
Abstract
Diarrhoeal disease kills about 1.5 million human beings per year across the continents. The enterotoxigenic Escherichia coli (ETEC) pathotype has been noted as a major cause of diarrheal disease in human and livestock. The aim of this study is to identify broad-spectrum molecular targets in bacteria and broad-spectrum lead compounds (functional inhibitors) with high efficacy and no significant adverse implication on human systems, in relevance to diarrhea therapy through computational approaches which include phylogenetics, target prediction, molecular docking, and molecular flexibility dynamic simulations. Three molecular target genes, murA, dxr, and DnaE, which code for uridine diphosphate-N-acetylglucosamine-1-carboxyvinyltransferase, 1-deoxy-D-xylulose-5-phosphate reductoisomerase, and deoxyribonucleic acid polymerase III alpha subunit, respectively, were found to be highly conserved in 7 diarrhea-causing microbes. In addition, 21 potential compounds identified showed varied degree of affinity to these enzymes. At free energy cutoff of −8.0 kcal/mol, the highest effective molecular target was DNA polymerase III alpha subunit (PDB ID: 4JOM) followed by UDP-N-acetylglucosamine-1-carboxyvinyltransferase (PDB ID: 5UJS), and 1-deoxy-D-xylulose-5-phosphate reductoisomerase (PDB ID: 1ONN), while the highest effective lead compound was N-coeleneterazine followed by amphotericin B, MMV010576, MMV687800, MMV028694, azithromycin, and diphenoxylate. The flexibility dynamics of DNA polymerase III alpha subunit unraveled the atomic fluctuation which potentially implicated Asp593 as unstable active site amino acid residue. In conclusion, bacteria DnaE gene or its protein is a highly promising molecular target for the next generation of antibacterial drugs of the class of N-coeleneterazine.
Introduction
Diarrheal disease has been ranked as the second leading cause of mortality of children. There are about 1.7 billion cases per year of diarrheal disease in children below the age of 5 across the continents. The death rate by diarrhoeal disease is about 1.5 million per year, which include 620 thousand children below the age of 5, and 320 thousand adults above the age of 70.1,2 The occurrence of childhood mortality in developing countries due to diarrheal diseases has been found to be between 9% and 34%. 1
Diarrhea may be inflammatory, secretory, osmotic, or neurogenic. There are many pathogens that cause diarrheal infection which are broadly classified as viral (rotaviruses, noroviruses, astroviruses, and enteric adenoviruses), bacterial (Campylobacter jejuni, Salmonella typhi or paratyphi, Clostridium difficile, Helicobacter pylori, Shigella flexneri, Vibrio cholera, and Shiga toxin (Stx)-producing Escherichia coli), and parasitic (Cryptosporidium parvum, Entamoeba histolytica, and Giardia duodenalis [also known as G. intestinalis or G. lamblia]) infections.1,3,4
Enterotoxigenic E. coli (ETEC) remains a major cause of diarrhea-associated mortality and morbidity of infants, young adults, and adults in endemic areas. 5 The indicative mark of ETEC was found to be the occurrence of either 1 or both of 2 enterotoxins, namely the heat-labile toxin (LT) and the heat-stable toxin (ST).5,6 Shiga-toxin-producing enterohemorrhagic E. coli (EHEC), enteropathogenic E. coli (EPEC), and enteroinvasive E. coli (EIEC) are other subtypes of E. coli which cause diarrhea in human. 6 Different enterotoxins are produced by a variety of enteric pathogenic organisms, including diarrhoeagenic E. coli, V. cholerae, Vibrio mimicus, Yersinia enterocolitica, Citrobacter freundii, and Klebsiella pneumoniae. The ETEC with ST biomarker has been noted as a major cause of diarrheal disease in human and livestock globally.6,7
The drawbacks of current antidiarrheal therapies include limited efficacy and concern about their safe use for pediatrics which makes significant percentage of diarrheal incidence. 1 Therefore, general antimicrobial agents such as fluoroquinolones (ciprofloxacin or levofloxacin) have been the mainstay of therapy, while other administered drugs include azithromycin, ceftriaxone, trimethoprim-sulfamethoxazole (cotrimoxazole), metronidazole, tinidazole, nitazoxanide, mebendazole, or paromomycin. 4 The potential targets for diarrhea therapy include intestinal calcium-sensing receptor (CaSR), 1 type III secretion system (T3SS), 8 tight junctions (TJs), 9 and replication initiation (virulence cascade).10,11
Multidrug-resistant bacteria continue to emerge, and there is urgent need for the development of more specific therapeutic agents that effectively treat the severity of diarrheal infection without any related gastrointestinal motor effects or other side effects. Computational methods such as homology modeling, molecular docking, and dynamic simulations, have been successfully used to study potential compounds against cholera toxin and bacteria virulence,12-14 Salmonella-induced diarrhea 15 and cryptosporidiosis. 16 Computer-aided docking simulation has been proposed as one of the target-based methods for the discovery of T3SS virulence blockers. 17 Therefore, the aim of this study is to identify broad-spectrum molecular targets in bacteria and broad-spectrum lead compounds (functional inhibitors) with high efficacy and no significant adverse implication on human systems, in relevance to diarrhea therapy through computational approaches. Identification of novel molecular targets and lead compounds will be advantageous for the treatment of diarrhea disease by overcoming the rising antibiotic resistance in bacteria.
