
Abstract
Rhodobacter capsulatus is a purple non-sulfur bacteria widely used as a model organism to study bacterial photosynthesis. It exhibits extensive metabolic activities and demonstrates other distinctive characteristics such as pleomorphism and nitrogen-fixing capability. It can act as a gene transfer agent (GTA). The commercial importance relies on producing polyester polyhydroxyalkanoate (PHA), extracellular nucleic acids, and commercially critical single-cell proteins. These diverse features make the organism an exciting and environmentally and industrially important one to study. This study was aimed to characterize, model, and annotate the function of a hypothetical protein (Accession no. CAA71016.1) of R capsulatus through computational analysis. The urf7 gene encodes the protein. The tertiary structure was predicted through MODELLER and energy minimization and refinement by YASARA Energy Minimization Server and GalaxyRefine tools. Analysis of sequence similarity, evolutionary relationship, and exploration of domain, family, and superfamily inferred that the protein has S-adenosylmethionine (SAM)-dependent methyltransferase activity. This was further verified by active site prediction by CASTp server and molecular docking analysis through Autodock Vina tool and PatchDock server of the predicted tertiary structure of the protein with its ligands (SAM and SAH). Normally, as a part of the gene product of photosynthetic gene cluster (PGC), the established roles of SAM-dependent methyltransferases are bacteriochlorophyll and carotenoid biosynthesis. But the STRING database unveiled its association with NADH-ubiquinone oxidoreductase (Complex I). The assembly and regulation of this Complex I is mediated by the gene products of the nuo operon. As a part of this operon, the urf7 gene encodes SAM-dependent methyltransferase. As a consequence of these findings, it is reasonable to propose that the hypothetical protein of interest in this study is a SAM-dependent methyltransferase associated with bacterial NADH-ubiquinone oxidoreductase assembly. Due to conservation of Complex I from prokaryotes to eukaryotes, R capsulatus can be a model organism of study to understand the common disorders which are linked to the dysfunctions of complex I.
Introduction
Evolution of Earth’s biosphere has largely limited the primitive role of anoxygenic phototrophs, which once performed the fixation of entire global carbon, and brought about their spatial distribution.1,2 The curiosities were revealed by the efforts of Erwin von Esmarch in 1887 and Hans Molisch in 1907. They first demonstrated the presence of anoxyphototrophs including Rhodobacter capsulatus, previously known as Rhodopseudomonas capsulata. 3 It is a gram-negative, photosynthetic, purple non-sulfur bacterium (PNSB). The individual cells are spherical, ovoid, filamentous, or rod-shaped. However, the organism exhibits comprehensive morphological properties and distinguishing features such as “zigzag” or straight chain arrangement and both flagellum-dependent and flagellum-independent motility. At present, different ecosystems around the world harbor this prokaryote, most commonly in freshwater.4-6
The completely sequenced genome of R capsulatus contains a 3.74-Mb chromosome and a 133-kb plasmid with a median GC% of 66.6. According to the reported data, 84.1% of the open reading frames (ORFs) within the genome encode proteins which have defined functional roles, whereas 16.6% ORFs putatively code for hypothetical proteins (HPs). 7 By definition an HP is a predicted product expressed from an ORF whose translation has not been shown and functional relevance yet remains uncharacterized. 8 Even though X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy are the most authenticated methods to resolve the structures of biological macromolecules, attempts have been made for direct characterization from sequence information due to rapidly growing laboratory datasets and accessible computational methods. Nowadays, plenty of bioinformatic tools are available in the public domain, which have made it possible to elucidate the structural details and functional roles of HPs.9,10 In this study, an effort has been made to characterize a hypothetical protein (CAA71016.1) from R capsulatus, propose a 3-dimensional (3D) structure, and annotate its functional role as S-adenosylmethionine (AdoMet or SAM)-dependent methyltransferase (MTase) through in silico proteomics approaches.
Class I MTase is a major structural family of methyl transferring enzymes which use SAM as a cofactor and act on diverse substrates, particularly free amino acids, proteins, nucleic acids, and small bioorganic compounds.
11
In common with many other organisms, R capsulatus has harnessed this enzymatic principle in different biochemical pathways. Notable examples include the bchM gene product S-Adenosyl-
As previously demonstrated, bioinformatic analysis can be a feasible approach to build de novo protein models, predict new functions as well as biochemical properties, and enrich the proteome. It reduces time and labor for an indispensable wet laboratory analysis. 22 Considering the environmental and socioeconomic landscapes of R capsulatus, in silico characterization of HPs can guide us to profoundly understand its behavior and develop new strategies for its application, which may unlock a gateway for a sustainable future.
Materials and Methods
The workflow of this study is presented in Figure 1.

Flowchart of methodology. NCBI indicates National Center for Biotechnology Information.
