
Abstract
Introduction
Methyltransferases are a large group of proteins, with different subclasses having defined functions.
Materials and Methods
Sequence Retrieval
Initially, we searched the NCBI (http://www.ncbi.nlm.nih.gov/) protein database for proteins containing methyltransferase-like sequences. The hypothetical protein PF0847 (gi|18977219|) of
Physicochemical Properties Analysis
The ProtParam (http://web.expasy.org/protparam/) 11 tool of ExPASy was used for the analysis of the physiological and chemical properties of the targeted protein sequence. The properties including aliphatic index (AI), GRAVY (grand average of hydropathy), extinction coefficients, isoelectric point (pI), and molecular weight were analyzed using this tool.
Homology Identification and Domain Analysis
The PSI-BLAST program of NCBI database (http://blast.ncbi.nlm.nih.gov/Blast.cgi) was used for searching the homology of PF0847 with the non-redundant database. For the domain analysis, we used the Pfam (http://pfam.sanger.ac.uk/) program of the Sanger Institute. 12
Multiple Sequence Alignment (MSA) and Phylogenic Tree Construction
For the identification of the sequence conservation among different species and strains, MSA was done with BioEdit biological sequence alignment editor, 13 and the phylogenetic tree was also constructed by Jalview 2 tool. 14
Structure Prediction
The secondary structure of the protein was predicted by PSIPRED server of UCL Department of Computer Science (http://bioinf.cs.ucl.ac.uk/psipred/), 15 and the tertiary structure was predicted by MODELLER 16 through HHpred17,18 tools of the Max Planck Institute for Development Biology.
Model Quality Assessment
Finally, the quality of the predicted structure was determined by PROCHECK 19 and QMEAN6 20 programs of ExPASy server of SWISS-MODEL Workspace. 21
Protein-Protein Interaction Analysis
Protein residues interact with each other for their accurate functions. Here we used STRING (http://string-db.org/), a database of known and predicted protein interactions that works through physical and functional associations derived from genomic context, high-throughput experiments, coexpression and previous knowledge. This database quantitatively integrates interaction data from above sources. Currently, this database covers 5,214,234 proteins from 1133 organisms. 22
Active Site Detection
Active site of the protein was determined by the computed atlas of surface topography of proteins (CASTp) (http://sts.bioengr.uic.edu/castp/), 23 which provides an online resource for locating, delineating, and measuring concave surface regions on three-dimensional structures of proteins. These include pockets located on protein surfaces and voids buried in the interior of proteins. This provides an important means for the prediction of the interacting sites on protein with the ligand molecules.
Docking Analysis
Docking analysis was performed by Molegro Virtual Docker (MVD) of CLC bio lab. Docking is performed in an integrated environment for studying and predicting how ligands interact with macromolecules. This offers high-quality docking that depends on a novel optimization technique. 24 The combined binding of the target protein PF0847 with SAM (S-adenosyl-L-methionine), genticin and 16S rRNA A-site was obtained using PyMOL (The PyMOL Molecular Graphics System, Version 1.5.0.4, Schrödinger, LLC).
Results
Analysis of Physicochemical Properties and Homology Searching
Different physicochemical properties of the hypothetical protein PF0847 were analyzed using ProtParam analysis tool (Table 1). The 248 amino acids containing protein was estimated to possess a molecular mass of 27,905.5 and isoelectric pH at 9.36.
ProtParam tool analysis result.

Non-redundant database was searched for protein sequences homologous with PF0847, and some of the homologs found are listed in Table 2. The Pfam server identified conserved domain in our targeted protein. MSA was done among the homologs from Table 2, and the output is shown in Figure 1. Using the same data, a phylogenic tree was constructed as shown in Figure 2.
Similar proteins obtained from non-redundant database.


MSA among different methyltransferase proteins with the target protein at the top row (Sources for the sequences: Row 2 –

Phylogenetic tree showing average distance among different methyltransferase proteins and the target protein.
Structure Analysis and Model Quality Assessment
PSIPRED server was used to predict the secondary structure of the protein (Fig. 3). Tertiary structure of the protein was modeled by MODELLER (Fig. 4). Quality assessment of the predicted tertiary structure was obtained from PROCHECK through “Ramachandran plot” where we found 93.4% amino acid residues within the most favored region (Table 3 and Fig. 5A). The quality of our model was further checked by QMEAN6 server where the model was placed inside the dark zone and considered good (Fig. 5B). Active site of our targeted protein was analyzed by CASTp (Fig. 6). The amino acid residues of the active site were also determined.

Predicted secondary structure of the protein PF0847.

Predicted three-dimensional structure of the protein PF0847.
Ramachandran plot statistics of the protein PF0847.

