
Abstract
Most recently, monkeypox virus (MPXV) has emanated as a global public health threat. Unavailability of effective medicament against MPXV escalates demand for new therapeutic agent. In this study, in silico strategies were conducted to identify novel drug against the A36R protein of MPXV. The A36R protein of MPXV is responsible for the viral migration, adhesion, and vesicle trafficking to the host cell. To block the A36R protein, 4893 potential antiviral peptides (AVPs) were retrieved from DRAMP and SATPdb databases. Finally, 57 sequences were screened based on peptide filtering criteria, which were then modeled. Likewise, 31 monkeypox virus A36R protein sequences were collected from NCBI protein database to find consensus sequence and to predict 3D protein model. The refined and validated models of the A36R protein and AVP peptides were used to predict receptor-ligand interactions using DINC 2 server. Three peptides that showed best interactions were SATPdb10193, SATPdb21850, and SATPdb26811 with binding energies −6.10, −6.10, and −6.30 kcal/mol, respectively. Small molecules from drug databases were also used to perform virtual screening against the A36R protein. Among databases, Enamine-HTSC showed strong affinity with docking scores ranging from −8.8 to 9.8 kcal/mol. Interaction of target protein A36R with the top 3 peptides and the most probable drug (Z55287118) examined by molecular dynamic (MD) simulation. Trajectory analyses (RMSD, RMSF, SASA, and Rg) confirmed the stable nature of protein-ligand and protein-peptide complexes. This work suggests that identified top AVPs and small molecules might interfere with the function of the A36R protein of MPXV.
Keywords
Introduction
The eradication of smallpox from human populations was one of the 20th century’s most remarkable victories in medicine. One of the grisly incompatibilities of this achievement is the emergence of Monkeypox (MPX) with significant mortality and morbidity. 1 The recent surge in human monkeypox cases outside of their normal ecological range, the possibilities for advance spread through the transmission to naive population 2 as half of the world’s population has no immunity against orthopoxviruses, 3 and the lack of established surveillance have intensified the level of concern for this emerging zoonosis. 4 The zoonosis human monkeypox (MPX) is caused by monkeypox virus (MPXV) which belongs to the genus Orthopoxvirus, family Poxviridae, and sub-family Chordopoxvirinae.
MPXV was first unearthed during an outbreak in captive monkeys at a Danish laboratory in 1958.5,6 Congo Basin (CB) and West African (WA) are the two distinct clades of MPXV. 7 The CB clade primarily infects people residing in the Congo Basin.8,9 Between 1996 and 1997, more than 400 incidences were reported in Democratic Republic of the Congo (DRC) by CB clade followed by other outbreaks with hundreds of cases in the 2000s.9-11 Since 2016, DRC continues to report the most cases each year mainly in children aged below 10 years,4,5,12,13 and clustering of cases was reported in some particular areas within countries and within families. 14
In West Africa, only sporadic cases were revealed between 1970 and 1981. The largest known outbreak of monkeypox virus to date by WA clade with 122 confirmed or probable cases were recorded in 17 states of Nigeria in 2017-2018. 15 The characteristics of the 2017-2018 outbreak by WA clade suggest that the strains of MPXV in West Africa could prolong epidemic events, although West Africa was previously thought to be at low risk of a human monkeypox endemic. 8
Human monkeypox prevalence apart from Africa was first reported in the United States in 2003.2,9,16 The recent detection of MPX cases in each parts of the earth is an unprecedented and a big concern for global health security.
The majority of the clinical manifestations of human monkeypox infection mirrors those of smallpox but differs from it epidemiologically.14,17-19 A distinctive 2-day prodrome, apparent by fever, headache, muscle aches, backache, and malaise, reveals in most patients before the development of the rash. 9 Lymphadenopathy, a key distinguishable feature of human monkeypox which is not characteristic of smallpox,14,20 occurs in many victims 1 to 2 days before the inception of the rash.
MPXV is an enveloped double-stranded DNA virus with an approximate genome size of 196 858 base pairs. 21 Unlike many other DNA viruses, the replication cycle of poxviruses takes place in the cytoplasm of infected cells rather than in the nucleus. 22 Poxviruses enter cells by different mechanisms depending on whether they are intracellular mature virus (IMV) or extracellular enveloped virus (EEV). The IMV is encircled by a single membrane, which enters the cell by either direct fusion with the plasma membrane or endocytosis. By contrast, the EEV is composed of taking IMV enclosed within a second lipid membrane and forms shedding with the external membrane outside of the cell and then fusion of the IMV membrane with the host cell membrane. 23 Upon fusion with the host cell membrane, virus loses membrane to release its core into the cytoplasm that is transported on microtubules for moving deeper into the cell, where transcription and translation are occurred to form immature virions (IV), which are later processed to compose IMV.23,24 IMV particles are encased by a double membrane to constitute intracellular enveloped viruses (IEVs) that are further moved to the cell periphery on microtubules. The external IEV membrane attaches with the plasma membrane to expose a cell-associated extracellular virus (CEV) at the cell surface. Polymerization of an actin tail underneath the CEV drives the virus into the surrounding cell, or the virus is released as an EEV.23,25,26 According to Münter et al, 27 IEV transportation to the cell periphery necessitates the microtubule motor kinesin-1, which binds the integral viral membrane protein A36R. Surprisingly, the same protein gets localized beneath the cell-associated extracellular virus (CEV), detaches from kinesin-1, and shifts to signaling actin polymerization following tyrosine phosphorylation by Src and Abl family kinases. 27 Phosphorylation on Y112 and Y132 residues in A36R generates binding sites for the Src homology 2 (SH2)—domains of Nck and Grb2 adaptor proteins, respectively, that functionally attach to the actin binding proteins WIP and N-WASP. 24 N-WASP stimulates the nucleation of actin filaments making connections with actin and the Arp2/3 complex, a seven-subunit complex that drives dynamic actin assembly during migration, adhesion, or vesicle trafficking.28-30
These studies make it prominent that blocking the A36R protein of poxviruses prevents the actin polymerization thus warding off the viral migration, adhesion, and vesicle trafficking to the host cell. All the viruses of the genus Orthopoxvirus encompass A36R protein functioning in the same manner. 24 Therefore, to prevent the actin polymerization of the host cell the A36R protein of MPXV is thought to be a more promising drug target.
Peptides have evolved as extremely selective and potent signaling molecules that bind to specified cell surface receptors, or ion channels to exert powerful physiological effects. Their potential pharmacological profile and inherent features represent an exceptional starting point for the development of advanced therapeutics. 31 As a drug, peptides are prominent for their exorbitant affinity, specificity to a respective target and efficacy, at the same time, safety and tolerability in human that estimate assorted supremacy over small molecules.32-35 In 2011, US Food and Drug Administration (FDA) corroborated peptide-based drug against the Hepatitis C virus and multiple peptides are experiencing phase trials targeting Influenza virus, Hepatitis B and D virus. Peptide therapeutics against Chikungunya virus, Zika virus, and Dengue virus are exhibiting antiviral activity. 36
There was no authorized treatment against human MPXV before 2019. Dryvax, a smallpox vaccine, was practiced for both smallpox and monkeypox treatment, 37 developed various influential adverse effects in vaccinated people and persons in contact with the vaccine.38,39 Later information revealed that the prime mode of protection against MPX managed by the existing non-attenuated smallpox immunization is interfered by Abs. 40 Considering drawbacks of the current treatment, efforts were made to identify plausible small molecule as well peptide-based therapeutic agents against the A36R protein of MPXV conducting virtual screening strategies combined with molecular docking.
