliomas.26–29 How cytoskeletal remodeling alters LRP-1 cell surface availability and functions in ligand internalization have not yet been explored. Herein, we used Concanavalin-A (ConA), a lectin regulating MT1-MMP cell surface proteolytic functions30,31 as well as MT1-MMP catalytic independent inflammation and autophagy cell signaling,32,33 to trigger molecular alterations of the cytoskeleton34,35 and assessed its impact on LRP-1 ligand internalization functions.
Experimental Procedures
Materials
Electrophoresis reagents were purchased from Bio-Rad. HyGLO chemiluminescent HRP antibody detection reagents were from Denville Scientific Inc. Micro bicinchoninic acid protein assay reagents were from Pierce. The MMP inhibitor Ilomastat and the anti-LRP-1 light chain monoclonal antibody (mAb) (5A6) were purchased from EMD Millipore. Angiopep-2 and α2-macroglobulin were gifts from Angiochem Inc. The antibody against murine LRP heavy chain (8G1) was from Calbiochem, the anti-COX-2 antibody (610203) was from BD Biosciences, and the anti-glyceraldehyde 3-phosphate dehydhogenase (GAPDH) (Ab8245) and anti-ubiquitin (Ab7780) antibodies were from Abcam. The R-phycoerythrin (PE)-conjugated mouse antibodies against human CD91 and IgG1 κ isotype were from BD Biosciences. Horseradish peroxidase-conjugated donkey anti-rabbit and anti-mouse IgG secondary antibodies were from Jackson ImmunoResearch Laboratories. The anti-MT1-MMP hinge region antibody (M3927), ConA, cytochalasin-D (CytoD), nocodazole, furin inhibitor II, tofacitinib, SB203580, PP2, U0126, acetyl-11-keto-beta-boswellic acid, sodium dodecyl-sulfate (SDS), and bovine serum albumin (BSA) were from Sigma-Aldrich.
Cell culture
The human U87 glioblastoma cell line (American Type Culture Collection, HTB-14) was maintained in Eagle's minimum essential medium (EMEM, Wisent, 320-006CL) containing 10% (v/v) calf serum (HyClone Laboratories, SH30541.03), 1 mM sodium pyruvate (Sigma-Aldrich Canada, P2256), 100 units/mL penicillin, and 100 mg/mL streptomycin (Wisent, 250-202-EL). Cells were incubated at 37°C with 95% air and 5% CO2.
Total RNA isolation, cDNA synthesis, and real-time quantitative PCR
Total RNA was extracted from cell monolayers using TriZol reagent (Life Technologies, 15596-018). For cDNA synthesis, 2 μg of total RNA were reverse-transcribed using a high-capacity cDNA reverse transcription kit (Applied Biosystems, 4368814). cDNA was stored at -80°C prior to PCR. Gene expression was quantified by real-time quantitative PCR using iQ Sso Fast EvaGreen Supermix (Bio-Rad). DNA amplification was carried out using a CFX connect Real Time System (Bio-Rad), and product detection was performed by measuring binding of the fluorescent dye EvaGreen to double-stranded DNA. The QuantiTect primer sets were provided by Qiagen: LRP-1 (Hs_LRP1_1_SG QT00025536), MT1-MMP (Hs_Mmp14_1_SG QT00001533), Toll-like receptors (TLR)-2 (Hs_TLR2_1_SG, QT00236131), TLR-6 (Hs_ TLR6_1_SG, QT00216272), GAPDH (Hs_GAPDH_2_SG QT01192646), and β-actin (Hs_Actb_2_SG QT01680476). The relative quantities of target gene mRNA compared against two internal controls, GAPDH and β-actin RNA, were measured by following a δCT method employing an amplification plot (fluorescence signal vs. cycle number). The difference (δCT.) between the mean values in the triplicate samples of target gene and those of GAPDH and β-actin mRNAs were calculated by the CFX Manager software version 2.1 (Bio-Rad), and the relative quantified value was expressed as 2-δCT.
Transfection method and RNA interference
Cells were transiently transfected with 10 nM siRNA against MT1-MMP (Hs_MMP14_6 HP validated siRNA; QIAGEN SI03648841), TLR-2 (HS_TLR2_1_SG QIAGEN QT00236131), TLR-6 (HSTLR6_1_SG QIAGEN QT00216272), or scrambled sequences (AllStars Negative Control siRNA; QIAGEN, 1027281) using Lipofectamine 2000 (Invitrogen, 11668). Every specific gene knockdown was evaluated by reverse transcription (qRT)-PCR as described above.
