
Abstract
Schistosomiasis, otherwise known as bilharzia or snail fever, is a disease that usually affects poor people and people exposed to poor sanitation. The disease affects over 200 million people worldwide annually. Schistosomiasis has been treated using a single drug, praziquantel, since the 1970s and this is resulting in schistosomes becoming resistant. Therefore, there is an urgent need to develop new antischistosoma drugs and vaccines. This study focuses on identifying potential antischistosomal compounds from the plant Salvia fruticosa. We virtually screened a library of 163 S fruticosa compounds by docking against Schistosoma mansoni sulfotransferase (SmSULT) using the PyRx software. Docking scores ranged from −4.7 to −9.3 kcal/mol. Compounds with binding affinity of −7.6 or stronger were subjected to drug-likeness assessments using the DataWarrior software. We also employed the PAINS removal tool to filter off false-positive results. Twelve compounds passed the drug-likeness screen, and these were subjected to in silico toxicity predictions to determine their mutagenic, tumorigenic and reproductive potential. Seven compounds were predicted to be nontoxic. After considering the toxicity analysis results and drug scores of the compounds, we identified rosmarinic acid and hispidulin as qualifying for further evaluation as potential drugs against schistosomiasis. Free energy calculations using the fastDRH webserver and molecular dynamics simulations using CABS-flex showed that the receptor-ligand complexes for the 2 lead compounds are stable under physiological conditions. We recommend that rosmarinic acid and hispidulin be used as hit compounds for the development of potential antischistosomal drugs.
Introduction
Schistosomiasis (also called bilharzia) is a neglected tropical disease with a significant socioeconomic impact and a global public health concern. 1 According to conservative estimates, schistosomiasis affects over 230 million people worldwide.2,3 The disease is caused by trematode flukes of the Schistosoma genus. 4 The 3 main species infecting humans are Schistosoma mansoni, Schistosoma japonicum and Schistosoma haematobium.5,6 Of these, S mansoni is considered a significant public health threat because of its chronic evolution. 7 Unfortunately, there is no vaccine available against human schistosomes.8-10 Praziquantel (PZQ) is the only drug recommended to treat schistosomiasis.11-13 This is despite the drug being vulnerable to drug resistance and has limited effectiveness against the juvenile stage of the parasite.14,15 Therefore, there is an urgent need for alternative treatment approaches, to avert a potential health crisis.1,16
The complexity and diversity of natural products offer a wide range of potential compounds for drug discovery. 1 Since ancient times, medicinal plants have been used to treat a variety of diseases and, 80% of the population in developing countries still rely on herbal remedies to meet their primary health care requirements.17,18 To date, several plants are being used in folklore medicine for treatment of schistosomiasis and these include Cucurbita maxima, 16 Salvia fruticosa, 19 Ziziphus spina-christi, 19 Zingiber officinale, 20 Piper nigrum, 20 and garlic. 21 Research on plant extracts has identified several chemical classes, including terpenoids, steroids, glycosides, anthraquinones and tannins, that show potential in killing S mansoni worms. 22 According to recent publications, over 50% of newly approved medications come from natural products.22,23 This highlights the significance of plant secondary metabolites in drug discovery and the need to find novel drugs with low toxicity.
Identification of drug candidates through computational screening approaches is a promising and cost-effective strategy that plays a vital role in drug design and discovery. 24 To discover new and affordable treatments for schistosomiasis, computer-aided drug design (CADD) approaches have been employed to develop novel schistosomicidal agents.16,25-27
In this study, molecular docking, drug-likeness prediction, toxicity prediction, binding free energy calculations, and molecular dynamics (MD) simulations were applied to identify potential lead candidates from a library of S fruticosa compounds. In structure-based CADD, it is crucial to identify a suitable target onto which a lead drug molecule can bind. Studies have identified Schistosoma mansoni sulfotransferase (SmSULT) to be one of the attractive drug targets for rational drug design to treat schistosomiasis.28,29 Sulfotransferases catalyse the transfer of a sulfuryl group from a sulphate donor to substrates. 29 Therefore, inhibition of the SmSULT is fatal to the organism. The past effectiveness of oxamniquine against S mansoni infections in Brazil has validated SmSULT as a viable drug target. 30
We employed molecular docking to find potential S. fruticosa compounds with a good binding affinity with SmSULT. Binding free energy calculations of the selected molecules provided further in silico evidence of the strong affinities between the ligands and the target. The drug-likeness and in silico toxicity prediction were carried out to identify compounds that are ‘drug-like’ and that do not have toxic side effects. In addition, MD simulations were employed to observe the structural stability of the protein-ligand complexes in real-time during the simulation. Overall, our in silico study has identified 2 potential ligands with good inhibitory potential against SmSULT, and therefore, these compounds are worthy to be assayed against the target enzyme or against the whole worms to validate the in silico findings.
Materials and Methods
The complete workflow for the identification of S fruticosa compounds with potential schistosomicidal activity is shown in Figure 1.