Sequence retrieval
Hypothetical proteins (HPs) of R capsulatus were searched in the NCBI Protein database (https://www.ncbi.nlm.nih.gov/protein/) using the keyword “Hypothetical proteins (Rhodobacter capsulatus).” From the resultant hits, a HP (Accession no. CAA71016.1, GI|2182083|) was randomly selected for the study and its sequence was retrieved in FASTA format for further analysis. In addition, a sequence-based peptide search was also performed in the UniProt database (https://www.uniprot.org/peptidesearch/) to inspect whether the protein is redundant. 23
Analysis of physicochemical properties
The physicochemical properties of the selected HP were studied using the ProtParam tool (https://web.expasy.org/protparam/) on the ExPASy server. This online tool executes theoretical measurements such as molecular weight, amino acid composition, total number of positive and negative residues, theoretical pI, instability index (II), aliphatic index (AI), extinction coefficient, and grand average of hydropathicity (GRAVY) value. 24
Sequence analysis and homology identification
Looking for the structural homologs and sequence similarity in different genomics and proteomics-based databases is the most basic step for the function prediction of a hypothetical or an uncharacterized protein. 25 The most frequently used tool for studying sequence similarity is the Basic Local Alignment Search Tool (BLAST) (https://blast.ncbi.nlm.nih.gov/Blast.cgi). In relation to the previous statement, a similarity search for proteins was performed using NCBI’s BLASTp algorithm 26 against a non-redundant database to make the preliminary prediction about the function of the query protein.
Functional domain and family/superfamily prediction
HPs can be classified into families and superfamilies based on their sequence feature, domain, or motif architecture and functional similarities through automated and manual curation. For this reason, different databases use different algorithms to make a prediction from an unknown protein sequence. 27 Thereby, for classification and precise functional annotation, we have used multiple sequence alignment (MSA)-based servers such as Pfam, 28 SUPERFAMILY, 29 and Conserved Domain Database (CDD) 30 ; domain profile-based Conserved Domain Architecture Retrieval Tool (CDART) 31 ; and an integrative database InterProScan. 32 In each case, default parameters were considered.
Multiple sequence alignment and phylogenetic analysis
At first, several protein sequences having annotated similar functionality were retrieved from the NCBI protein database. Molecular Evolutionary Genetics Analysis X (MEGA X) software 33 was used to carry out the MSA and phylogenetic analysis between the targeted HP and fetched dataset. The progressive ClustalW algorithm 34 was applied for the MSA analysis. Furthermore, a phylogenetic tree was also constructed using the similar sequence alignment to show the evolutionary distance among the related proteins. For this purpose, we have considered the default parameters (WAG model) with 500 bootstrap replications. Statistically, the WAG model is based on maximum-likelihood (ML) methods. It incorporated the best attributes of previously proposed matrices and provided an optimal result, hence was our preferable choice. 35
Structure prediction
PSIPRED server (http://bioinf.cs.ucl.ac.uk/psipred/) of UCL Department of Computer Science was used to predict the secondary (2D) structure of the targeted HP. It uses 2 feed-forward neural networks and PSI-BLAST algorithm for analysis. 36 The tertiary (3D) structure was designed using MODELLER 37 through the HHpred 38 tool of the Max Planck Institute for Developmental Biology.
Structure refinement and energy minimization
YASARA Energy Minimization Server 39 was used to attain a minimum energy arrangement of the constructed 3D structure of the HP. Subsequently, the minimized 3D structure was further optimized using GalaxyRefine. 40 After analyzing all the potential structures generated by GalaxyRefine, arguably the one having the best quality and performance was selected.
Model quality assessment
Evaluation of the energy minimized and refined 3D structure was done by PROCHECK, 41 ERRAT, 42 and Verify3D 43 modules of the SAVES server (https://saves.mbi.ucla.edu/). The ExPASy server (https://www.expasy.org/) of the Swiss Institute of Bioinformatics (SIB) incorporates different bioinformatic tools. Between these resources, the SWISS-MODEL Structure Assessment tool and QMEAN tool were collaboratively used to estimate the QMEAN Z-score and global quality of the model. In the QMEAN server, both QMEAN 44 and QMEANDisCo 45 scoring functions were considered. To further consolidate the global quality score, the result generated by the ModFOLD server 46 was taken into account.
Active site prediction
The active site of the protein was identified by Computed Atlas of Surface Topography of proteins (CASTp) (http://sts.bioe.uic.edu/castp/index.html). The web server interlinks protein’s structural and sequence information using the Protein Data Bank (PDB), UniProt, and SIFTS database for timely residue-level annotations. 47 This tool also predicted the active residues which were further validated by analyzing the protein-ligand interactions of the docked complex.
Subcellular localization and function prediction
A protein’s optimum performance depends on the regional environment which dictates its interaction patterns and biological networks. Therefore, predicting the subcellular localization is one of the important steps in specifying the cellular function of a hypothetical or uncharacterized protein. 48 Prediction of the gene ontology (GO) 49 and protein topology 50 display more extensive framework of its molecular function, biological process, and location. Tools used for these objectives were CELLO2GO, 49 CELLO v.2.5, 51 PSORTb, 52 PSLpred, 53 SOSUIGramN, 54 Gneg-PLoc, 55 BUSCA, 56 PRED-TMBB, 57 TMHMM, 50 and HMMTOP 58 tools. The ProFunc 59 and PredictProtein 60 servers were used to validate the function of the hypothetical protein predicted by the CELLO2GO tool.