Gelatin zymography
Gelatin zymography was used to assess the extracellular levels of proMMP-2 and MMP-2 activities. Briefly, an aliquot (20 μL) of the culture medium was subjected to SDS-polyacrylamide gel electrophoresis (SDS-PAGE) in a gel containing 0.1 mg/mL gelatin (Sigma-Aldrich Canada, G2625). The gels were then incubated in 2.5% Triton X-100 (Bioshop, TRX506.500) and rinsed in deionized distilled water. Gels were further incubated at 37°C for 20 hours in 20 mM NaCl, 5 mM CaCl2, 0.02% Brij-35, 50 mM Tris-HCl buffer, pH 7.6, and then stained with 0.1% Coomassie Brilliant blue R-250 (Bioshop, CBB250) and destained in 10% acetic acid and 30% methanol in water. Gelatinolytic activity was detected as unstained bands on a blue background.
Immunoblotting procedures
Following treatments or transfection, U87 cells were washed with phosphate buffered saline (PBS) and lysed with lysis buffer (50 mM Tris-HCl, pH 7.4, 120 mM NaCl, 5 mM ethylenediaminetetracetic acid (EDTA), 0.5% Nonidet P-40, 0.1% Triton) in the presence of phosphatase and protease inhibitors on ice for 30 minutes. Cell debris was pelleted by centrifugation for 10 minutes at high speed. Protein concentration was quantified using a micro bicinchoninic acid protein assay kit (Thermo Fisher Scientific Inc.). Proteins (30 μg) from control and treated cells were separated by SDS-PAGE. After electrophoresis, proteins were electrotransferred to polyvinylidene difluoride membranes, which were then blocked for one hour at room temperature with 5% nonfat dry milk in Tris-buffered saline (150 mM NaCl, 20 mM Tris-HCl, pH 7.5) containing 0.3% Tween-20 (TBST; Bioshop, TWN510-500). Membranes were further washed in TBST and incubated with the indicated primary antibodies (1/1,000 dilution) in TBST containing 3% BSA and 0.1% sodium azide (Sigma-Aldrich Canada, S2002), followed by an one-hour incubation with horseradish peroxidase-conjugated donkey anti-rabbit (Jackson ImmunoResearch Laboratories, 711-035-152) or goat anti-mouse IgG (Jackson ImmunoResearch Laboratories, 115-035-062) at 1/2,500 dilutions in TBST containing 5% nonfat dry milk. Immunoreactive material was visualized by enhanced chemiluminescence (Amersham Pharmacia Biotech, RPN3004).
Binding and uptake assays of angiopep-2 and α2-macroglobulin
Cells were incubated with 250 nM Alexa488-α2-macroglobulin/Ringer HEPES or Alexa488-angiopep-2/Ringer HEPES or Ringer HEPES alone for one hour at 37°C in the dark and washed three times with PBS/BSA (5%)/EDTA (2 nM). Fluorescence was then measured in the FL1-A channel using a C6 Accuri flow cytometer (BD Biosciences).
Confocal fluorescent microscopy assays of angiopep-2 uptake and LRP-1 expression
Cells were incubated with 50 nM Alexa488-angiopep-2 in EMEM without phenol red for 18 hours at 37°C, 5% CO2. Cells were fixed in 4% formaldehyde (Fisher Scientific) for 20 minutes. Immunostaining was performed under nonpermeabilizing conditions for one hour with the anti-LRP Heavy Chain antibody (2 μg/mL) in 1% BSA/PBS/NaN3, followed by Rhodamine Red-X donkey anti-mouse IgG (Invitrogen, Canada). A solution of 10 μg/mL 4’,6-diamidino-2-phenylindole (DAPI) diluted in PBS was used to stain the nuclei. Fluorescence was then monitored by confocal microscopy using a Nikon Eclipse Ti confocal microscope and NIS Elements software.
LRP-1 cell surface immunophenotyping
Cells were collected and resuspended in a solution of binding buffer. Then, the cells were incubated with either a PE mouse α-human CD91 or a mouse IgG1 κ isotype control antibody for one hour at room temperature in the dark. Cells were washed three times with PBS. Fluorescence was then examined by flow cytometry in the FL2-A channel with a C6 Accuri. The results obtained were quantified as the difference between the geometric means of LRP-1-PE and PE isotype control antibodies.