Materials and Methods
Methodological overview is demonstrated in Figure 1.

Methodological overview.
Peptide Preparation
Peptide retrieving
Potential 3459 antiviral peptides (AVPs) were retrieved from the SATPdb v1.0. 41 Among the 3459 AVPs, 331 sequences were excluded as those were in modified forms. Similarly, 2042 AVPs were extracted from the data repository of antimicrobial peptides (DRAMP v3.0). 42 From 3128 and 2042 AVP sequences, unique AVP set was created using Venn diagram43,44 for further analysis.
Peptide screening based on physicochemical properties
Physical and chemical parameters of the curated AVP sequences were determined using Expasy’s Protparam 45 in Linux operating system. Peptide filtering criteria set in accordance with physicochemical frameworks are shown in Table 1. Accordingly, the AVPs in the rage of 5 to 22 amino acids in length were selected. The peptides having instability index value over 40, and in vivo or in vitro half-lives less than 16 h, were removed. In addition, peptides having aliphatic index value ranging from 71.13 to 143.54 were kept in record. Also, positive grand average of hydropathicity (GRAVY) value containing AVPs were eliminated.
Peptide filtering criteria based on length, instability index, aliphatic index, half-life, and grand average of hydropathicity (GRAVY).

In silico structural modeling and validation
From amino acid sequences, 3 dimensional (3D) structures were modeled using the bio tools PEP-FOLD 3, 53 and predicted structures were validated using structural analysis and verification server (SAVES v6.0) and protein structure analysis (ProSA-web) program. 54
Energy minimization of the modeled structure
From the 3D structures, water molecules were removed and hydrogen atoms were added using AutoDock tools (Version 1.5.6). 55 The energy of the final cleaned structures was minimized by implementing Swiss-PdbViewer v4.1.0. 56
Antigenicity and allergenicity prediction
To determine allergenicity like antigenicity and immunogenicity of AVPs, AllergenFP v1.0 57 and AllerTOP v2.0 58 tools were used. Toxicity of the peptides were determined using ToxinPred bio tools. 59
Protein Preparation
Retrieval of A36R protein sequences and multiple sequence alignment
The monkeypox A36R protein sequences were retrieved from the NCBI protein database. 60 Molecular Evolutionary Genetics Analysis (MEGA v11) software 61 was applied to perform multiple sequence alignment (MSA) of the protein sequences. MUSCLE algorithm with UPGMA cluster method was used for MSA.
3D structure prediction of the target receptor
The 3D structure of the monkeypox virus A36R protein (NCBI GenBank ID: QJQ40287.1) was developed using two different bioinformatics tools, namely, Contact-guided Iterative Threading ASSEmbly Refinement (C-I-TASSER) 62 and RaptorX63-65 as no crystal structure of the A36R protein was found in the protein data bank (PDB) and UniProt. Homology modeling of A36R protein was also conducted and the sequence data of A36R protein was compared with another protein having both a known sequence and a known structure (PDB hit: 1sl6A). The highest similarity identity to a known protein template is 16.67%, a value less than 40%, so A36R protein structure could not be built using homology modeling. Therefore, ab initio 3D structure was modeled using C-I-TASSER and RaptorX servers.
Refinement and validation of the modeled 3D structure of the target receptor
The predicted PDB format of the monkeypox virus A36R protein was refined exploiting 3Drefine bio tools, 66 and the refined structures were further rectified taking advantage of GalaxyWEB server.67,68
Validation of the modeled 3D structure of the target receptor
The refined PDB model of the A36R protein was validated by structural analysis and verification server (SAVES v6.0) and MolProbity. 69 ERRAT, 70 Ramachandran Plot,71,72 and overall G-factors71,72 from SAVES v6.0 server were used to measure the maximum quality of the predicted structures.
Energy minimization of the target receptor
Water molecules of the refined A36R protein were deleted to avoid untoward molecular interaction, and hydrogen atoms were attached using MGL Tools (Version 1.5.6). The energy minimization of A36R protein was performed using Avogadro software 73 with the support of MMFF94 force field, setting up 500 steps, steepest descent algorithm and 10e-7 convergence, respectively. The minimized protein 3D structure was used for molecular docking and dynamics simulation.
Prediction of active sites
The ligand-binding pocket, ie, inhibitory site of the A36R protein, was determined applying the Computed Atlas for Surface Topography of Proteins (CASTp 3.0) (Figure 2A), 74 DoGSiteScorer (Figure 2B), 75 and DeepSite (Figure 2C), 76 respectively. AutoDock Vina77,78 was also used to generate the grid box size 30, 30, 30 for x, y, z axes, grid center −38.4 × 54.2 × 0.2, respectively, and 0.375 Å spacing.
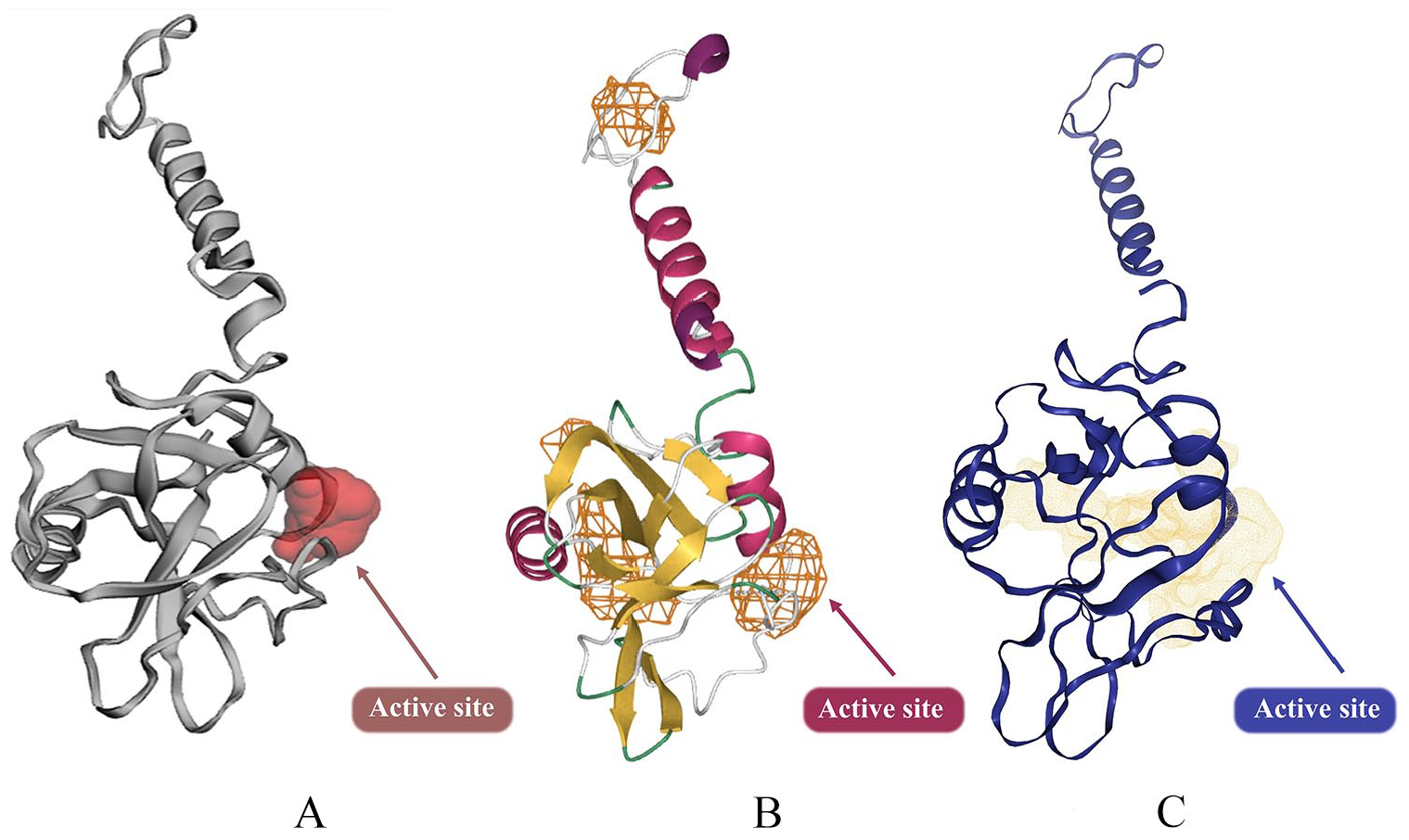
3D structure of A36R and its predicted active site: (A) Showing the inhibitory site by using CASTp, (B) DeepSite, and (C) DoGSiteScorer. CASTp indicates Computed Atlas for Surface Topography of Proteins.