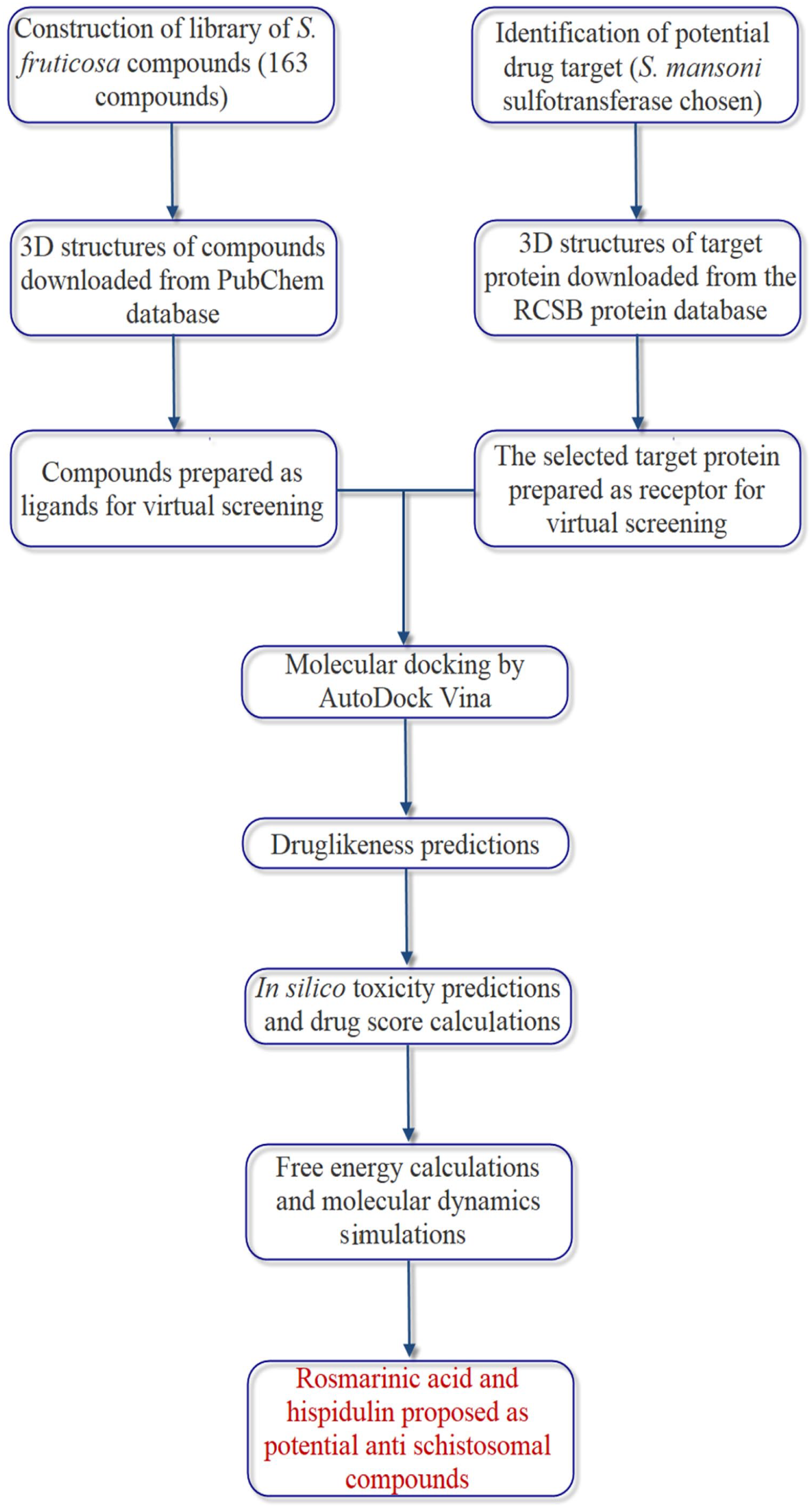
Schematic representation of the steps of the methodology.
Computational tools
All software were run on an HP 2170p Intel(R) Core(TM) i5-3427U CPU @ 1.80 GHz, 4 GB RAM, and 464 GB hard disk space. Windows 10 Professional was used as the Computer Operating System.
Ligand preparation
Salvia fruticosa compounds were downloaded from the Lotus Natural Compounds 31 database (https://lotus.naturalproducts.net) by typing the words ‘Salvia fruticosa’ in the Lotus search box. The 3-dimenisional (3D) structures of ligands were downloaded from the PubChem 32 database (https://pubchem.ncbi.nlm.nih.gov/) in simple data file (.sdf). The ligands were prepared using the Open Babel plugin of Python Prescription (PyRx) v0.8 33 (https://pyrx.sourceforge.io/). Before molecular docking, energy minimisation of ligands was done using the universal force field (uff) to achieve a better relaxation of atoms. Energy minimisation was achieved with the algorithm set to conjugate gradient and 200 steps. The energy-minimised ligands were then converted to the PDBQT file format, ready for docking.
Protein preparation
The crystal structure of sulfotransferase in complex with oxamniquine (PDB ID: 4MUB, Resolution: 1.75 Å) was retrieved from Protein Data Bank (PDB) (https://www.rcsb.org/). All ligands were deleted before the protein was prepared using the Dock Prep module of UCSF Chimera v1.17.1 34 (https://www.cgl.ucsf.edu/chimera/). The Dock Prep steps included the deletion of water molecules, adding missing hydrogen atoms, adding polar charges, and completing missing side chains using the Dunbrack 2010 rotamer library.