Fluorescent microscopy
U87 cells were seeded onto coverslips where they were treated with ConA, CytoD, or nocodazole. Subsequently, cells were either incubated and/or immunolabeled followed by fixation. Images were acquired using a Nikon Eclipse Ti confocal microscope and NIS Elements software. Images were then deconvoluted and, where indicated, colocalization analysis was carried out with AutoQuant X software.
Statistical data analysis
Data are representative of three or more independent experiments. Statistical significance was assessed using Student's unpaired t-test. Probability values of less than 0.05 were considered significant and an asterisk (*) identifies such significance in the figures.
Results
Cytoskeleton remodeling alters LRP-1 trafficking and cell surface expression
The lectin ConA was used to investigate whether cytoskeleton remodeling affected LRP-1 trafficking and recycling process to the cell surface in U87 glioblastoma cells. While untreated cells expressed basal levels of MT1-MMP and of the 85-kDa LRP-1 subunit, ConA-activated U87 cells resulted in a loss of the 85-kDa transmembrane LRP-1 subunit from whole cell lysates upon a 24-hour dose–response with ConA (Fig. 1A). As expected, ConA treatment also led to the induction of cyclooxygenase (COX)-2 expression31,32 and was correlated with both MT1-MMP proteolytic processing into its 43-kDa species and proMMP-2 activation (Fig. 1A).30,31,36 Densitometric analysis of immunoblots showed that LRP-1 decrease in expression correlated with the ConA-mediated effects on COX-2 induction and MT1-MMP activation (Fig. 1B). Following this, a time-course assay was performed with various concentrations of ConA. A loss in LRP-1 expression occurred within 30 minutes of treatment with ConA (Fig. 1C, upper panel), and LRP-1 expression was completely abolished from lysates by 12 hours (Fig. 1C, lower panel). Having established that the LRP-1 expression declined in cell lysates, we questioned whether a mature 600-kDa LRP-1 still trafficked and compartmentalized at the cell surface. We performed immunophenotyping by flow cytometry to assess the rate at which LRP-1 was being internalized from and recycled to the cell surface. U87 cells were incubated for the indicated times with ConA and nonpermeabilized cells immunomarked as described in the “Experimental Procedures” section. Following 15 minutes of ConA treatment, cell surface LRP-1 had decreased to 25% of that seen with the control treatment and to 10% of control at 2 hours of treatment (Fig. 1D, closed circles). LRP-1 gene expression, as assessed by qRT-PCR, was unaltered (Fig. 1D, open circles). Altogether, the combined data lead us to hypothesize that mis-trafficking of LRP-1 to the cell surface possibly leads to its compartmentalized degradation.
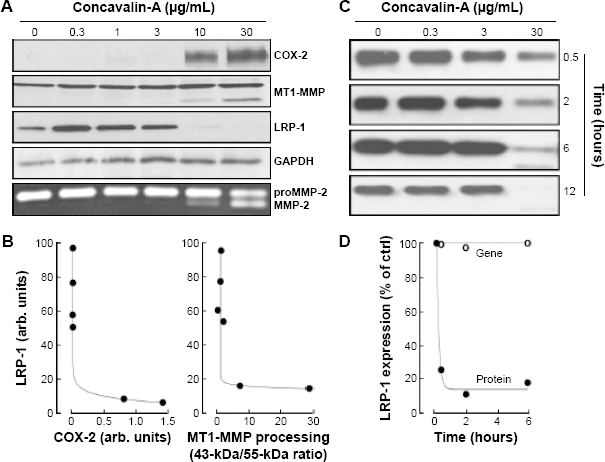
ConA triggers a rapid decrease of LRP-1 expression in U87 cells. (A) Serum-starved U87 cells were treated for 24 hours with various concentrations of ConA. Cell lysates were isolated and processed for the immunodetection of COX-2, MT1-MMP, LRP-1 (85-kDa subunit), and GAPDH. Conditioned media were harvested in order to assess proMMP-2 activation using gelatin zymography (bottom panel). (B) Representative densitometry analysis of LRP-1 to COX-2 expression (left panel) and LRP-1 to MT1-MMP proteolytic processing (43-kDa form/55-kDa form ratio, right panel). (C) serum-starved U87 cells were treated with various concentrations of ConA. Cell lysates were isolated, and immunodetection of LRP-1 for the indicated times performed. (D) Representative cell surface immunophenotyping of LRP-1, as measured by flow cytometry (closed circles), and assessment of LRP-1 gene expression as measured by qRT-PCR (open circles) are presented relative to time of treatment with ConA 30 μg/mL.