Docking analysis
Molecular docking is performed to study and predict intermolecular interactions between ligands and macromolecules, using open-source software and web servers. 61 To further validate the probable function of our HP of interest, separate docking analyses were performed between the HP and 2 different ligand molecules, S-adenosylmethionine (SAM) and S-adenosylhomocysteine (SAH). Ligand structures were fetched from PDB (https://www.rcsb.org/). 62 Afterward, the hypothetical protein-ligand docking was performed using AutoDock Vina through PyRx 63 and PatchDock server. 64
Protein-protein interaction analysis
Protein network databases aim to integrate possible protein-protein interactions (PPIs) and present them under a network topology, from which a conclusion about shared functional features of a HP can be drawn. The STRING database evaluates both functional and physical associations. It currently features 24.6 million proteins 65 and aims to cover 14 000 organisms by the year 2021. 65 It was used in our analysis because of its larger coverage. The results obtained from STRING database were further validated by protein-protein docking analysis through HADDOCK v2.4, 66 HDOCK, 67 ClusPro 2.0, 68 and AutoDock Vina. 63 The tertiary structures of NuoF, NuoG, NuoI, NuoJ, and NuoH were obtained using SWISS-MODEL server 69 before docking analysis. Multiple docking tools were used to obtain high confidence about the findings.
Results and Discussion
Sequence retrieval
The HP (Accession no. CAA71016.1, GI|2182083|) of R capsulatus fetched from the NCBI database contains 257 amino acids. The retrieved sequence was further searched in UniProt which is a comprehensive, high-quality, and freely accessible resource of protein sequence along with functional information. The database entries showcased the protein to be non-redundant which might have a significant role. Further information collected from the NCBI database is listed in Table 1.
Retrieval of the hypothetical protein from the NCBI database.

Abbreviation: NCBI, National Center for Biotechnology Information.
Physicochemical properties of the protein
Both physical and chemical properties of the HP can be estimated by analyzing the analogous properties of individual amino acids or the N-terminal residue of the protein. From the results obtained from the ProtParam tool, the HP was found to have a molecular weight of 28 971.14 Da. The theoretical pI value of a molecule is the pH at which that particular molecule carries no net electrical charge and it is also feasible to comprehend the protein charge stability. The calculated theoretical pI value of 6.84 indicated the protein to be negatively charged and considered as an acidic protein. The II is a measurement of primary structure–dependent protein stability under in vitro conditions. It is expected that an II value less than 40 (<40) would predict the protein to be stable and a value greater than 40 (>40) would predict the protein to be unstable. The II value of the HP is computed to be 36.35, which classified the protein to be stable. 24 A protein’s AI is known as the relative volume occupied by aliphatic side chains (alanine [Ala], valine [Val], isoleucine [Ile], and leucine [Leu]). It signifies the maintenance of a thermostable structure. The computed AI value of the HP was 75.91, which indicated that the protein is stable over a wide temperature range. 70 For a peptide or protein, the GRAVY score is defined as the total of the hydropathy values divided by the number of residues in the query sequence, where all the amino acids are taken into consideration. It was computed to be −0.335. The extinction coefficient is an expression of a proportionality constant in the Beer-Lambert law. It estimates the amount of light that is absorbed by proteins at a particular wavelength. 71 It was calculated to be 47 900 for our query protein. The high extinction coefficient indicated the presence of a high amount of tyrosine, tryptophan, and cysteine. 24 Besides, all the physicochemical properties of our HP are listed in Table 2. These properties will be useful for experimental handling of the protein.
Physicochemical parameters of the hypothetical protein (CAA71016.1).

Sequence similarity, alignment, and phylogenetic tree
The BLASTp results of the HP against non-redundant databases showed significant homology with other methyltransferase proteins, precisely with class I SAM-dependent methyltransferase from different species. The fetched methyltransferase proteins from BLASTp results for MSA are listed in Table 3. The MSA depicted the sequence similarity in between the targeted hypothetical protein and other methyltransferase proteins (Figure 2). Phylogenetic analysis was carried out for further confirmation of homology identification and to find out the evolutionary distance among our target protein and aligned methyltransferase proteins. The phylogenetic tree was constructed based on the alignment and BLASTp result, which showed similar concept about the HP (Figure 3).
Data from BLASTp result against nonredundant protein sequences.

Abbreviations: BLAST, Basic Local Alignment Search Tool; SAM, S-adenosylmethionine.

MSA among different methyltransferase proteins and targeted hypothetical protein using ClustalW algorithm by MEGA X software. MEGA X indicates Molecular Evolutionary Genetics Analysis X; MSA, multiple sequence alignment.

Evolutionary analysis of different methyltransferase proteins with the target protein (CAA71016.1 Rhodobacter capsulatus). The phylogenetic tree follows WAG replacement matrices which is based on maximum-likelihood (ML) methods. The branch lengths reflect the degree of divergence of each sequence.
Domain, family, and superfamily prediction
The results obtained from NCBI Conserved Domain (CD) Search, CDART, Pfam, SUPERFAMILY, and InterProScan revealed that the HP sequence was found to have methyltransferase domain. The protein belongs to the Methyltransf_24 family and the S-adenosyl-
Protein domain, family, and superfamily analysis.

Abbreviations: NCBI, National Center for Biotechnology Information; SAM, S-adenosylmethionine.