Model quality assessment. (

Active site determination of the protein PF0847. (
Biological Function Analysis
Using our analysis thus far on the protein under study, we relied on molecular docking to find out the probable ligand. Molegro Virtual Docker docked the selected ligand SAM with both the hypothetical protein and the reference protein (3P2 K: D) with grid lines

SAM ligand (red stick) docked in the active site of proteins. (
Comparative docking study.

To visualize the protein–protein interaction network of the protein PF0847, STRING was employed, and the obtained network is shown in Figure 8. Continuing with the STRING results, we found that geneticin bound to the eubacterial 16S rRNA A-site (PDB code: 1MWL) binds with the active site of the target protein PF0847 (Fig. 9). As we could not find an archaeal rRNA A-site 3D structure entry on the database, we tried to look for the similarity between eubacterial and archaeal rRNA with MSA (Fig. 10).

STRING network representing the predicted functional partners of the protein PF0847.
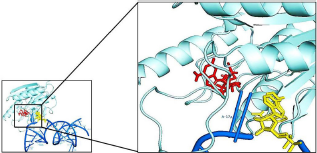
A-site of 16S rRNA (blue) bound to the protein PF0847 (cyan). The zoom view shows that geneticin (yellow) binds with the protein very close to the SAM (red) binding site.

MSA of the 16S rRNA A-site from different organisms (gi|444303952 was taken from
Discussion
Physicochemical properties of the protein were calculated by the ProtParam server including AI, instability index (II), pI, extinction coefficient and average hydropathicity. The AI is the relative volume occupied by the side chains of amino acids (alanine, leucine, valine and isoleucine). Increase in AI denotes increased thermostability of the globular proteins. 25 The calculated II of our protein was 25.90, which means it is stable in test tube condition. 26 The extinction coefficient indicates the light absorption capacity. 27 pI denotes protein net charge. Most of the calculations in this server demonstrate protein stability, because the stability is related to its proper function. 28 PSI-BLAST against non-redundant database revealed 98% similarity with methyltransferase protein. It also found similarity with putative RNA methyltransferase and adenine-specific methylase protein. Pfam server identified mostly conserved methyltransferase domain from 79 to 205 amino acid residues. MSA among the related proteins showed higher conservancy with methyltransferase domain and with the whole protein sequences too. Phylogenetic tree also expressed evolutionary relationship among different methyltransferase-related proteins of both archaeal and eubacterial origins. It also indicated that the target protein PF0847 had some evolutionary relation with eubacterial methyltransferases, even though they were very distant.
The proposed secondary structure predicted by PSIPRED has a good confidence of prediction. Tertiary structure was modeled by MODELLER with multiple templates to cover the whole sequence. Quality of the model was assessed by PROCHECK and is represented by Ramachandran plot (Fig. 5A). According to the plot statistics, 93.4% residues are in the most favored regions [A, B, L], 6.1% residues are in the additional allowed regions [a, b, l, p], and 0.0% residues are in the disallowed regions – a statistics that reveals a good model. QMEAN6 server assessment (Fig. 5B) result showed that the Z score of the predicted model was 0.18, which indicates a high-quality model. Active site of the protein predicted by CASTp server (Fig. 6) gives insight about the active site cleft and the amino acid residues that interact with different ligands.
STRING interaction network revealed that our targeted protein (PF0847) interacted with four different proteins for its functioning. The protein flpA (PF0059) from
Molegro Virtual Docker (MVD) performed docking between SAM ligand and our targeted protein within an integrated environment. The ligand SAM was fetched from an antibiotic-related methyltransferase protein (PDB code 3P2K: D). SAM ligand docked with both reference and targeted proteins active site (Fig. 7), and the docking results revealed that for both the bindings, the binding energy was similar (Table 4). RMSD value is an indication of how significant the computer-derived docking is, and smaller values indicate better docking. The RMSD values for the docking of SAM ligand to PF0847 and the reference protein were very close, which suggests a significant binding of SAM with PF0847.
From the insights of the STRING interaction network, we found that our targeted protein also binds with the geneticin bound to the eubacterial 16S rRNA A-site (Fig. 9). It has been reported that
Conclusion
The study was designed to predict the three-dimensional structure and biological function of the hypothetical protein PF0847 of
Author Contributions
ARO and SAIA conceived and designed the experiments. ARO and TPJ analyzed the data. ARO wrote the first draft of the manuscript. ARO, TPJ, and SAIA agreed with manuscript results and conclusions. ARO and SAIA jointly developed the structure and arguments for the paper. SAIA made critical revisions and approved the final version. All authors reviewed and approved the final manuscript.
Footnotes
As a requirement of publication the authors have provided signed confirmation of their compliance with ethical and legal obligations including but not limited to compliance with ICMJE authorship and competing interests guidelines, that the article is neither under consideration for publication nor published elsewhere, of their compliance with legal and ethical guidelines concerning human and animal research participants (if applicable), and that permission has been obtained for reproduction of any copyrighted material. This article was subject to blind, independent, expert peer review. The reviewers reported no competing interests.