LRP-1 decrease is independent of MT1-MMP catalytic activity but correlates with ConA-mediated MT1-MMP proteolytic processing
To explore the link between ConA and LRP-1, we next examined whether MT1-MMP was involved in mediating the degradation of LRP-1 in ConA-activated U87 cells. ConA is classically used to trigger MT1-MMP-mediated activation of proMMP-2, which is further reflected through MT1-MMP proteolytic activation.31,32 The broad-acting MMP catalytic inhibitor Ilomastat was thus used and validated through the inhibition of ConA-mediated proMMP-2 activation. Neither proMMP-2 activation nor generation of the proteolytically processed MT1-MMP 43-kDa species were observed in Ilomastat-treated cells (Fig. 2A). This effect was emulated by the efficient silencing of MT1-MMP using RNA interference (Fig. 2B), followed by treatment with ConA (Fig. 2C). Furthermore, we tested whether furin-mediated LRP-1 maturation to the cell surface was involved. We found that inhibition of the proprotein convertase furin, which is implicated in the maturation of LRP-1 into its 85– and 515-kDa subunits, did not prevent the decrease in the 85-kDa LRP-1 subunit expression, though partially inhibiting MT1-MMP proteolytic processing into its 43-kDa species (Fig. 2A). This observation potentially suggests that, upon cytoskeleton disruption, a pool of intracellular LRP-1 protein trafficking is rather directed toward degradation than recycled to the cell surface.
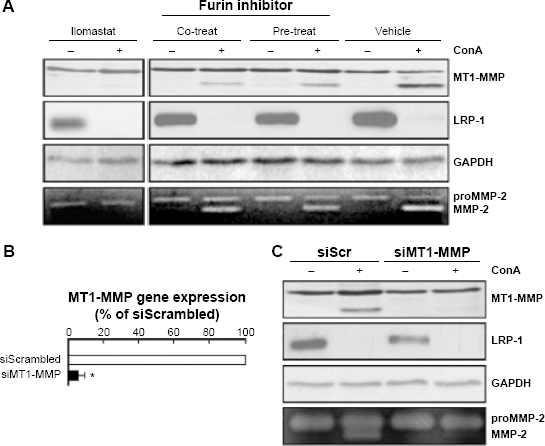
ConA-induced LRP-1 degradation is independent of MT1-MMP catalytic or proteolytic maturation. (A) Serum-starved U87 cells were treated (or not) for 24 hours with ConA (30 μg/mL) or with Ilomastat (25 μM) ± furin inhibitor II (50 μM) (as a cotreatment, or as a 24-hour pretreatment). Conditioned media were harvested to assess proMMP-2 activation by gelatin zymography (bottom images). (B) U87 cells were transiently transfected with siScrambled (siScr) or siMT1-MMP and transfection efficacy confirmed by qRT-PCR, then (C) followed by treatment for 24 hours with or without ConA (30 μg/mL). Following treatments, cell lysates were isolated for immunodetection of MT1-MMP, LRP-1 (85-kDa subunit), and GAPDH. Conditioned media were harvested to assess proMMP-2 activation by gelatin zymography (bottom of panel).
LRP-1 loss is mediated by both the ubiquitin-dependent proteasome system and lysosomes in ConA-activated U87 cells
As LRP-1 has been reported to be degraded by the ubiquitin-dependent proteasome system (UPS) and within lysosomes, 37 we next examined whether either of these systems was active in ConA-activated U87 cells. As almost complete disappearance of LRP-1 from cell lysates was observed within six hours of treatment, we chose to treat U87 cells for this duration with various doses of UPS inhibitors, MG132 or lactacystin, in the presence or absence of ConA. Inhibition of the UPS was confirmed by immunodetection of ubiquitin, and a significant rescue of LRP-1 was observed (Fig. 3A). Treatment with epigallocatechin 3-gallate (EGCG), a green tea catechin known to inhibit the UPS, 38 was unable to rescue the ConA-mediated LRP-1 decrease (Fig. 3B). To assess whether lysosomal degradation was involved, we treated coverslip-seeded U87 cells with ConA (30 μg/mL) for 0, 2, and 6 hours, after which we labeled both lysosomes and LRP-1 in order to evaluate whether these were colocalized. We found that, within two hours of treatment, significant colocalization between LRP-1 and lysosomes occurred, suggesting that the loss of LRP-1 is performed within this compartment (Fig. 3C).