Secondary and tertiary structure analysis
The secondary structure (2D) of the HP was predicted by PSIPRED server (Figure 4) with a good confidence of prediction. The tertiary structure (3D) was predicted by MODELLER using multiple templates having a probability greater than 99% (Figure 5). It was further energy minimized by YASARA energy minimization server. The energy calculated before energy minimization was −299 229.5 kJ/mol. After 2 rounds of energy minimization, it was changed to −128 802.2 kJ/mol. The score also improved from −3.37 to −0.65 after energy minimization. This indicated that the predicted 3D model became more stable after energy minimization compared to the initial one. This structure was further refined using the GalaxyRefine server and then the quality assessment of the model was carried out.

Secondary structure analysis by using PSIPRED server.

Illustration of predicted 3-dimensional structure of the hypothetical protein: (A) ribbon diagram and (B) surface diagram.
Ramachandran plot analysis (Figure 6A) results revealed that the most favored region, additional allowed region, generously allowed region, and disallowed region covered 93.8%, 5.3%, 0.0%, and 1.0% of residues, respectively. These results showed that majority of the amino acids follow a phi-psi distribution that is consistent with a right-handed α-helix. Hence, the protein adopts a flexible and stable structure. 72 The structure passed in the validation analysis by Verify3D and the graph (Figure 6D) showed that 89.20% of the residues have 3D-1D score ⩾0.2 on average. The overall quality factor predicted by the ERRAT server was 97.826, which indicated the model to be a good-quality structure as high-resolution structures produce values around 95% or higher on ERRAT. The graph (Figure 6C) generated on ERRAT showed that no residue crossed the 99% rejection limit which is also an indication of good-quality and high-resolution structure. The results obtained from the ModFOLD server showed that the structure have a P-value of 8.322E-4 and a global model quality score of 0.6722. The P-value indicates the confidence of the prediction of the model to be in CERT category. It designates the structure to be valid and indicates a very high confidence of prediction. The P-value less than .001 denotes that the model has less than a 1/1000 chance of being incorrect. The QMEAN4 value predicted by the QMEAN server was −0.57 and the value was transformed into a Z-score. It is depicted in the estimated absolute model quality graph (Figure 6B) where our protein model was in the dark region. It has a|Z-score| < 1 which infers the model scores to be expected from an experimentally determined structure of similar size. The global score of the protein structure was calculated to be 0.63 ± 0.05 which validated the global score predicted by the ModFOLD server.

Quality assessment of the predicted tertiary structure. (A) Ramachandran plot of modeled structure validated by PROCHECK program. (B) Graphical presentation of estimation of absolute quality of model with QMEAN. (C) Graphical representation of ERRAT value estimated overall quality factor of 97.826. (D) Graphical representation of the averaged 3D-1D scores of the amino acid residues of the tertiary structure determined by VERIFY3D server. PDB indicates Protein Data Bank.
Active site detection and docking analysis
The active site of the protein predicted by the CASTp server found that 25 amino acids are involved in the potent active site. The predicted active site of the protein with their amino acid residues is depicted in Figure 7. Further docking analysis between the HP and the ligands (SAM and SAH) was carried out considering the amino acids involved in the active site predicted by CASTp server. S-adenosylmethionine is an exigent molecule and the principle biological methyl donor, found in almost all living organisms. S-adenosylmethionine-dependent methyltransferase enzymes use SAM as methyl donor. 11 After donating the methyl group, SAM converts into SAH which acts as a potent competitive inhibitor of methyltransferase depending on the available concentration of SAM and SAH molecules in physiological condition. 73 The docking analyses were carried out through Autodock vina on the PyRx server. The binding affinity (kcal/mol) of SAM and SAH with the target protein was −7.1 and −6.7 kcal/mol, respectively (Table 5). It indicated a strong interaction of the ligands with the target protein. The interacting residues and the interactions of the ligands with the target protein are depicted in Figure 8. The molecular docking analysis was also carried out using the PatchDock server through interaction refinement with FireDock server. It also showed promising results (Table 5) indicating that the ligands bind efficiently with the target protein.
Docking study of the ligands to the target protein.


Active site of the hypothetical protein. (A) The sphere indicates the active site/pocket of the protein. (B) The marked amino acid residues construct the active site of the protein.

Molecular docking (targeted protein-ligand interactions). (A) 3D interaction between the targeted protein and ligand (SAM). (B) 2D interaction between the targeted protein and ligand (SAM). (C) 3D interaction between the targeted protein and ligand (SAH). (D) 2D interaction between the targeted protein and ligand (SAH). SAH indicates S-adenosylhomocysteine; SAM, S-adenosylmethionine.
Another set of docking analyses was performed without marking the active site amino acids, targeting the whole protein, using the Autodock vina on the PyRx server. It helped to reinspect the active site predicted by CASTp server and find out whether the ligands actually interact within the predicted active site or some other site of the protein (Table 5). The comparative analysis of active sites through docking showed that the ligands interact firmly with the protein within the pocket inferred by CASTp server and validated the active site detection to be a preferably precise prediction. The comparative active sites of interaction are depicted in Figure 9. Overall, the results obtained from these docking analyses strongly justify the precision of prediction of the target protein to be a SAM-dependent methyltransferase.