Molecular docking
The PDB structure of the A36R protein as the target receptor and the peptides as the ligands were provided as input in the DINC 2.0 server,79-82 which is a parallelized meta-docking method for the incremental docking of large ligands (using AutoDock Vina).
Virtual screening of A36R protein
TACC, a virtual drug discovery portal, was used to conduct virtual screening. The AutoDock Vina77,78 application recommended that our target structure A36R be imported in PDB format and subsequently converted to PDBQT format. Aside from the target structure, the center and size of the search space, as well as the parameters of the designated virtual libraries for screening from the set of accessible compound libraries, were also provided for the initiation of the virtual screening experiment. The grid box’s dimensions and coordinates were center x = −38.4, center y = 54.2, center z = 0.2, size x = 30, size y = 30, and size z = 30. The exhaustiveness was adjusted to 10 for molecular docking simulations. In high-throughput virtual screening of the A36R, the ZINC-in-trials (9270 molecules), ZINC-fragments (546 003 fragments), and Enamine-HTSC (3 467 770 molecules) libraries were employed. The ZINC library in-tails, on the other hand, contains medications that are now being tested in clinical trials. 83
Pharmacokinetic profile (ADMET) evaluation of the hit compounds from virtual screening
A preliminary predictive in silico pharmacokinetic study was conducted with ADMETlab 2.0, 84 which includes absorption (Papp Caco-2 permeability, human intestinal absorption), distribution (plasma protein binding [PPB], blood-brain barrier[BBB]), metabolism (P450 CYP3A4 inhibitor, P450 CYP3A4 substrate, P450 CYP2 C9 inhibitor), elimination (half-life time [T1/2], clearance rate [CL]), and toxicity (hERG toxicity, AMES toxicity, hepatotoxicity, respiratory toxicity).
Molecular dynamics (MD) simulation
To appraise the ligand’s effectiveness in suppressing A36R of the monkeypox virus, MD simulations of the protein–ligand (drug molecule) complex were undertaken. We conducted the MD simulation as described in our earlier work and few other studies.85,86 GROMACS (v2021) 87 with the company of the CHARMM General Force Field (CGenFF) 88 following energy minimization was used to accomplish the MD simulation. To develop the system, an in-house python script was used which used Chimera 89 dock prep functionality to cleanse the protein and to attach missing residues exploiting the Dunbrack rotamer library. Hydrogens were physically put into the protein employing the GROMACS pdb2gmx function, whereas hydrogens were manually placed into the ligand using custom force field settings. The GROMACS tool was imposed to create the protein topology for A36R, and parameterization of the ligand topology was achieved by applying the LigParGen program. 90 The LigParGen bio tool accomplished optimization using the CGenFF, and the charge model (CM1A) parameters were also developed. The system was put in the center of a cubic box of a TIP3P water model with a minimum distance of 1.0 nm between the wall and any component of the protein set up before the start of the experiment. Normalization of the solvated system was done by adding Na+ (sodium) and Cl– (chloride) to a 0.15 M ionic strength aqueous solution. Canonical (NVT) and constant-temperature, constant-pressure (NPT) ensemble were used to perform system equilibration for 100 picoseconds (ps). The trajectories were also constructed in every 2 femtoseconds (fs) and recorded every 1 ps. The minimized system was progressively heated to appropriate temperatures for 100 ps within an isothermal ensemble by soft coupling with the Berendsen thermostat (NVT). 91 To avoid drift away from the protein during equilibration, ligand location limitations were applied before running NVT simulations. All bonds were restricted using the LINCS algorithm. 92 Periodic boundary conditions (PBC) were applied with a constant number of particles in the system, constant pressure, and constant temperature simulation requirements (NPT). The NPT ensemble was executed at a temperature of 300 K. The system was combined with a Parrinello-Rahman barostat 93 for equilibrating at 1 bar pressure for 100 ps. The Particle Mesh Ewald (PME) method was used to process the electrostatic interactions. With a cutoff of 1.0 nm, the short-range van der Waals cutoff (rvdW) interactions were calculated. Production simulations with limitations were run for 10 ns. The LINCS algorithm was taken to limit all bond lengths. 92 DELL workstation with Intel Xenon processor (10 C, 20 T), 256 GB RAM; running Fedora (v31) Linux operating system was employed to run MD. The root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), hydrogen bond (H-bond), solvent accessible surface area (SASA) (using van der Waals Volumes and Radii), and distance analysis were computed using Gromacs. 94 Trajectory plots and figures were developed to exploit an in-house python script with Matplotlib 95 and NumPy 96 library as well as implementing R version 3.6.3 97 and Peptides library. 98
Result
Peptide selection
We obtained 4893 AVP sequences for scrutiny by Expasy’s Protparam (Proteomics Protoc. Handb., 2005), after excluding 277 common AVP set from either database and 331 revised versions from SATPdb. Among 4893 AVPs, 57 sequences exceeded the selection benchmarks of length, instability index, aliphatic index, half-life, and grand average of hydrophobicity, respectively (Supplementary Table A.1). This selection procedure had a high resemblance with the peptide filtering technology reported by Chang and Yang, 2013. 51
Validation analysis of the predicted 3D structures
Five crystal structures were received against each insertion of sequences and overall 285 3D models were attained from PEP-FOLD 3 bio tools. 53 After implementing 5 models individually to SAVES v6.0 server and ProSA-web program, 54 1 model was validated because of showing maximum percentage of residues in the most favored region of Ramachandran Plot and exhibited significant ERRAT score, weighty G-factors and notable ProSA-web Z score. Thus, 57 models were finalized those revealed the highest ERRAT score, greater core region of Ramachandran plot, and appropriate G-factors and Z-score, respectively (Table 2).
Peptide structures validation through SAVES v6.0 result and ProSA-web Z-Score.