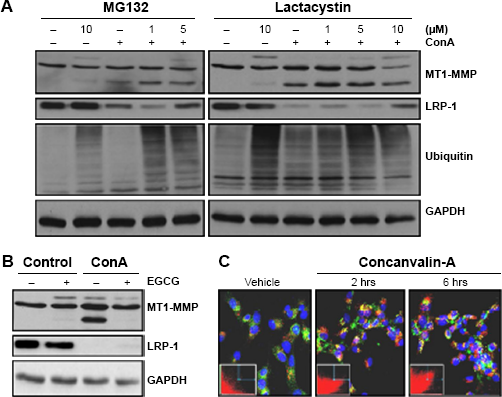
LRP-1 degradation in ConA-activated U87 cells is mediated both by lysosomes and by the UPs. U87 cells were treated for six hours, in the presence or absence of ConA (30 μg/mL) with (A) the UPs inhibitors MG132 (left panels) or lactacystin (right panels) or (B) EGCG (25 μM). In all conditions, cell lysates were isolated for immunodetection of MT1-MMP, LRP-1 (85-kDa subunit), GAPDH, and ubiquitin (panel A only). (C) U87 cells were seeded onto coverslips where they were treated (or not) with ConA (30 μg/mL) for two and six hours. Cells were then incubated with LysoTracker (red), fixed, and stained for LRP-1 (green). Images were acquired using a Nikon Eclipse Ti confocal microscope and using NIS Elements software. Deconvolution and colocalization analysis was carried out with AutoQuant X software. Colocalization analysis revealed Pearson's correlation coefficients (t = 0 hours, 0.59; t = 2 hours, 0.99; and t = 6 hours, 0.99) and 2D histogram plots of colocalization inset (green, y-axis (LRP-1); red, x-axis (Lysotracker)).
LRP-1 decreases in ConA-activated U87 cells is independent from ConA-mediated cell signaling
Having ruled out MT1-MMP as a mediator of the observed ConA effects (Fig. 2), we next tested whether ConA receptors, TLR-2, and TLR-6 were involved. Blocking TLR-2 in vivo was shown to attenuate experimental hepatitis induced by ConA in mice. 39 In parallel, TLR-6 gene silencing was found to abrogate ConA-induced CSF-2 and CSF-3 transcriptional regulation (Fig. 4A). 40 RNA interference allowed us to silence the expression of TLR-2 and TLR-6, which was followed by a six-hour treatment with ConA; this did not abrogate the ConA-induced LRP-1 degradation (Fig. 4B). We next inhibited several intracellular signaling pathways that could play a role in ConA signaling. However, despite some modulation observed in LRP-1 immunodetections, it appeared that this rapid regulation and almost complete loss in LRP-1 was independent from the aforementioned mechanisms (Fig. 4C).
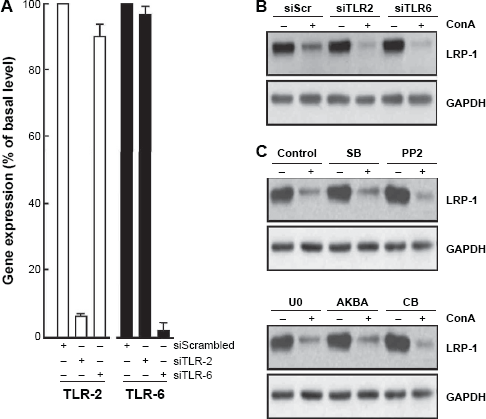
LRP-1 degradation in ConA-activated U87 cells does not require cell surface-mediated cell signaling. (A) U87 cells were transiently transfected with 10 nM of siScr, siTLR-2, or siTLR-6, and transfection efficacy assessed by qRT-PCR. (B) Following ConA treatment, all cell lysates were used for immunodetection of LRP-1 (85-kDa subunit) and GAPDH. (C) U87 cells were treated for six hours, in the presence or absence of ConA (30 μg/mL) in combination with the JAK/STAT inhibitor tofacitanib (CB, 30 μM), the MAPK inhibitor SB203580 (SB, 10 μM), the Src inhibitor PP2 (PP2, 10 μM), the MEK inhibitor U0126 (U0, 20 μM), or the MAPK inhibitor Acetyl-11-keto-beta-boswellic acid (AKBA, 20 μM).