Molecular docking
Autodock Vina 35 integrated within the PyRx software was used for molecular docking simulations. Salvia fruticosa phytocompounds were docked into the active site of sulfotransferase. A grid box was generated using the receptor grid generation module of PyRx, and the dimensions were adjusted to ensure that the active site is enclosed. The grid box was centred at X = 108.24, Y = 7.98 and Z = 16.57 with the size (Å) of 45.87 × 27.67 × 33.55. The co-ordinates were determined using DeepSite 36 (https://www.playmolecule.com), a binding pocket predictor that uses neural networks. The best protein-ligand complexes were selected according to the binding energy and root-mean-square deviation (RMSD) values in each trial. Visualisation of the protein-ligand complexes was done using LigPlot+ v2.2 37 (https://www.ebi.ac.uk/thornton-srv/software/LigPlus/download.html) and PyMOL (https://pymol.org).
Validation of the molecular docking protocol
To validate the molecular docking protocol, we used the re-docking method. Briefly, the pose of the ligand co-crystallised with the receptor protein was compared with the pose of the ligand docked in the active site of this same protein. Then, the RMSD was used to compare the average distance between the ligand subjected to molecular docking and the co-crystallised ligand. In this method of validation, the docking protocol is valid if the RMSD is less than 2.0 Å. 38 The RMSD calculation was done using the BIOVIA Discovery Studio Visualiser (http://accelrys.com/products/collaborative-science/biovia-discovery-studio/) software.
Drug-likeness analysis
The drug-likeness of compounds was evaluated by DataWarrior v5.5.0. 39 The Lipinski Rule of Five parameters (RO5) 40 (molecular weight ⩽ 500, lipophilicity/clogP ⩽ 5, H-Donors ⩽ 5 and H-Acceptors ⩽ 10), which is an empirical rule of thumb was used to filter the compounds for their drug-likeness properties. Compounds that satisfy the RO5 are considered to have good solubility and permeability properties. 41 To remove false positives from the screened library, the Pan Assay Interference Compounds (PAINS) were filtered out using the PAINS Remover 42 tool (https://www.cbligand.org/PAINS).
Toxicity prediction
The ORIS Property Explorer (https://www.organic-chemistry.org/prog/peo/) was used to predict the toxicity (mutagenic, tumorigenic and reproductive effects) and drug score of the compounds that had passed the drug-likeness assessment.
Molecular mechanics/Poisson-Boltzmann (generalised Born) surface area calculations
The webserver, fastDRH 43 (http://cadd.zju.edu.cn/fastdrh/), was used to calculate the free energies of protein-ligand complexes using the molecular mechanics/Poisson-Boltzmann (generalised Born) surface area, MM/PB(GB)SA, methods. The force fields for the receptor and ligands were ff19SB (with OPC water model) 44 and GAFF2, 45 respectively. Binding free energy calculations are considered more accurate than molecular docking as it considers the solvation effect. 46
Molecular dynamics simulation
CABS-flex 2.0 47 was used to evaluate the conformational stability of the top-ranked protein-ligand complexes. CABS-flex offers rapid and accurate protein flexibility simulations (10 ns), which correlate with protein flexibility data generated by the nuclear magnetic resonance.47,48 Simulations were executed with default parameters.
Results and Discussion
Molecular docking
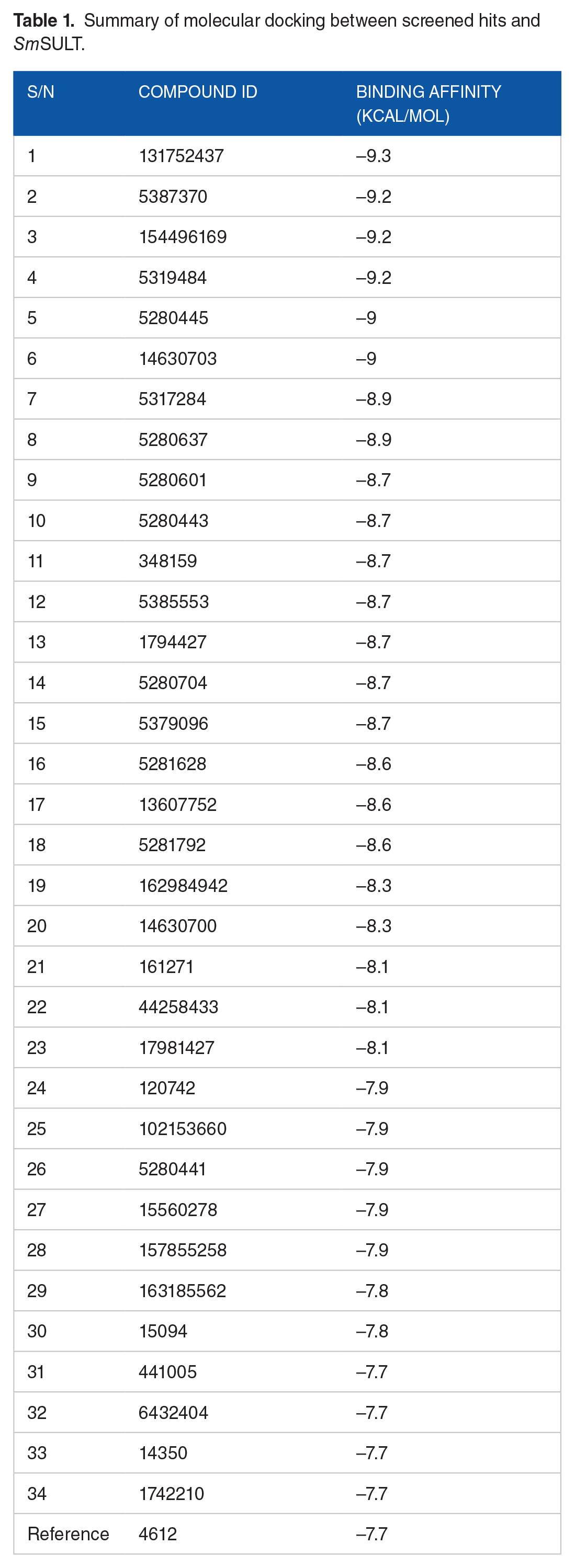
Molecular docking is an established method used in structure-based computer-aided drug discovery. Docking is employed in virtual screening to identify compounds likely to bind to a target protein. This can, therefore, reduce the number of compounds that need to be experimentally tested. In this study, 163 S fruticosa compounds were docked against SmSULT and these gave binding affinities ranging between −4.7 and −9.3 kcal/mol. In our hands, oxamniquine, which we used as a reference drug, had a binding affinity of −7.7. We, therefore, used this value as a cut-off point for virtual screening. Based on that criterion, 34 compounds (Table 1) were selected for further evaluation.