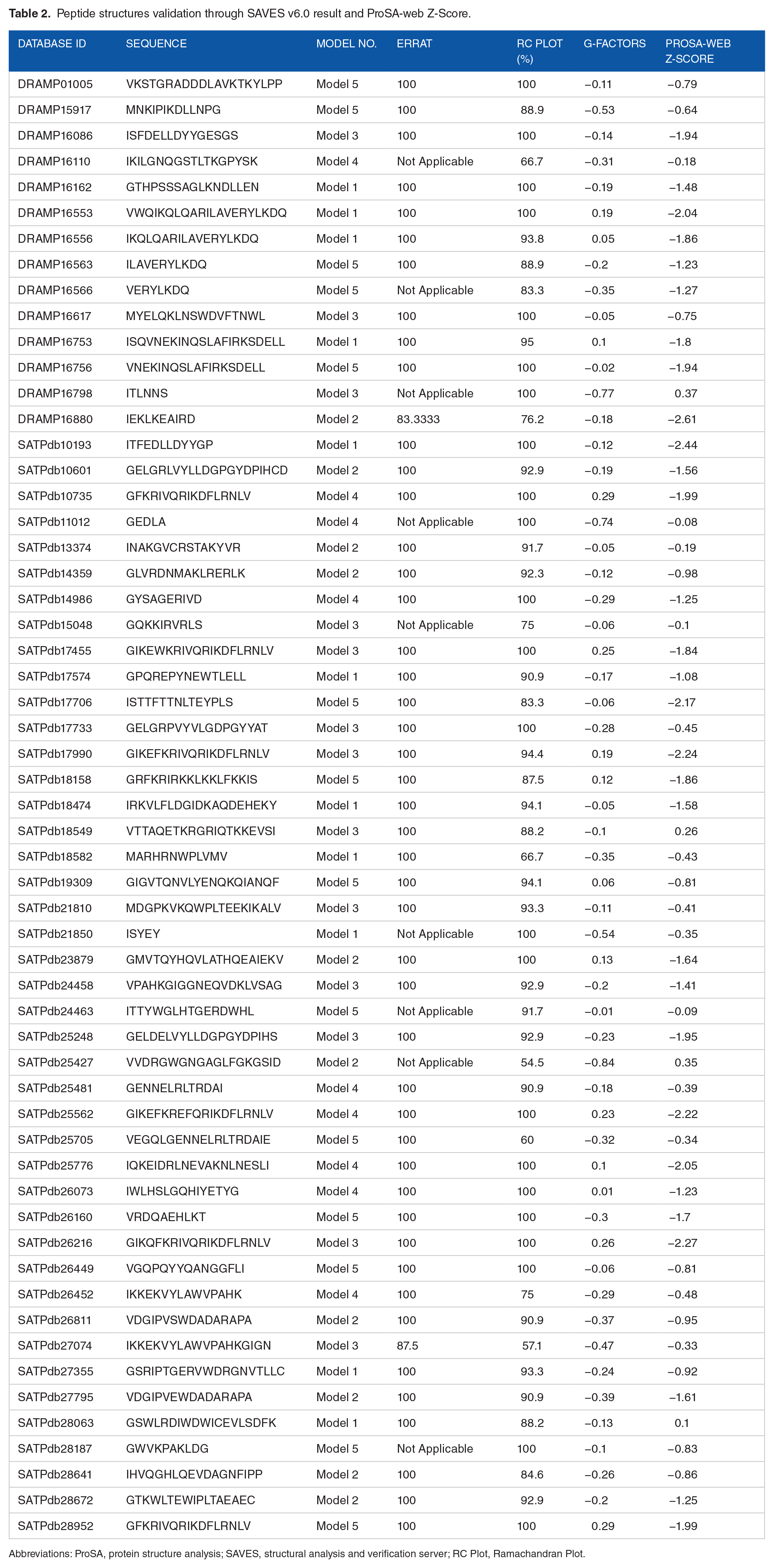
Abbreviations: ProSA, protein structure analysis; SAVES, structural analysis and verification server; RC Plot, Ramachandran Plot.
Multiple sequence alignment analysis of central and western strain of monkeypox virus (MPV) A36R protein
Total 31 monkeypox virus A36R protein sequences of both West African and Central African strain were retrieved from NCBI protein database. 60 The sequences comprised 168 amino acids in length. MSA (Supplementary Fig. A.1) execution of 31 MPXV A36R protein sequences exhibited mutation in the 3 sequences at 1 point (glycine instead of arginine) compared with others and these mutated sequences carrying 11 patients were isolated in Central Africa and named Zaire 96-I-96 strain or Monkeypox virus Z1 strain. 99 Therefore, we excluded mutation containing 3 sequences (Accession: AAL40604.1; NP_536573.1 and CAC69954.1) and selected West African strain as our final A36R protein sequence. Finally, NCBI GenBank ID holding QJQ40287.1 sequence 100 from West African strain was chosen to predict 3D structure.
Structural modeling and validation analysis of monkeypox virus (MPV) A36R protein
Predicted 5 models of C-I-TASSER generated 25 refined 3D structures after employing 3Drefine bio tools, and 1 model among 25 was found as the finest because it displayed optimum residues in acceptable region of RC Plot and particular quality factor (ERRAT) compared with others. Further refinement was performed inserting this finest model to GalaxyWEB program which developed 5 more 3D rectified structures. The superior model out of five 3D rectified structures was certified based on SAVES v6.0 outcome and MolProbity statistics. According to SAVES v6.0 outcome, the utmost model of the C-I-TASSER showed 86.1% residues in the most key zone of Ramachandran plot with estimating G-factors value −0.06, exhibited ERRAT score 93.08, and demonstrated 75.60% of Verify3D, respectively. Summary statistics of MolProbity demonstrated 89.76% residues in the satisfactory zone of RC plot for the best model of C-I-TASSER server (Supplementary Table A.3). Similar approach was implemented to establish the top model from the estimated 5 models of RaptorX server. RC plot for the best refined model of RaptorX server showed 87.3% of residues in most preferred area with G-factors value 0.04, exhibited overall quality factor (ERRAT) 94.90, and fewer than 80% of the amino acids have scored ⩾0.2 in the 3D/1D profile. Also, MolProbity statistics displayed 95.18% residues in the preferred region of RC plot for the finest model of RaptorX server (Supplementary Table A.3). Table 3, Supplementary Table A.3 and Supplementary Fig. A.2 reveal validation statistics which indicated that RaptorX server predicted better A36R protein model of MPXV than C-I-TASSER program and can be used for further peptide-based molecular docking analysis.
Ramachandran plot statistics, ERRAT, and Verify3D scores for the A36R protein generated models using C-I-TASSER and RaptorX server.

Abbreviation: C-I-TASSER, Contact-guided Iterative Threading ASSEmbly Refinement.
Ramachandran plot: Residues in most favored regions [A, B, L].
Ramachandran plot: Residues in additional allowed regions [a, b, l, p].
Ramachandran plot: Residues in generously allowed regions [∼a, ∼b, ∼l, ∼p].
Ramachandran plot: Residues in disallowed regions.
Ramachandran plot: Overall G-factors.
Overall quality factor generated by ERRAT server.
Averaged 3D-1D score generated by Verify3D server.
Active site analysis of A36R protein
CASTp, DoGSiteScorer and DeepSite bio tools identified the active groove of the A36R protein of MPXV. There was detection around 12 active points of the A36R protein; those are PRO85, ASP86, THR87, ARG88, LEU90, ARG91, PHE94, ILE121, ASP122, ILE123, SER124, and TYR144 (Supplementary Fig. A.3). These amino acids of the A36R protein were meticulously evaluated to analyze molecular docking.