Altered LRP-1 trafficking is triggered upon ConA-mediated alterations in actin cytoskeleton integrity
Having been unable to identify receptor-mediated signaling pathway linking ConA to the internalization and degradation of LRP-1, we explored ConA's ability to rather directly disrupt the cytoskeleton as a cause of the observed effects. In order to compare ConA to other cytoskeleton-disrupting agents, U87 cells were treated with CytoD, an inhibitor of actin polymerization, and nocodazole, an inhibitor of microtubule polymerization. We found that CytoD, in contrast to nocodazole, emulated the effects caused by ConA in cell lysates (Fig. 5A). The internalization of LRP-1 from the cell surface, as measured by flow cytometry, was again emulated by CytoD but not by nocodazole (Fig. 5B). An immunofluorescent assay of CytoD- and nocodazole-treated U87 cells failed to show colocalization between LRP-1 and lysosomes, although a decrease in LRP-1 fluorescence was observed by microscopy (Fig. 5C) when cells were treated with CytoD, but not with nocodazole (Fig. 5D). This suggests that, although common actin cytoskeleton perturbations are involved between the CytoD and ConA actions, differential compartmentation processes regulate ConA-mediated LRP-1 decreased expression. Furthermore, one can also hypothesize that LRP-1's incapacity to recycle back to the cell surface triggers its intracellular proteolytic degradation.
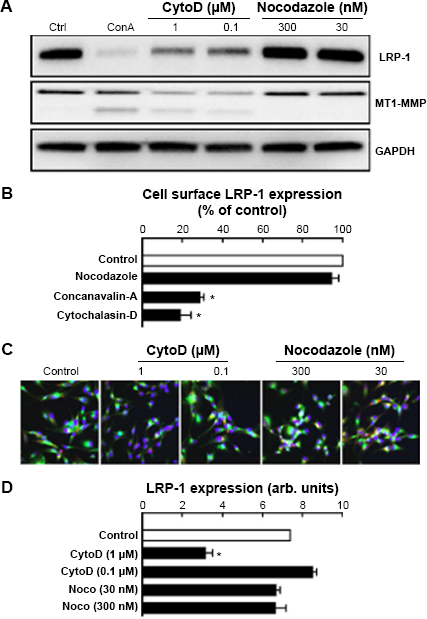
Disruption of the actin cytoskeleton is responsible for LRP-1 internalization and degradation. (A) U87 cells were treated for six hours with ConA (30 μg/mL), CytoD (1 and 0.1 μM), or nocodazole (300 and 30 nM). Cell lysates were then isolated for immunodetection of LRP-1 (85-kDa subunit), MT1-MMP, and GAPDH. (B) U87 cells were treated for two hours with ConA (30 μg/mL), CytoD (1 μM), or nocodazole (300 nM). Cells were then harvested for immunophenotyping of cell surface mature 600-kDa LRP-1 by flow cytometry. (C) U87 cells were seeded onto coverslips and treated with CytoD (1 and 0.1 μM) or nocodazole (300 and 30 nM) for six hours. Cells were then incubated with LysoTracker (red), fixed, and stained for LRP-1 (green), or for the nucleus with DAPI (blue). Images were acquired with a Nikon Eclipse Ti confocal microscope and using NIS Elements software, deconvolution and colocalization analysis was carried out with AutoQuant X software. (D) Mean cell surface LRP-1-associated fluorescence was measured using ImageJ software and divided by number of nuclei (arb. units).
Reduced uptake of LRP-1 ligands results from LRP-1 loss from the cell surface
Having established a rapid loss of LRP-1 from the cell surface, we next questioned whether this would lead to any ligand-mediated internalization functional impairment. Thus, we carried out cellular uptake assays with physiological and synthetic LRP-1 ligands in ConA-activated cells. Following five minutes of incubation with ConA, the cellular uptakes of both Alexa488-angiopep-2 and Alexa488-α2-macroglobulin decreased by ~60% (Fig. 6A). To assess whether the observed degradation of cell surface LRP-1 correlated with the increasing concentrations of ConA, we carried out an uptake assay with Alexa488-angiopep-2 following two hours of ConA treatment (Fig. 6B). We observed a significant decrease in uptake by up to ~60% when treated with 30 μg/mL ConA. To further support our findings, we treated U87 cells with varying concentrations of ConA for two hours, after which we incubated the cells with Alexa488-angiopep-2 and labeled cell surface LRP-1 (Fig. 6C). Quantification of these markers showed that the uptake of Alexa488-angiopep-2 correlated directly with LRP-1 cell surface expression, whereas high cell surface LRP-1 status resulted in high angiopep-2 internalization (Fig. 6D).