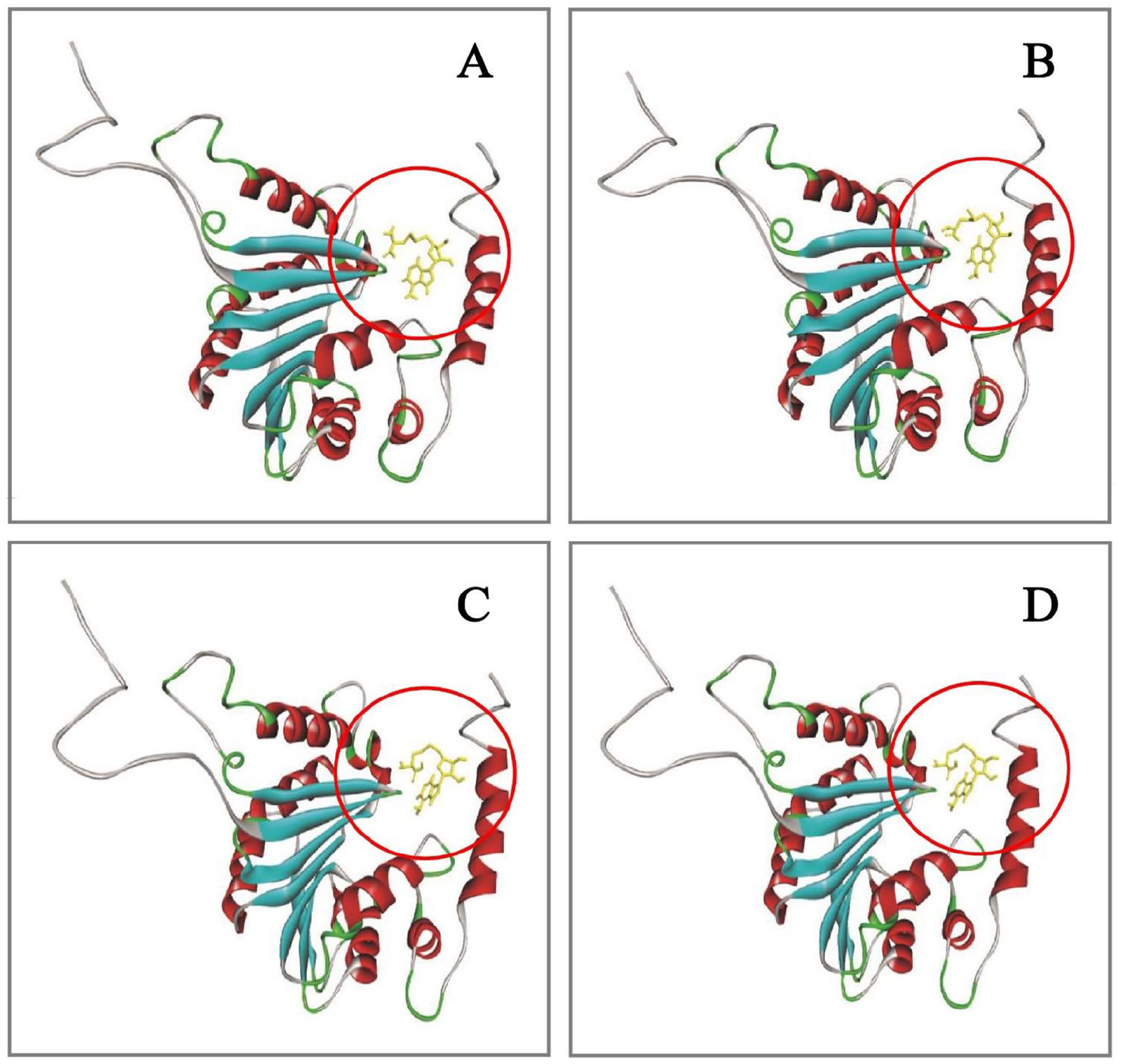
The comparative analysis of active site of the protein. The ligand(s) docked inside the same pocket (Circled) in all of the 4 cases indicating toward the precise active site determination by CASTp server. (A) Protein-ligand (SAM) docking analysis after marking the active residues. (B) Protein-ligand (SAM) docking analysis without marking the active residues. (C) Protein-ligand (SAH) docking analysis after marking the active residues. (D) Protein-ligand (SAH) docking analysis without marking the active residues. CASTp indicates Computed Atlas of Surface Topography of proteins; SAH, S-adenosylhomocysteine; SAM, S-adenosylmethionine.
Subcellular localization nature and functional annotation
The subcellular localization prediction of a protein involves finding out where the protein actually resides within a cell. Subcellular localization predicted by the CELLO2GO and CELLO v2.5 server revealed that the protein is predicted to be localized in the cytoplasm of the cell. The result was further validated by PSORTb, PSLpred, SOSUIGramN, Gneg-PLoc, BUSCA, and PRED-TMBB tools which also predicted the protein to be a cytoplasmic protein (Table 6). The TMHMM and HMMTOP servers predicted the absence of transmembrane helices. The absence of transmembrane helices overrules the possibility of the HP to be a transmembrane protein. Gene ontology results from CELLO2GO tool predicted the molecular function of the protein and its involvement in biological processes. The tool revealed that the major molecular function of the target protein is to impart methyltransferase activity. It also predicted that the protein is mainly involved in the biosynthetic process. Besides, the protein also has a probability to have involvement in protein complex assembly, cellular component assembly, and macromolecular complex assembly. The ProFunc and PredictProtein servers also validated the result by predicting our query protein as a methyltransferase protein.