Molecular docking analysis of A36R against AVPs
All 57 models were used to dock against the A36R protein of MPXV. The docking energy from each peptide is tabulated in Supplementary Table S2. The most conducive binding scores of 10 peptide models were recorded in Table 4. Furthermore, we selected the best 3 favorable peptides among the 10 peptides for non-bonded structure analysis. The docking scores for top 3 peptide models were −6.10, −6.10 and −6.30 kcal/mol, respectively, for SATPdb10193, SATPdb21850, and SATPdb26811 peptides.
The binding energy of the top 10 peptide molecules. The more negative energy signifies the favorable binding.
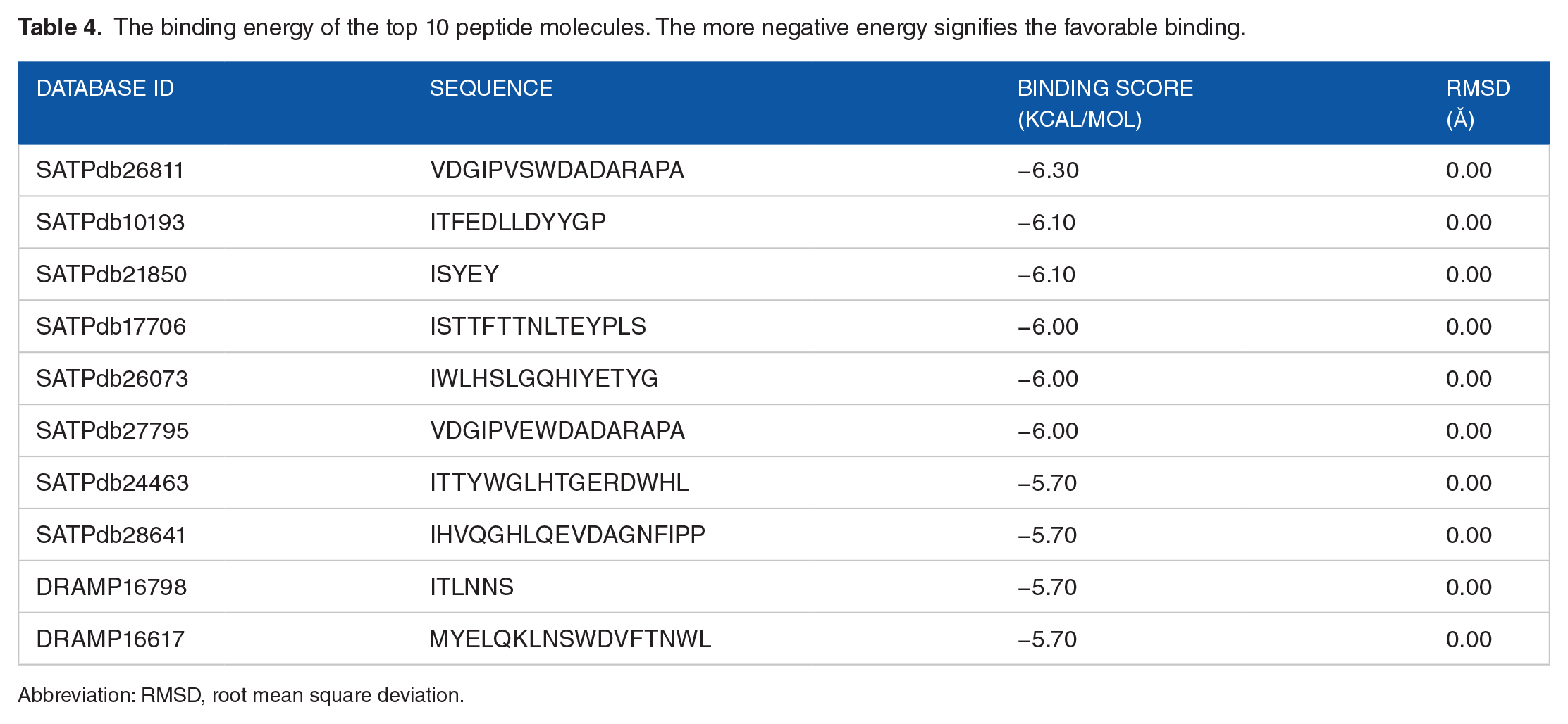
Abbreviation: RMSD, root mean square deviation.
Docking interaction between A36R and AVPs
The non-bonded interactions between the peptides and the A36R protein of MPXV are shown in Figure 3. The SATPDb10193 peptide formed 6 hydrogen bonds with the A36R protein of MPXV at THR87, ARG91, SER124, ARG88, TYR10, and ARG88 and each of the amino acid residues had bond distance around 3.0 Å. This interaction was also stabilized by 2 electrostatic bonds at ARG88, and ASP8, 7 hydrophobic bonds at TYR54, VAL92, HIS89, PHE94, VAL92, LYS119, and ILE121, and 4 unfavorable bonds at GLU4, ASP5, ASP8, and PRO12 (Figure 3 and Table 5). The SATPdb21850 peptide and the A36R protein interaction created 6 hydrogen bonds at ASP122, SER124, GLY147, THR87, ASP120, and ILE121 showing bond distance about 2.5 Å. This complex additionally generated 1 electrostatic bond at GLU4, 2 hydrophobic bonds at ARG91 and PHE94, and 4 unfavorable bonds at TYR5, GLU4, ARG91, and SER124 (Figure 3 and Table 5). The SATPdb26811 and the A36R protein complexation developed 10 hydrogen bonds at THR87, LYS125, ASP55, THR127, ASP122, ARG88, SER124, LYS125, and ILE123 securing approximate bond distance 3.0 Å. The SATPdb26811 and the A36R protein were also bound by 6 hydrophobic bonds at ARG91, VAL92, LEU90, LEU126, HIS89, and ILE121, and 4 unfavorable bonds at ARG88, LYS148, ASP55, and ASP122 (Figure 3 and Table 5).