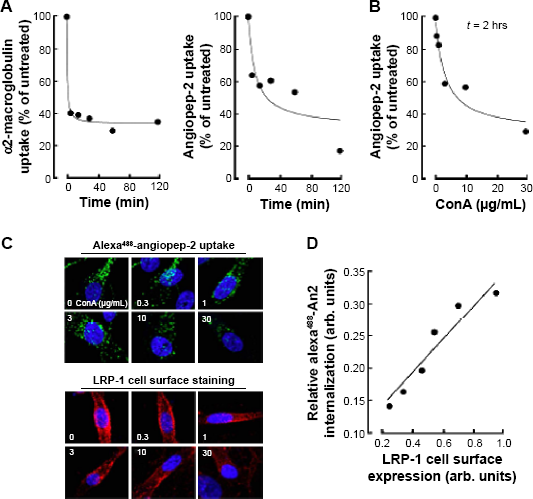
ConA-induced LRP-1 internalization and degradation leads to a reduced uptake of LRP-1 ligands. U87 cells were treated with ConA (30 μg/mL) for up to 120 minutes, followed by incubation with Alexa488-labeled LRP-1 ligands (A) α2-macroglobulin or (B) angiopep-2 (left panel). Uptake of ligands was measured by flow cytometry. (B) U87 cells were treated with various concentrations of ConA (0–30 μg/mL) for two hours, following which angiopep-2 uptake was assessed. (C) U87 cells were plated on coverslips and treated with various concentrations of ConA for two hours, then subsequently incubated with Alexa488-labeled angiopep-2 (top panels, green). Fixed cells were stained for LRP-1 (bottom panels, red) and images were acquired with a Nikon Eclipse Ti confocal microscope using NIS Elements software. Images shown are from a representative experiment. (D) Relative angiopep-2 internalization as a function of LRP-1 cell surface expression. Linear regression (R 2 = 0.92).
Discussion
In the current study, we questioned whether cytoskeleton remodeling, as it dynamically occurs in invading cells, altered endocytic processes such as those that regulate LRP-1 recycling and cell surface availability. We found that LRP-1 rapidly exited the cell surface of ConA-activated U87 glioblastoma cells, a phenomenon that was unrelated to MT1-MMP's catalytic activity. However, LRP-1 decreases correlated with ConA-induced MT1-MMP intracellular proteolytic activation and with induction of the inflammation biomarker COX-2 expression. Consequently, a decrease in cell surface LRP-1 resulted in diminished binding to its physiological α2-macroglobulin or synthetic angiopep-2 ligands. Our study implies that cytoskeleton integrity is required for LRP-1 expression and recycling to the plasma membrane and strengthens the pivotal requirement of cell surface LRP-1 functions in the vectorized transport of therapeutic angiopep bioconjugates into brain cancer cells.
Although the intracellular compartments involved in endocytic recycling processes remain to be well defined, LRP-1's tethering to the actin cytoskeleton and focal adhesion sites is an aspect of our study, which confirms its absolute requirement for activated α2-macroglobulin or angiopep-2 internalization processes to efficiently occur. Furthermore, we demonstrate that LRP-1's expression and function can be significantly regulated upon ConA-mediated cytoskeleton reorganization, which mimics the migrating/invading cell phenotype.11,41 As a consequence, the mis-trafficking of endosomal LRP-1 proteins may indeed affect cell migration, a process that is essential for development, tissue remodeling, and wound healing, as well as LRP-1 ligand recycling functions altered in many abnormal pathological states.42,43 To migrate directionally, cells indeed require to coordinate temporal and spatial cytoskeleton rearrangements through actin polymerization and focal adhesion turnover in order to generate the forces required for directional movement and cell surface protein availability. 44 Among the processes that could regulate LRP-1 cell surface availability, the recycling endosome is an organelle in the endocytic pathway where plasma membrane proteins are internalized by endocytosis and processed back to the cell surface for reuse. This allows the cell to maintain constituents of the plasma membrane on cell surfaces. 45 Evidence using brefeldin A, a vesicular trafficking inhibitor that trapped MT1-MMP within the cell, demonstrated similar induction of endoplasmic reticulum (ER) stress than ConA. 31 While nocodazole was ineffective, CytoD, a potent inhibitor of actin polymerization, reduced LRP-1 expression to a level similar to that seen with ConA. Possible nonspecific effects of CytoD, which may alter various other processes within the cell, must be acknowledged here. The use of latrunculin B, a specific inhibitor of cytoskeleton polymerization, 46 may ultimately help in demonstrating that LRP-1 degradation is indeed mediated by cytoskeletal disruption. This is now mentioned in the “Discussion” section of the revised manuscript. Interestingly, silencing of MT1-MMP prevented ConA from inducing both ER stress and COX-2 expression, but was unable to prevent ConA-mediated LRP-1 attenuation (this study). Inhibition of furin-dependent MT1-MMP proteolytic processing by ConA was also ineffective at reversing the LRP-1 decrease in our hands, although a potential mature ~600 kDa LRP-1 unprocessed form may have been expected to be observed and required to be further investigated.