Subcellular localization analysis.
Protein-protein interaction analysis
STRING is a web-based database of known and predicted PPIs that includes direct and indirect associations. Protein-protein interaction network analysis obtained from this database revealed that our HP of interest has interaction with other proteins, some having experimentally known functions and some whose functions are not yet experimentally annotated (Figure 10). Our targeted protein has a strong predicted interaction with NuoF (NADH-quinone oxidoreductase subunit F) and also has a moderate interaction with NuoH (NADH-quinone oxidoreductase subunit H), NuoI (NADH-quinone oxidoreductase subunit I), NuoG, and NuoJ. Besides, the protein has also interaction with several proteins having functions which are not yet annotated. NuoF, NuoH, NuoI, NuoG, and NuoJ are among the 14 subunits of Complex I of R capsulatus. 74 Two motifs in the NuoF subunit are likely to be involved in the binding of NADH and FMN. 75 NuoG subunit may ligate an extra iron-sulfur (FeS) cluster required for the assembly of Complex I.76,77 NuoH subunit is one of the most conserved subunits in Complex I. It is located in the membranous part and assists Complex I assembly. 78 Whereas, subunit NuoI is essential for the connection between the membranous domain and peripheral domain, in Complex I. 79
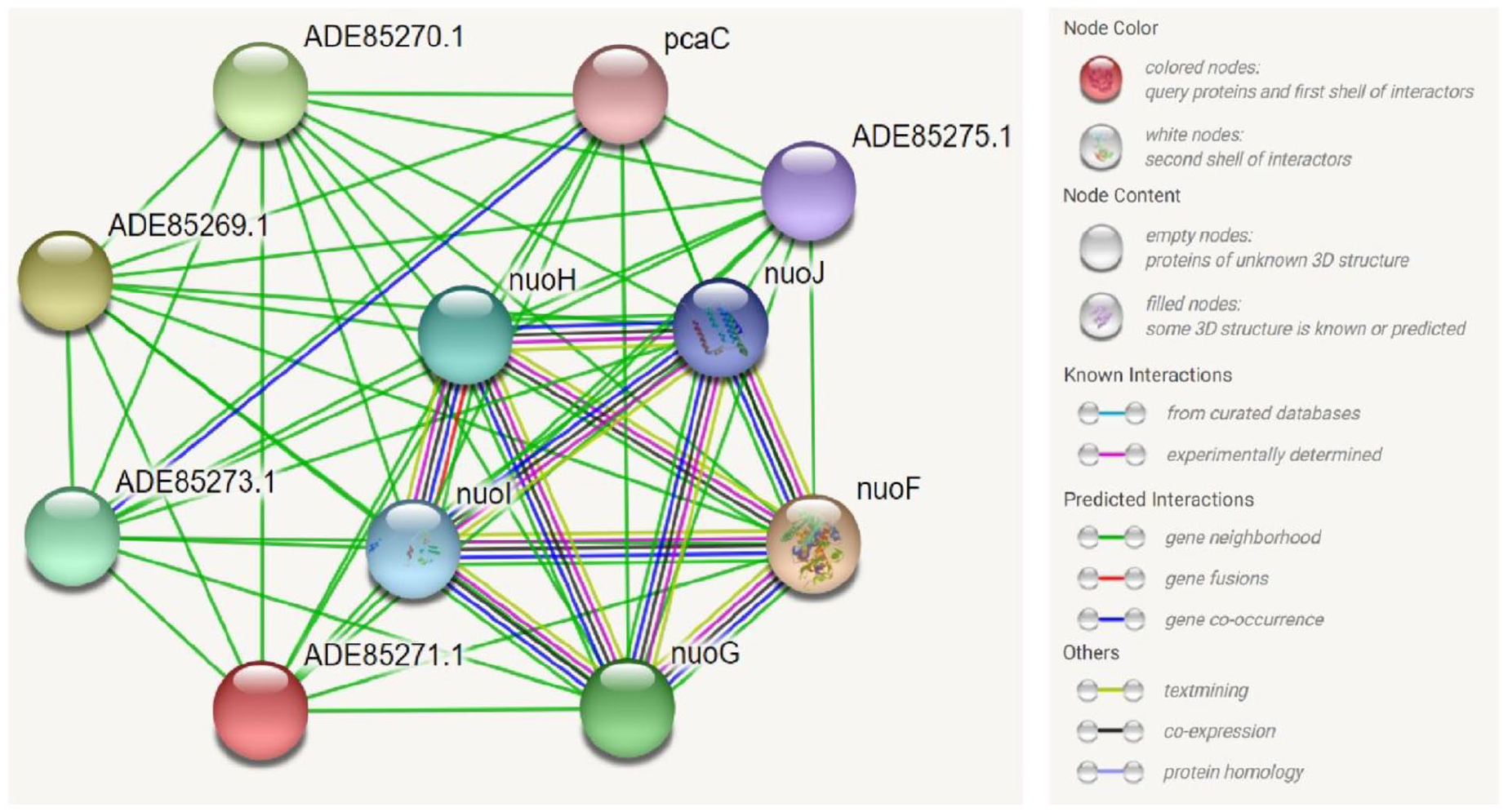
STRING network analysis of the target hypothetical protein (ADE85271.1) depicting the interactions with other proteins.