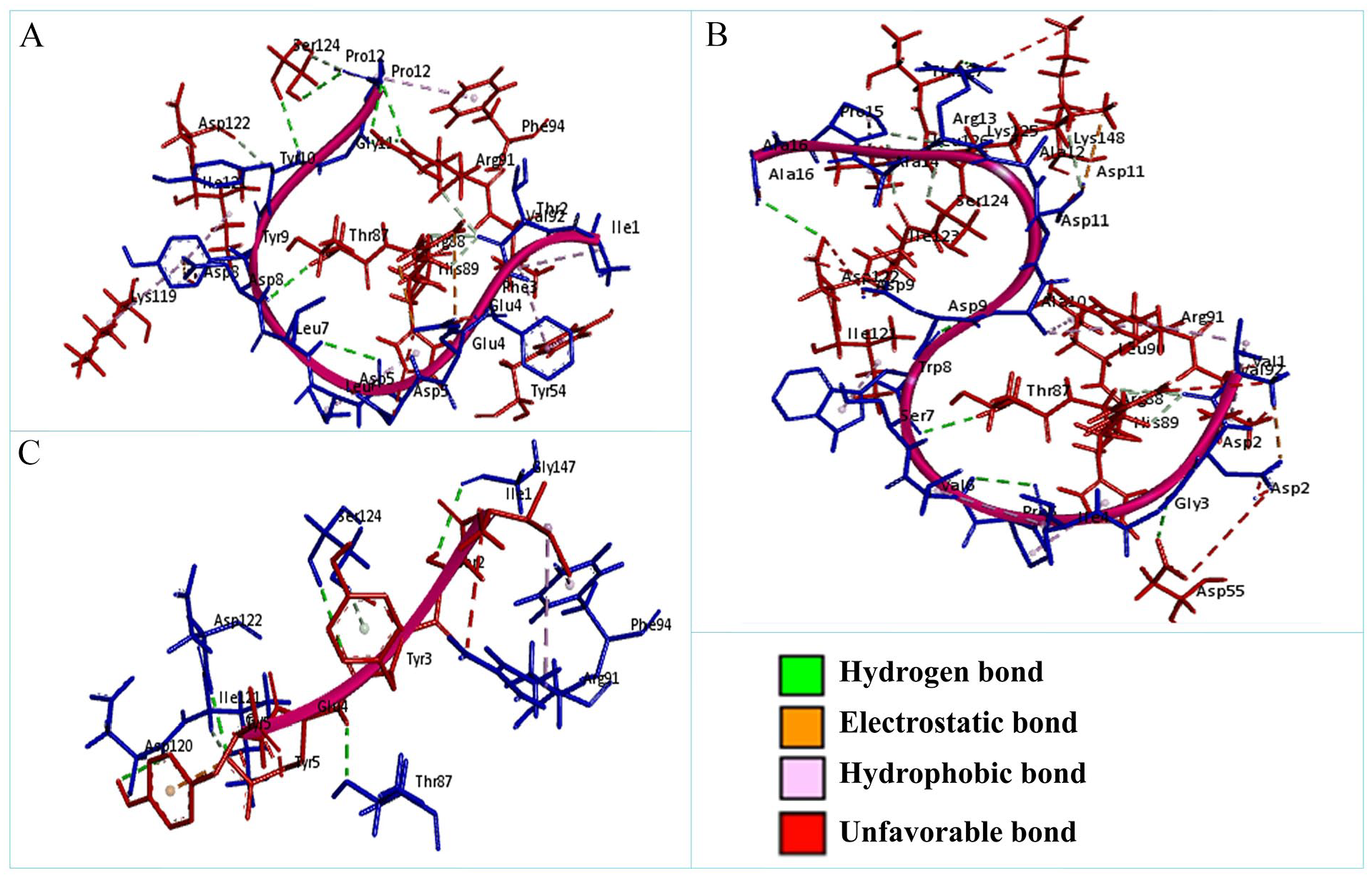
The non-bonded interaction of the SATPdb10193, SATPdb21850, SATPdb26811 peptides and the A36R protein of MPXV. Here, (A), (B), and (C) delineate the binding interactions of the SATPdb10193, SATPdb21850, and SATPdb26811 peptide molecules with the A36R protein of MPXV after 0 ns time. MPXV indicates monkeypox virus.
Non-bonded interactions of the SATPdb10193, SATPdb21850, and SATPdb26811 peptides with the A36R protein of MPXV.

Abbreviation: MPXV, monkeypox virus.
Virtual screening analysis of A36R against ZINC trial, ZINC fragments, and Enamine HTSC databases
Virtual screening against 3 ligand databases was used to look for specific interactions with the protein A36R (ZINC Trial, ZINC Fragments, and Enamine HTSC databases). Via virtual screening, a list of hits was formed, which was subsequently ordered by docking score (Figure 4). A lower score indicates that the ligand and the target protein have more favorable energy interactions. The top 1000 ligands with the strongest binding affinity for the therapeutic target protein A36R were reported in each drug library evaluated. The highest binding affinities were identified in ZINC Fragments (−6.9 to −7.8 kcal/mol), while the lowest binding affinities were found in the Enamine HTSC library (−8.8 to −9.8 kcal/mol) based on the docking scores. The top 100 compounds from the 3 libraries were considered for further physicochemical features and drug likeliness evaluation based on their binding affinities against A36R. The top 100 compounds had docking scores ranging from −9.3 to −9.8 kcal/mol, and all of the molecules were obtained from the Enamine HTSC database.

Drugs according to their binding affinities from virtual screening analysis: (A) Docking scores of top 1000 drugs from ZINC Trial library ranging from −9.2 to −7.1 kcal/mol, (B) Binding affinities of top 1000 drugs from ZINC Fragments compound library ranging from −7.8 to −6.9 kcal/mol (C) Affinities of top 1000 drugs obtained from Enamine HTSC database ranging from −9.8 to −8.8 kcal/mol.
Pharmacokinetics (ADMET) characteristics analysis of top virtually screened molecules
Early estimation of physiologically based pharmacokinetic parameters such as absorption, distribution, metabolism, excretion, and toxicity (ADMET) have become a significant factor in minimizing the likeliness of clinical therapeutic failure. 101 The pharmacokinetics (ADMET) and drug likeliness qualities of all 100 medicines were examined, and the top 3 compounds for inhibiting the A36R protein of the monkeypox virus were screened. Two compounds (Z55287118 and Z97653013) exhibited outstanding Caco-2 Permeability (>−5.15 cm/s) and all 3 drugs had strong human intestine absorption (HIA) characteristics, meeting the optimum HIA value of ⩾30%, according to ADMET evaluation (Table 6). Although neither drug was able to pass the blood-brain barrier (BBB), the 3 medicines did show considerable plasma protein binding (⩽90% optimum). They were also reported to be non-inhibitors of most cytochrome P450 enzymes, non-respiratory harmful, non-carcinogenic, non-corrosive, and non-irritating to the eyes. On the other hand, only Z97653013 was shown to be non-AMES toxic. All 3 compounds were orally bioavailable (Table 6, Figure 5A), which is an important consideration in the development of oral medicines. The 3 medicines exhibit moderate clearance (2.399, 1.881, and 7.616 mL/min/kg), despite their lack of a decent T1/2 value. Despite this, the molecular weights of the compounds ranged from 483.160 to 356.120 g/mol, and the LogP values were determined to be within the ideal range of 1 to 5. The Polar surface area (TPSA) was also determined to be below the <140 Å2 threshold, and all 3 agents followed the Lipinski Rule of Five.
Pharmacokinetic (ADMET) and drug likeliness evaluation of hit virtually screened drugs.

Abbreviations: ADMET, absorption, distribution, metabolism, excretion, and toxicity; BBB, blood-brain barrier; CL, clearance rate; HIA, human intestine absorption; PPB, plasma protein binding; TPSA, total polar surface area.

Bioavailability radar, binding modes, molecular interaction, and Trajectory plots of MD simulation of the top hit compounds: (A) Oral-bioavailability profile of 3 hit ligands, where the yellow area indicates the upper limit, pink area denotes lower limit, and the blue lines signifies our compound properties; (B) hit ligands’ binding modes with receptor; (C) interactions of the hit compounds with receptor amino acid residues. The color bars represent the type of interaction formed between ligand and the receptor. (D) Plot of root mean square deviation (RMSD) (first column panel), root mean square fluctuation (RMSF) (second column panel), solvent accessible surface area (SASA) with respect to time (picoseconds) (third column panel), and radius of gyration (Rg) (fourth column panel) during MD simulation of A36Rreceptor in complex with the hit ligand Z55287118(red color) and unbound protein (green color). MD indicates molecular dynamic.