The use of pharmacological endocytosis and lysosomes inhibitors, as well as proteasome inhibitors (as performed in Fig. 3 with the use of MG132 and lactacystin), may in the future studies help in our data interpretation regarding LRP-1 recycling processes. Of the recent molecular players demonstrated to link vesicular trafficking processes to cancer cell migration and invasiveness, the functions of the Rab family of small GTPases in regulating vesicular transport has raised intriguing mechanistic insights. 47 For instance, a Rab 11-dependent recycling pathway was reported to regulate α2-macroglobulin/LRP1-induced cellular migration of Müller glial cells by a mechanism that involved MT1-MMP intracellular trafficking to the plasma membrane. 48 Furthermore, a specific subset of RabGTPases was found to control cell surface exposure of MT1-MMP, extracellular matrix degradation, and three-dimensional invasion of macrophages. 49 Whether any of these Rab proteins are also involved in LRP-1 internalization is unknown. However, such evidence points to the existence of possible crosstalk between these processes. LRP-1 internalization and recycling back at the cell surface from early endosomes is a rapid (0.5 minutes) and high capacitive process, 50 and it becomes reasonable to hypothesize that disruption of these networks may rather lead to compartmentalized degradation of LRP-1, possibly by the UPS and via lysosome. Interestingly, EGCG, a green tea catechin known to inhibit several ConA- and MT1-MMP-mediated processes,22,40,51 and which also inhibited some ubiquitin-proteasome properties, 52 had no effect on the ConA-mediated LRP-1 decrease. Given that neither TLR-2 silencing, TLR-6 silencing, nor inhibition of multiple signaling pathways resulted in any reversal of LRP-1 decrease, we conclude that LRP-1 decreases are unrelated to any cell surface ConA-induced signaling pathway but rather caused through ConA's capacity to alter cytoskeleton integrity and, hence, trafficking/ recycling processes.
Finally, ConA is a plant lectin that has also been used for its properties in inducing a plethora of cellular events including cell proliferation and cell death/survival, as well as molecular biomarkers expression such as cytokines secretion,40,53 nitric oxide synthesis, 54 inflammatory COX-2 expression,30–32,36 autophagy BNP-3 expression,33,55 and activation and transcription of MT1-MMP.56,57 ConA has thus begun to be explored as a potential therapeutic entity due to its ability to induce both autophagy and apoptosis in human cancer cell models including hepatoma, glioblastoma, melanoma, and breast cancer cells.54,58–60 It is thus considered as a potential antineoplastic in preclinical or clinical trials for cancer therapeutics. Interestingly, it has been inferred that LRP-1 could mediate toxin-induced autophagy and apoptosis in a human gastric epithelial cell model. 61 Due to ConA's potential as a therapeutic avenue, it will be useful to understand how it collectively affects MT1-MMP and LRP-1 regulation in tumor cells.
Abbreviations
blood–brain barrier
concanavalin-A
cyclooxygenase-2
cytochalasin-D
extracellular matrix
epigallocatechin 3-gallate
endoplasmic reticulum
low-density lipoprotein receptor-related protein-1
matrix metalloproteinase-2
membrane type-1 matrix metalloproteinase
Toll-like receptor
ubiquitin-dependent proteasome system.
Author Contributions
Conceived and designed the experiments: KH and BA. Analyzed the data: SBN, JP, DB, KH, and BA. Wrote the first draft of the manuscript: SBN. Contributed to the writing of the manuscript: SBN, KH, and BA. Agree with manuscript results and conclusions: SBN, JP, DB, KH, and BA. Jointly developed the structure and arguments for the paper: SBN, JP, DB, KH, and BA. Made critical revisions and approved final version: SBN, JP, DB, KH, and BA. All authors reviewed and approved of the final manuscript.
Footnotes
Acknowledgments
BA holds an Institutional Research Chair in Cancer Prevention and Treatment. JP is a Natural Sciences and Engineering Research Council of Canada (NSERC) awardee.