Summary of molecular docking between screened hits and SmSULT.
Validation of molecular docking accuracy
The molecular docking protocol was validated by cross-docking the co-crystallised ligand (sulfotransferase) using the same parameters as those applied for the S fruticosa compounds against the crystallographic structure of SmSULT (PDB ID: 4MUB). As the calculated value is below 2 Å (Figure 2), we can conclude that the docking method used in our work is valid.

Validation of molecular docking by superimposition of SmSULT co-crystallised ligand (blue) and the re-docked ligand (lime green).
Drug-likeness analysis
Drug-likeness analysis is the calculation of the chances of a molecule becoming an oral drug regarding bioavailabity. 49 Some compounds have good binding affinity with target receptors but, however, fail in clinical trials at advanced stages of drug discovery due to their lack of drug-likeness properties. 50 The desirable properties are derived from the Lipinski RO5 which states that drug-like compounds have Log P ⩽ 5, number of hydrogen bond acceptors ⩽10, the number of hydrogen bond donors’ ⩽5 and a molecular weight ⩽500. 51 The 34 S fruticosa ligands that demonstrated good binding affinity with SmSULT were analysed for drug-likeness using DataWarrior to assess their physiochemical properties. Of the 34, only 14 compounds violated none of the Lipinski RO5 criteria (see Table 2).
The physiochemical properties of screened hits and the reference drug.

Besides the RO5, we also evaluated the screened compounds for PAINS. Compounds that belong to PAINS have promiscuous behaviour as they react nonspecifically with many biological targets rather than specifically affecting 1 desired target.42,52 Thus, compounds with a substructure of PAINS are unsuitable to be lead compounds for drug discovery.53-55 As shown in Table 2, of the 14 compounds that passed the RO5 screen, only 2 compounds (CID_5280445 and CID_5317284) were filtered out. This means that the remaining 12 ligands are not likely to produce false positives in high-throughput screen tests.
Toxicity risk prediction
In silico toxicity analysis is an important step as it helps identify potential toxicities early in the drug discovery process, saving time and resources. 56 In this study, in silico toxicity analysis was carried out for the compounds that passed both the drug-likeness criterion and the PAINS filter (Table 3). Toxicity risk was calculated to determine the mutagenicity, tumorigenicity reproductive effects and dermatological effects (irritants) of the screened compounds. As shown in Table 3, the reference drug and 6 other compounds had no predicted toxic properties. In addition to toxicity assessments, OSIRIS calculated the drug scores of the screened hits. The drug score shows the probability of a compound to be a drug and it considers both the physiochemical and toxicity evaluation. The score value of 1 shows that the compound is a good drug candidate and shows that the compound has no possibility of being a drug. The toxicity and drug score results are shown in Table 3.
Toxicity prediction and drug scores of the screened hits and the reference drug.

Shortlisted compounds
After our rigorous virtual screening, drug-likeness assessments, toxicity tests and PAINS analysis, we remained with 5 possible potential inhibitors of SmSULT (Table 4).
Shortlist of screened hit compounds that passed the RO5, PAINS filter and the toxicity screen.