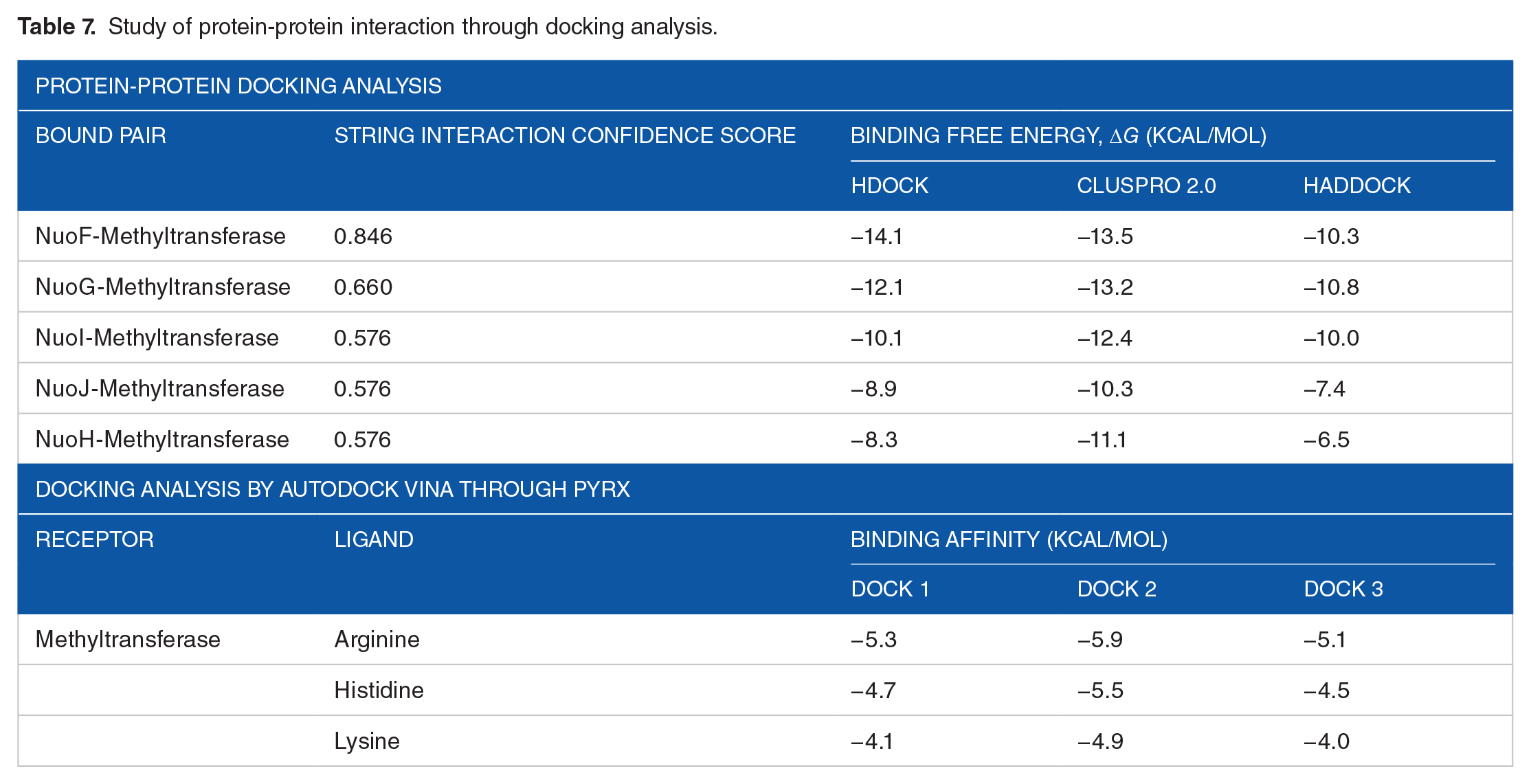
As a part of the nuo gene cluster, urf7 gene product encodes a SAM-dependent methyltransferase. Previously, the roles of this class of enzymes associated with bacterial mitochondrial complex I have been addressed both for prokaryotes and eukaryotes. 74 The results obtained from STRING database were further evaluated by protein-protein docking analysis. It revealed that NuoF has the highest binding affinity with the targeted HP (Predicted SAM-dependent Methyltransferase). The subunit NuoG showed strong binding affinity after NuoF. The other 3 subunits (NuoI, NuoJ, NuoH) showed relatively lower binding affinity than NuoF and NuoG (Table 7). The outcome of the protein-protein docking analysis aligned with the confidence score obtained from STRING database presented in Table 7.
Study of protein-protein interaction through docking analysis.