Molecular interactions of the selected top drug candidates with A36R protein
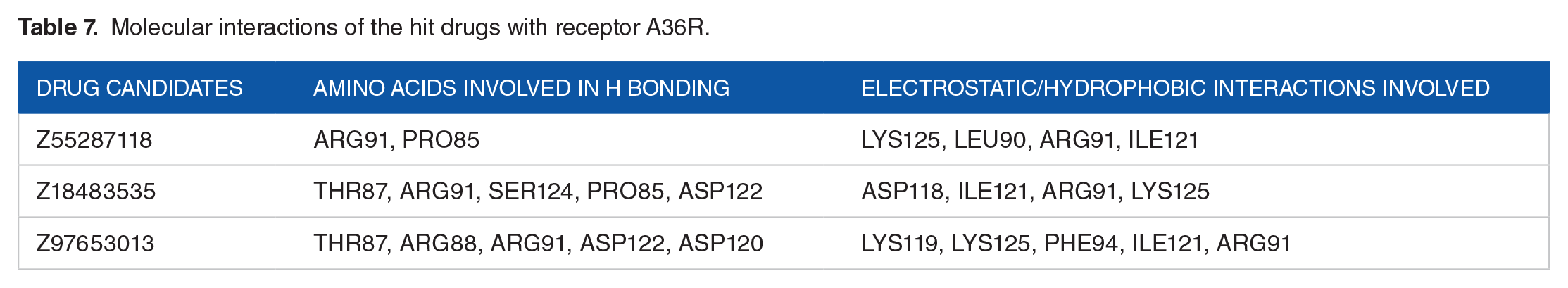
The docking interactions of the hit ligands (Z55287118, Z18483535,and Z97653013) with the A36R were visualized in detail with BIOVIA Discovery Studio 2021 (Table 7, Figure 5B and C). The molecules formed van der Waals interaction, conventional hydrogen bonds, and carbon hydrogen bonds with ARG91, PRO85, ARG91, SER124, ASP12, THR87, ARG88, ARG91, ASP122, and ASP120, whereas unfavorable acceptor-acceptor, pi-alkyl, and pi-anion interactions were generated with amino acids LYS125, LEU90, ARG91, ILE121, ASP118, LYS119, LYS125, PHE94, and ARG91, respectively.
Molecular interactions of the hit drugs with receptor A36R.
Molecular dynamics simulation analysis of the top hit compounds
The top-ranking drug Z55287118 and the 3 peptides SATPdb10193, SATPdb21850, and SATPdb26811 had the best docking affinity and binding free energy; therefore, they were considered for molecular dynamics (MD) simulation to determine their binding kinetics and stability with the A36R receptors. The energy minimization, NVT, NPT equilibration methodologies were used to simulate receptor-drug complexes using the CHARMM36 force field. The RMSD (after lsq fit) stability of the protein-drug and protein-peptide complexes demonstrated that after 2 ns of simulation, the system was consistently stabilized and inclined to remain in the plateau phase for the remainder of the time (Figure 5D, first column panel and Figure 6, first column panel). While the protein grew up quickly, the RMSD value remained steady in the range of 0.2 to 0.25 nm and 1.0 to 1.5 nm for drug and peptide, respectively. The protein-drug and protein-peptide complexes have the same RMSD pattern as the protein alone. The complexed RMSD was between 0.2 and 0.25 nm for protein-drug and 1.0 and 1.5 nm for protein-peptide. This indicates that the structures of the protein-ligand complex have already achieved a state of relative equilibrium. The complex RMSF gives each atom’s variance during the simulation (Figure 5D, second column panel and Figure 6, second column panel). The findings revealed that the binding site residues were less fluctuant for protein-drug and protein-peptide complexes, except SATPdb10193. On average, the RMSF values were 0.4 and 0.6 nm for protein-drug and protein-peptide, respectively. A protein’s Solvent Accessible Surface Area (SASA) is a characterization of the surface area accessibility of the complex during simulation (Figure 5D, third column panel and Figure 6, third column panel). The graph illustrates how SASA for protein-drug as well as protein-peptide complexes has changed throughout time. The drug-protein complex’s SASA is lower than the proteins on its own. It means the protein is attached to the ligand. Nevertheless, for peptide-protein complex, the SASA scores were higher than the proteins. When the protein structure is exposed to solvent, it partially unfolds, as seen by the small increase in SASA values with time. SASA levels that are reduced when medications attach to receptors, on the other hand, suggest that the protein has shrunk. The radius of gyration (Rg) was estimated as a measure of structural compactness (Figure 5D, fourth column panel and Figure 6, fourth column panel). The Rg value will most likely remain consistent if the protein is folded correctly. If the protein unfolds, Rg levels will change over time. The average Rg of the protein-ligand and protein-peptide complexes was 2.0 nm, and it remained fairly stable throughout time, excluding SATPdb10193. The Rg figure shows a small decline in the total Rg value of the protein when docked with the drug and peptides, indicating that receptors in conjunction with the medicines were compactly packed and displayed stable folding. All of the data from the MD simulation of a chosen docked structure confirmed the stability of protein-ligand complex, and for protein-peptide complexes, SATPdb21850 and SATPdb26811 showed better stability.

Trajectory plots of MD simulations: Plot of root mean square deviation (RMSD) (first column panel), root mean square fluctuation (RMSF) (second column panel), solvent accessible surface area (SASA) with respect to time (picoseconds) (third column panel), radius of gyration (Rg) (fourth column panel), and hydrogen bond (fifth column panel) during MD simulation of A36R in complex with peptides SATPdb10193, SATPdb21850, SATPdb26811 (red color), or unbound protein A36R (blue color). MD indicates molecular dynamic.
Allergenicity and toxicity prediction
The SATPdb10193 and SATPdb26811 peptides were characterized as probable non-allergen, whereas SATPdb21850 was identified as probable allergen by AllerTOP v. 2.0 bio tools. On the other hand, the SATPdb10193, SATPdb21850, and SATPdb26811 peptide molecules were non-allergic and non-toxic which were detected by AllergenFP v.1.0 and ToxinPred tools, respectively (Table 8).
The allergic and toxicity profile of the top three peptide molecules.