Interaction analysis of shortlisted best binders and the reference drug with SmSULT
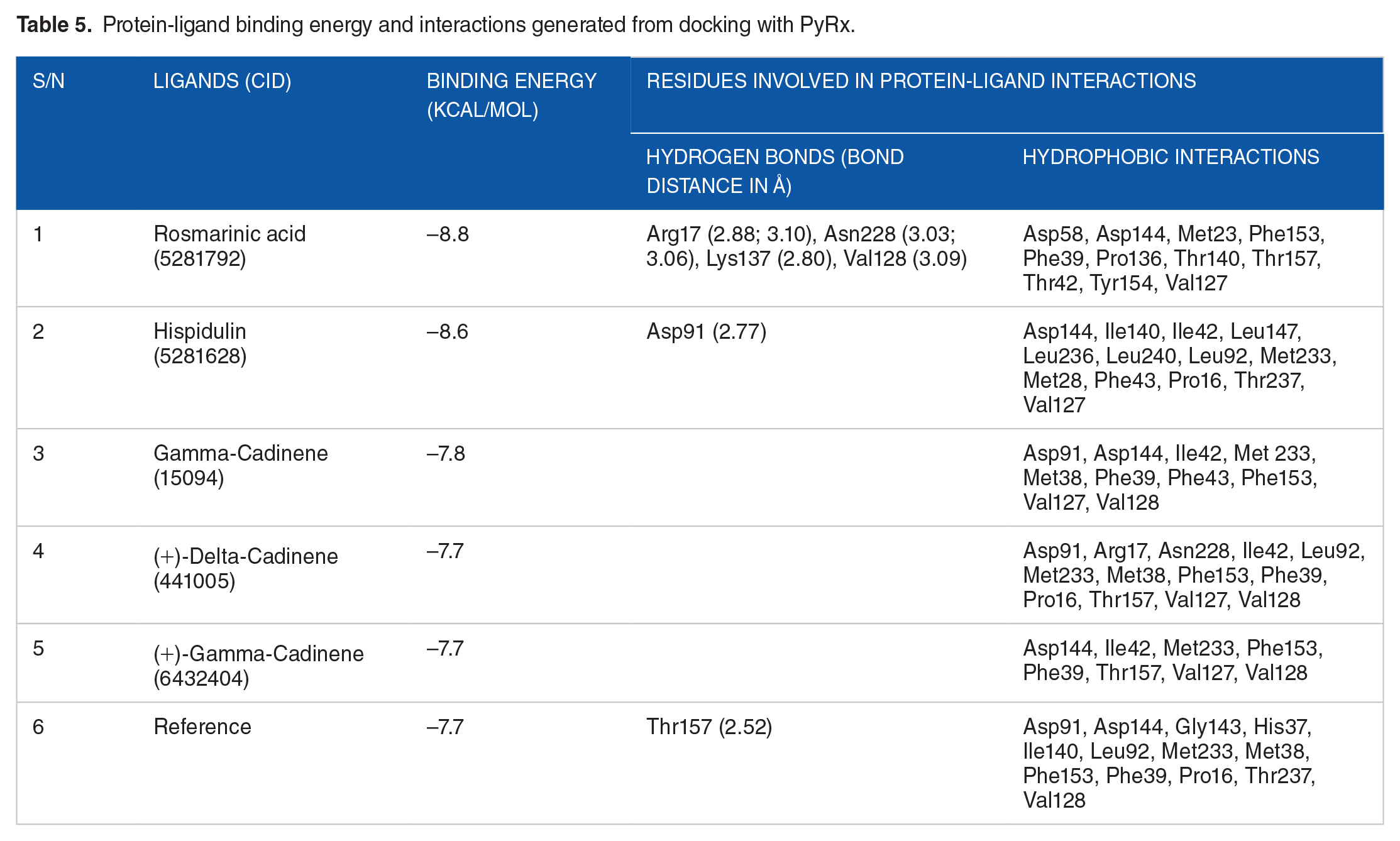
LigPlot+ and PyMOL were used to visualise the 2-dimenisional (2D) and 3D interactions of the protein-ligand complexes (Figures 5 to 8) produced from docking with Autodock Vina. The interaction results are summarised in Table 5. Rosmarinic acid (CID5281792) showed the highest binding affinity of −8.8 kcal/mol, forming 4 hydrogen bonds with Lys 137, Val 128, Asn 228, Arg 228, and 11 hydrophobic interactions with SmSULT as calculated by Autodock Vina. This was followed by hispidulin (CID5281628) which gave a binding energy of −8.6 kcal/mol, forming 1 hydrogen bond with Asp 91 and 13 hydrophobic bonds. The other 3 top compounds – gamma-cadinene (CID15094) (−7.8 kcal/mol), (+)-delta-cadinene (CID441005) (−7.7 kcal/mol), (+)-gamma-cadinene (CID6432404) (−7.7 kcal/mol) – did not form any hydrogen bond interactions with side chains. However, the compounds formed 10, 13 and 8 hydrophobic interactions, respectively.