Prior studies have noted that SAM-dependent methyltransferases are involved in regulation or subunit assembly of Complex I.74,80 In some lower and higher eukaryotes, the roles of methylation-dependent regulation in mitochondrial Complex I have been suggested to be associated with conserved amino acid residues, notably with arginine. However, the roles of histidine and lysine methyltransferases also have been documented.81-83 Further docking analysis by Autodock Vina through the PyRx server showed higher binding affinity of arginine than histidine and lysine with the HP (Table 7). The previously discussed protein-protein docking analysis also revealed maximally evident interaction of the HP with the arginine residues of NuoF and NuoG subunit (Figure 11). Considering all compelling evidences and significant results, it can be strongly theorized that the predicted SAM-dependent methyltransferase plays a noteworthy role in the regulation of Complex I assembly as a protein arginine methyltransferase (PRMT).
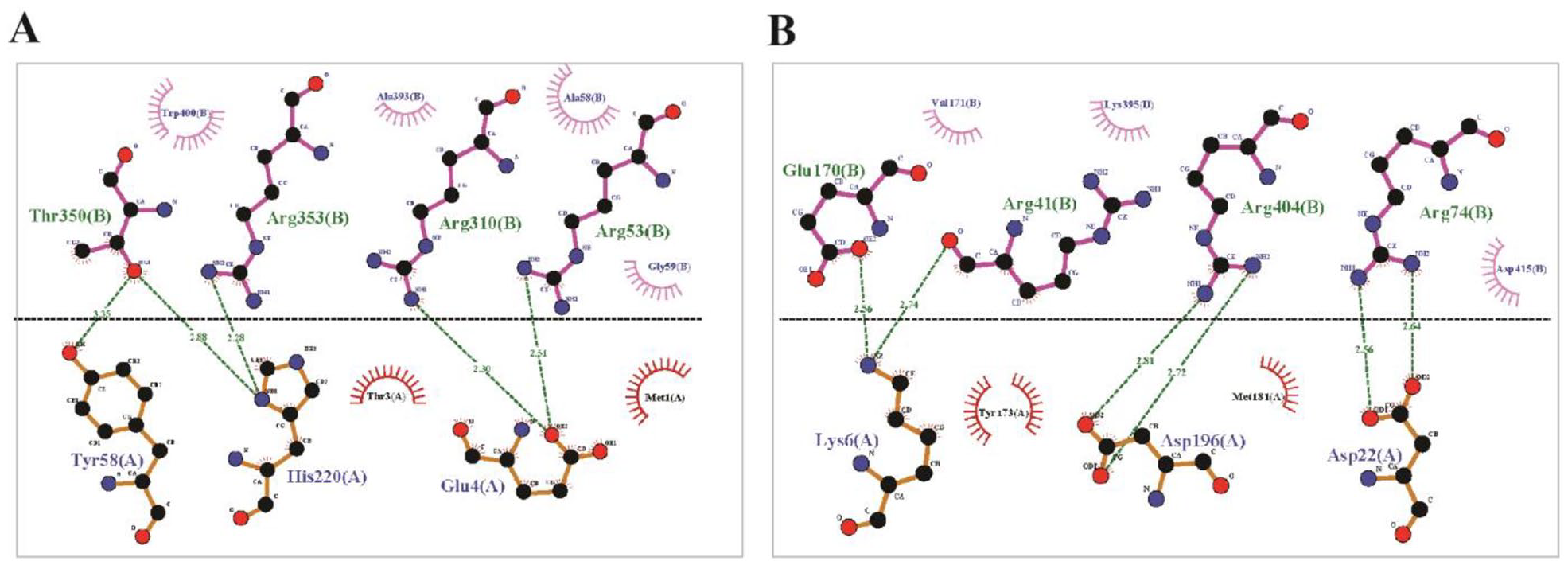
Protein-protein interaction through docking analysis. (A) Interaction between NuoF (chain B) and targeted HP (chain A). (B) Interaction between NuoG (chain B) and targeted HP (chain A). HP indicates hypothetical protein.
Conclusions
The study was designed to explore and annotate a hypothetical protein of an unknown function of R capsulatus through an in silico approach. Different computational tools and extensive bioinformatics workflow established its 3D structure and biological function. Our targeted hypothetical protein was predicted to be a SAM-dependent methyltransferase protein. The respective genes encoding different SAM-dependent methyltransferases are mostly responsible for catalyzing key steps in photosynthetic pigment biosynthesis. However, with the exception of this heavily studied role, the characterized protein of this study was predicted and proposed to be associated with the assembly of bacterial respiratory complex I. Throughout the process of evolution, the central subunits of complex I are conserved from prokaryotes to eukaryotes, including in humans. Deficiency in complex I is also associated with several human disorders. Most vigorous ones are associated with encephalomyopathy, Parkinson’s disease (PD), Down syndrome, etc. Previously, R capsulatus has been harnessed as a model organism to study for its commercial aspects. Due to high level of sequence conservation, establishing the structural and functional roles of unannotated protein as SAM-dependent methyltransferase can help to facilitate experimental studies and unfold new treatment strategies for critical human disorders.
Footnotes
Author Contributions
SMM conceived and designed the experiment. SMM, DD, and DMP carried out the primary investigation and literature review. SMM and DD performed data validation and formal analysis, interpreted the results, and wrote the manuscript. SMM, DD, and DMP primarily revised and edited the manuscript. MMR, MSR, and MRI reviewed the manuscript and supervised the study. All authors have read and agreed to submit the final version of the manuscript.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.