Discussion
Monkeypox virus has been focused by national and international media, public and scientific attention after its recent emergence reported on May 7, 2022 in the United Kingdom, 102 has created a pandemic situation because of its onward spread around the globe. Although different suggested drugs and vaccines are there, however, monkeypox infection is still challenging because no effective drugs or vaccines have been found that mitigate the risk of MPX infection significantly. For that, there is a necessity for rapid and effective development of active antiviral agents against MPXV. As conventional drug development procedures are time-consuming and costly, in silico–based drug design approaches were conducted using molecular docking that may promote to find out the new therapeutic agent against MPXV due to its fast and on target screening proficiency from a broad small molecule library or peptide database.103-105
The A36R protein of MPXV plays significant role for the polymerization of actin tail beneath the CEV that moves the virus into the neighboring cell or the virus is freed as an EEV. Blocking the A36R protein of poxviruses inhibits the actin polymerization; consequently, viral migration, adhesion, and vesicle trafficking to the host cell are thwarted. AVPs are thought as leading component that directly target the viral proteins or inhibit the development of viruses by targeting its specific regions or components. 36
Our study shows that most of the selected AVPs have strong interaction with the A36R protein. It was observed that their docking scores ranging from −4.10 to −6.30 kcal/mol (Supplementary Table A.2). However, we focused on the SATPdb10193, SATPdb21850, and SATPdb26811 peptides based on docking scores for further analysis because these AVPs exhibited the higher binding affinity (–6.10, −6.10 and −6.30 kcal/mol, respectively) compared with other AVPs. The SATPdb10193 peptide had several hydrogen, electrostatic, and hydrophobic binding interactions in the catalytic residues at THR87, ARG91, SER124, ARG88, PHE94, and ILE121. These interactions at the active groove of the protein may be responsible for the higher binding energy. Researcher suggests that hydrogen, electrostatic, and hydrophobic interaction at the active site of the target protein may involve to possible inhibition of the protein. 106 Similarly, multiple hydrogen and hydrophobic interaction in the active cavity residues at ASP122, SER124, THR87, ILE121, ARG91, and PHE94 may be responsible for the tight binding and better affinity which indicates possible blockage of the A36R protein by SATPdb21850. Moreover, the SATPdb26811 ligand showed the highest binding energy −6.30 kcal/mol while interacting with the A36R protein of MPXV. This might be result of more hydrogen and hydrophobic bonds with active site residues at THR87, ASP122, ARG88, SER124, ILE123, ARG91, LEU90, and ILE121.
A virtual screening evaluation of the A36R protein was also performed against the ZINC Trial library, ZINC Fragments library, and Enamine HTSC database by using Texas Advanced Computing Center’s Drug Discovery portal (TACC). Despite the fact that they offered the top 1000 molecules (Figure 4) from each chemical library, we chose the top 100 based on the greatest binding affinities, with the majority of the medications from Enamine HTSC having the highest binding affinities to A36R.
The projected spike in the number of commercialized new medicines has not occurred in comparison with the number of novel drugs created each year, which has been attributed to a number of factors, including the candidate products’ poor pharmacokinetic (PK) properties.107,108 As a result, effective absorption, distribution, metabolism, elimination/excretion, and toxicity (ADMET) screening criteria are required.101,109 All of the top 100 medications from the virtual screening study undergone ADMET analysis and were filtered out based on their ADMET characteristics. After screening, 3 (Z55287118, Z18483535, and Z97653013) of the 100 virtually screened compounds were eventually certified. The certified Z55287118, Z18483535, and Z97653013 molecules formed potent hydrogen, electrostatic, and hydrophobic bonds with pocket residues at ARG88, ARG91, PRO85, LEU90, ILE121, THR87, SER124, and ASP122 (Table 7). In addition, it is indisputable that Caco-2 permeability and Human Intestinal Absorption (HIA) are 2 of the most important indicators for drug absorption, and 2 of the compounds (Z55287118 and Z97653013), as well as all 3 molecules (Table 6), were shown to have satisfactory Caco-2 permeability and HIA, respectively. When the distribution properties (plasma protein binding and blood-brain barrier permeability) were investigated, almost all drugs had acceptable distribution throughout the body and conformed to the optimum values. A great oral medication should never block cytochrome p450 enzymes and be non-toxic. The majority of the compounds, on the other hand, showed good cytochrome p450 inhibition and toxicity profiles (have no Ames toxicity, non-hERG Blockers, non-respiratory toxic, and non-carcinogenic, non-irritating, and non-corrosive to the eyes). Despite this, all drugs demonstrated sufficient ADMET properties to be deemed therapeutic candidates.
As per Walters and Murcko, 110 drug-like compounds are molecules with functional groups and physical properties that are comparable to the majority of established therapeutics, and so may be regarded as compounds that are pharmacologically active or have therapeutic potential. According to Lipinski et al, 111 the compounds are “drug-like” if they show significant acceptable ADMET properties and pass Phase I clinical studies. As a first-step screening strategy, the rule of five has been frequently used to assess the druggability of new pharmaceutical substances. Oral bioavailability is determined by the logP value and total polar surface area (TPSA), and it is important to highlight that all of the drugs were orally bioavailable (Table 6). Furthermore, all of the drugs adhered to the Lipinski rule of five as well.
In this context, we find that AVPs SATPdb10193, SATPdb21850, and SATPdb26811 as well as small molecule compounds Z55287118, Z18483535, and Z97653013 have the potentiality to block the function of the A36R protein, and such findings can be further evaluated using with additional experiments.
Conclusion
The study was conducted to identify the lead molecules that particularly impede the function of the A36R protein of MPXV. For that, peptide and small molecule databases were used to determine novel drug candidates against the A36R protein. Modeling, validating, docking, and finally MD simulation findings suggest theSATPdb10193, SATPdb21850, and SATPdb26811 peptide molecules have strong interaction with the active point residues of the A36R protein of MPXV. Moreover, molecular docking and simulation results of the small molecule hit compounds demonstrate that Z55287118, Z18483535, and Z97653013 had high affinities with the vital active points of the A36R protein. However, these assessments, additionally future in vivo and in vitro clinical trials, can lead to the coherent and rigorous development of the peptide and molecular based inhibitors by targeting the A36R protein of MPXV.
Supplemental Material
sj-docx-1-bbi-10.1177_11779322221141164 – Supplemental material for Drug and Anti-Viral Peptide Design to Inhibit the Monkeypox Virus by Restricting A36R Protein
Supplemental material, sj-docx-1-bbi-10.1177_11779322221141164 for Drug and Anti-Viral Peptide Design to Inhibit the Monkeypox Virus by Restricting A36R Protein by Mohammad Mohasin Miah, Nuzhat Tabassum, Maliha Afroj Zinnia and Abul Bashar Mir Md. Khademul Islam in Bioinformatics and Biology Insights
Supplemental Material
sj-docx-2-bbi-10.1177_11779322221141164 – Supplemental material for Drug and Anti-Viral Peptide Design to Inhibit the Monkeypox Virus by Restricting A36R Protein
Supplemental material, sj-docx-2-bbi-10.1177_11779322221141164 for Drug and Anti-Viral Peptide Design to Inhibit the Monkeypox Virus by Restricting A36R Protein by Mohammad Mohasin Miah, Nuzhat Tabassum, Maliha Afroj Zinnia and Abul Bashar Mir Md. Khademul Islam in Bioinformatics and Biology Insights
Supplemental Material
sj-docx-3-bbi-10.1177_11779322221141164 – Supplemental material for Drug and Anti-Viral Peptide Design to Inhibit the Monkeypox Virus by Restricting A36R Protein
Supplemental material, sj-docx-3-bbi-10.1177_11779322221141164 for Drug and Anti-Viral Peptide Design to Inhibit the Monkeypox Virus by Restricting A36R Protein by Mohammad Mohasin Miah, Nuzhat Tabassum, Maliha Afroj Zinnia and Abul Bashar Mir Md. Khademul Islam in Bioinformatics and Biology Insights
Footnotes
Acknowledgements
We acknowledge high performance computing facility support from Centre for Bioinformatics Learning Advancement and Systematics Training (cBLAST) and Texas Advanced Computing Center (TACC) for MD analysis. We also acknowledge support of Biomolecular Research Foundation (BMRF), Dhaka, Bangladesh.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
Data and Software Availability
All data added: Tables, figures, and supplementary file and supplementary tables. In this research work, publicly available free mostly online and few offline software/tools were used. Necessary link and reference of the software/tools are provided in the method section.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.