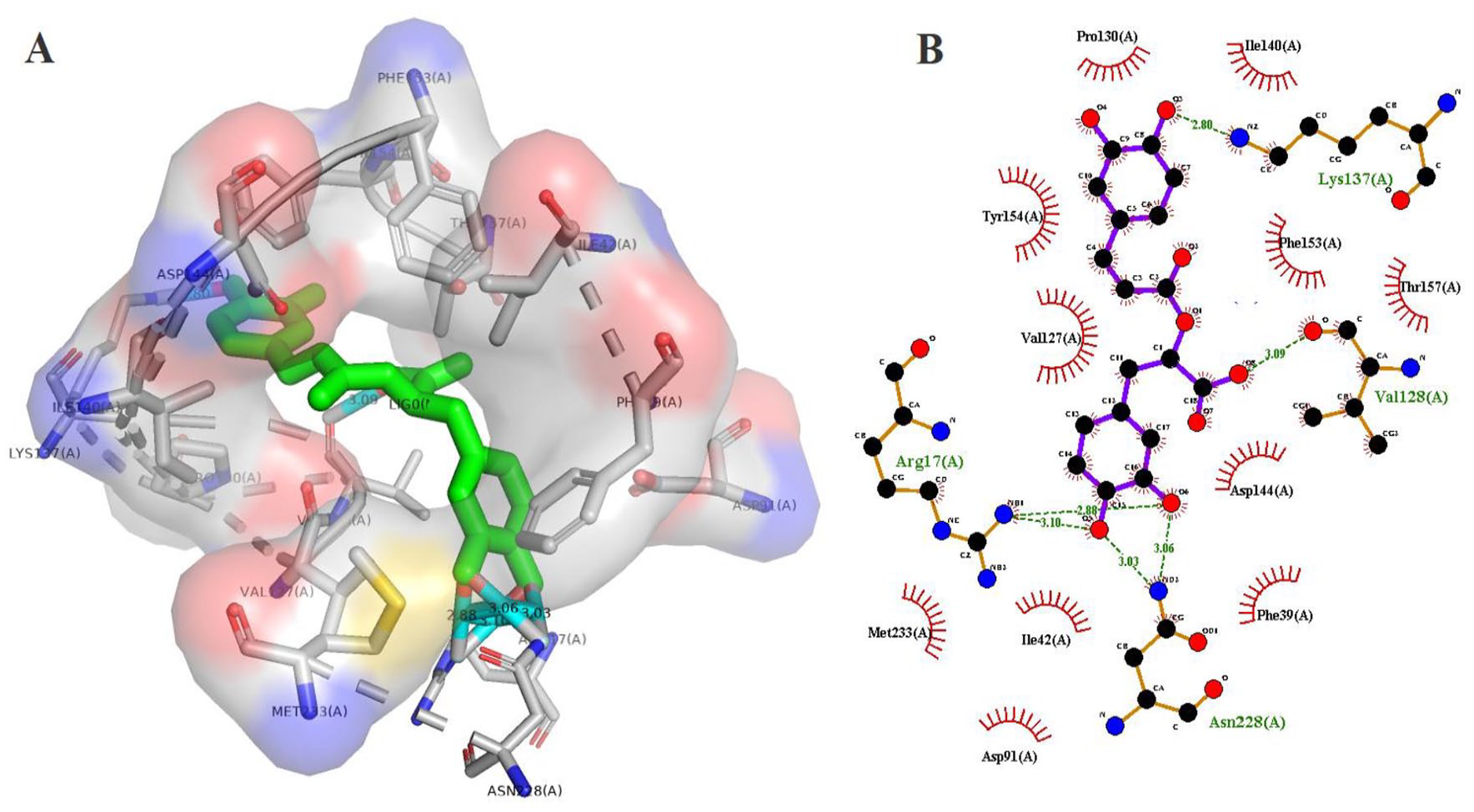
Hydrogen bonding and hydrophobic interactions of rosmarinic acid (CID5281792) with SmSULT. 3D (A) and 2D (B) representations of its interactions and binding pose within the SmSULT binding site.
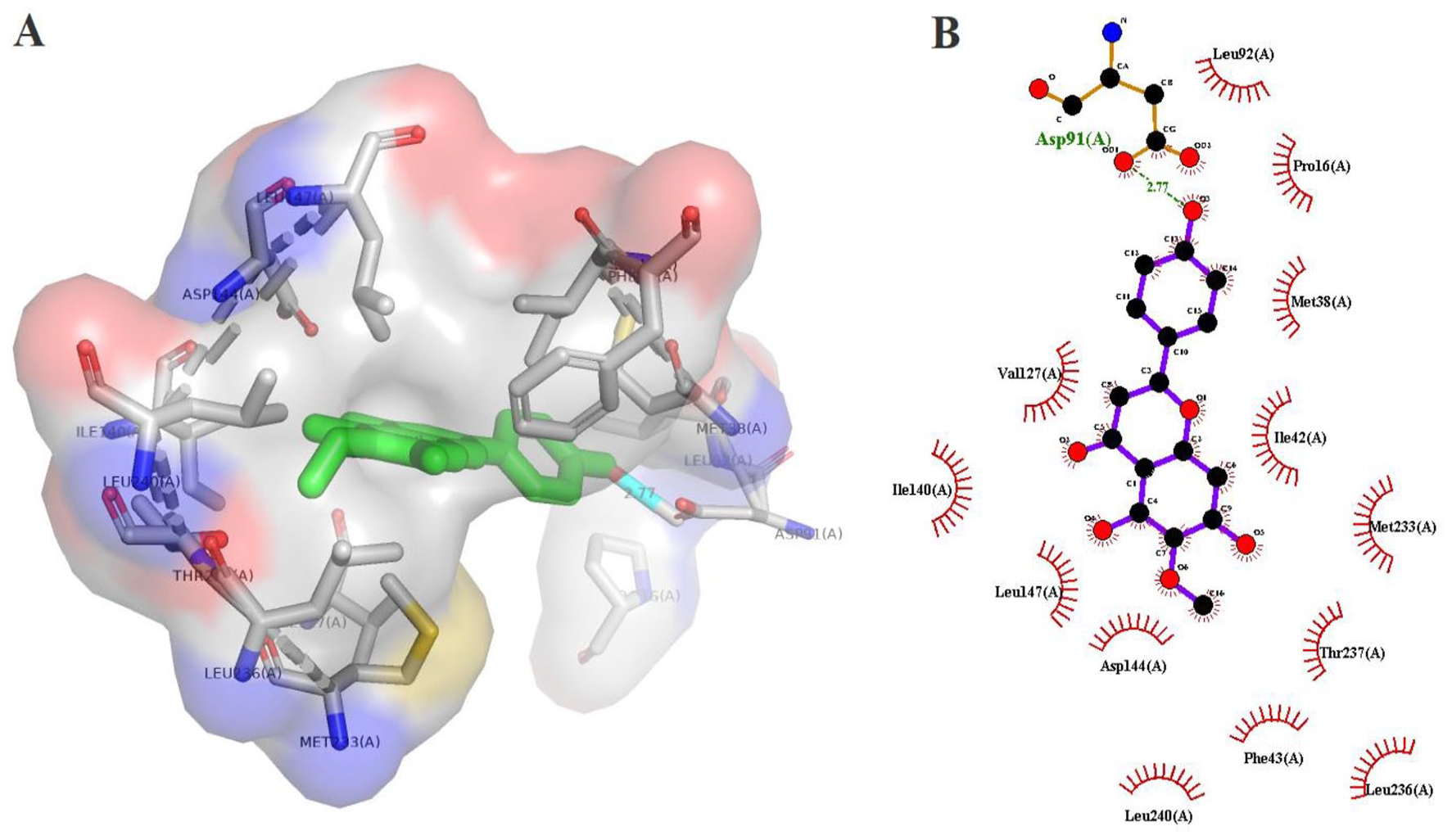
Hydrogen bonding and hydrophobic interactions of hispidulin (CID5281628) with SmSULT. 3D (A) and 2D (B) representations of its interactions and binding pose within the SmSULT binding site.

Hydrophobic interactions of gamma-cadinene (CID15094) with SmSULT. 3D (A) and 2D (B) representations of its interactions and binding pose within the SmSULT binding site.
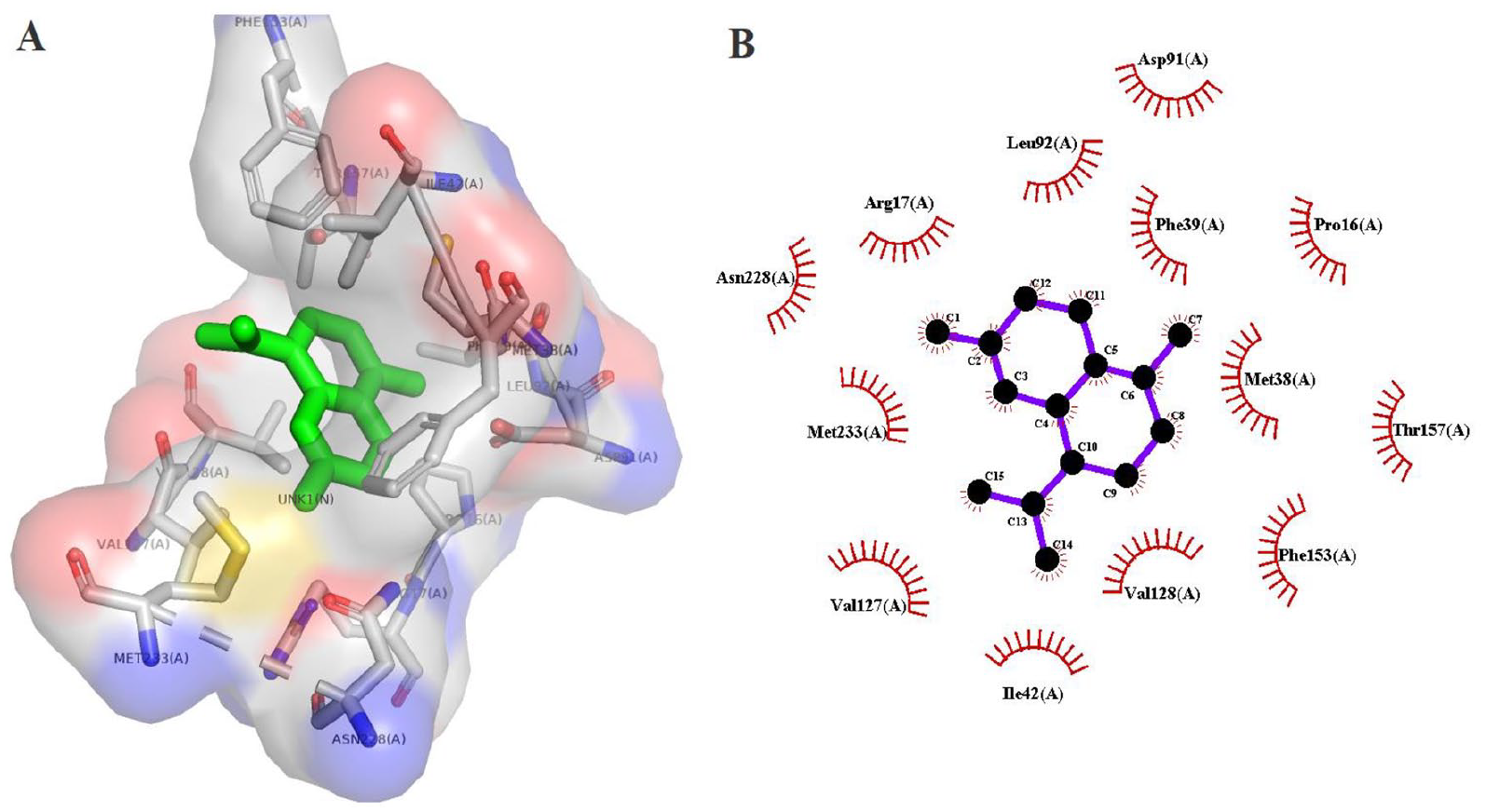
Hydrophobic interactions of (+)-delta-cadinene (CID441005) with SmSULT. 3D (A) and 2D (B) representations of its interactions and binding pose within the SmSULT binding site.

Hydrophobic interactions of (+)-gamma-cadinene CID6432404 with SmSULT. 3D (A) and 2D (B) representations of its interactions and binding pose within the SmSULT binding site.

Hydrogen bonding and hydrophobic interactions of the reference bond with SmSULT. 3D (A) and 2D (B) representations of its interactions and binding pose within the SmSULT binding site.
Protein-ligand binding energy and interactions generated from docking with PyRx.
The intermolecular hydrogen bonding between the protein and ligand plays an important role in stabilising the protein-inhibitor complex. 57 Therefore, hydrogen bonds are an important parameter to consider in computer-aided drug development. Ultimately, a combination of hydrogen bonds and many hydrophobic interactions are likely to result in a more desirable drug candidate. Our results showed that the top 2 high scoring compounds and the reference drug had extensive hydrophobic interactions with the SmSULT along with stable hydrogen bonding that anchor the compounds in the docked pocket.
Based on the interaction analysis results, we selected rosmarinic acid (CID5281792) and hispidulin (CID5281628) for further in silico analysis to calculate the binding free energy scores and stability of their respective docked complexes.
Molecular mechanics/Poisson-Boltzmann (generalised Born) surface area calculations
Molecular mechanics/Poisson-Boltzmann (generalised Born) surface area using fastDRH was used to calculate the binding free energy of selected docked complexes as relying only on the docking score does not adequately predict the binding affinity between the protein and the ligand. 58 As shown in Table 6, we found all complexes to have significant binding energy scores. In all the procedures used, rosmarinic acid had the highest negative binding free energy, followed by the reference ligand and then finally hispidulin. The negative binding energy scores shown for all the docked complexes suggest that the complexes are stable and warrant further investigations.
Binding free energy for the hit compounds and the reference drug in complex with 4MUB.

Molecular dynamics simulations
Unlike molecular docking that treats the receptor and the ligand as rigid molecules, MD simulations take into account the conformational changes of the receptor and the ligand, mimicking the real physiological conditions in the cell. 59 We, therefore, used CABS-flex 2.0 to evaluation of the root mean square fluctuation (RMSF) of the top-ranked protein-ligand complexes. Thus, the RMSF values of rosmarinic acid (CID5281792) and hispidulin (CID5281628) were compared with the reference drug. The RMSF values calculate the fluctuation of each amino acid residue as it interacts with the ligand throughout the simulation. The higher the RMSF value, the more flexible are the residues whereas low RMSF value indicates limited movements. For all the complexes studied, we observed that the RMSF variation for all the complexes was minimal, indicating less variability in the protein structure (Figure 9). For the reference drug, the minimum, maximum and average RMSF values of the SmPNP-ligand complex were 0.113, 4.352 and 0.905 Å, respectively. The minimum, maximum and average RMSF values of the SmPNP-rosmarinic acid (CID5281792) were 0.119, 3.717, and 0.833 Å, respectively and that of SmPNP-hispidulin (CID5281628) were 0.154, 3.893 and 0.913 Å, respectively, showing appreciably low residual fluctuations for the 2 complexes.

RMSF plots of rosmarinic acid (CID5281792), hispidulin (CID5281628) and the reference drug with the SmPNP (PDB ID: 4MUB) during MD simulation.
More importantly, the active pocket amino acid residue that interacted with the ligands during docking remained highly stable in the presence of both the screened compounds and the reference drug. Based on these results, we conclude that the protein-ligand complexes evaluated were stable in the physiological condition. Therefore, both rosmarinic acid and hispidulin are potentially potent inhibitors for SmPNP. The structures of the 2 compounds are shown in Figure 10.

2D structures of rosmarinic acid and hispidulin.
Conclusions
Praziquantel has been the only drug used for treating schistosomiasis, and it is slowly becoming less effective because of resistance of some schistosomes. Therefore, there is a need for immediate development of schistosomiasis drugs to prevent a potential health crisis. In this study, we employed various computational methods to identify novel S fruticosa compounds that can act as potential inhibitors for SmSULT, an important S mansoni drug target. These methods include molecular docking, drug-likeness analysis, toxicity predictions and MD simulations. Our rigorous, stepwise computational approach has led to the identification of rosmarinic acid and hispidulin as promising antischistosomal agents. These compounds exhibited good binding affinities to the target, with favourable drug-like properties and plausible toxicity profiles. We, therefore, propose that rosmarinic acid and hispidulin be considered a promising starting point for the development of new schistosomicidal compounds based on natural products.
Footnotes
Funding:
The author(s) received no financial support for the research, authorship and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Author Contributions
RS conceived and designed this study. All authors contributed to the primary investigation, formal analysis, data interpretation and to the writing of the manuscript. RS supervised the study. All authors have read and agreed to submit the final version of the manuscript.
Ethics approval
Ethics approval was not required for this study.