Materials and Methods
In silico preparation of ligands
An array of antidiarrheal compounds (experimental, investigational, and approved drug) as well as natural compounds were adapted from available scientific publications. The reference ligands used in the study were cycloserine (DB00260), fosfomycin (DB00828), cefmetazole (DB00274), cefazolin (DB01327), and azithromycin.4,18 Available structures of most of the compounds were obtained from the PubChem Compound Database in canonical SMILES format as well as Pathogen Box activity biological data structure and SMILES, 19 while unavailable structures were constructed using ChemSketch interface of ACDLabs (Freeware) 2015, version 2.5. The ligand structures generated were subjected to 3-dimensional (3D) optimization and saved in SMILES format. All file conversions required were performed using ChemSketch and PyMol, version 2.0.7 (Schrödinger Inc, NY, USA).
In silico prediction of targets and pharmacological properties
The predicted targets of the ligands were compared to those of the reference ligands used in this study. The screening was done using SwissTargetPrediction server, where Homo sapiens was selected as the target organism. 20 The ligands with similar pattern of targets as any of the reference ligands (Table 1) were selected for further analyses. The selected ligands were subjected to in silico ADME (Absorption, Distribution, Metabolism, and Excretion) screening on SwissADME server. 21 The ADME screening was performed at default parameters.
Typical schematic for the selection of study ligands with similar pattern to reference ligands in human targets.

Each *was assumed to be 10% probability on target from SwissTargetPrediction. Study ligands considered were within ±10% match with any of the reference ligands. In this scheme, S2 and S6 will be discarded from further analysis due to excessive probability and lack of match to any of the reference ligands, respectively.
In silico preparation of targets
Potential targets with relevance to diarrheal disease were extracted from available published literatures and screened manually. The protein sequence of each selected targets was obtained from UniProt database (www.uniprot.org) in FASTA format. Targets with adequate coverage in 7 diarrhea-causing microorganisms were selected for further analysis. Phylogenetic tree that showed the evolution of the selected targets was drawn after multiple sequence alignment using ClustalO server of the European Bioinformatics Institute (EBI) and visualized at https://phylo.io. The 3D structure of the selected target proteins was obtained from RCSB Protein Data Bank (PDB) database (www.rcsb.org/pdb) and the active site amino acid residues of the targets were noted.
Molecular docking studies
The molecular docking studies were carried out according to the method of Fatoki et al. 22 Briefly, all water molecules, hetero atoms, and multichains were removed from the crystal structure of the prepared targets using PyMol, version 2.0.7. Target proteins and ligands were prepared for docking using AutoDock Tools (ADT), version 1.5.6 23 at default settings, and the output file was saved in pdbqt format. Molecular docking program AutoDock Vina, version 1.1.2 24 was employed to perform the active site docking experiment. After docking, close interactions of binding of the target with the ligands were analyzed and visualized using ADT and PyMol, version 2.0.7.
Molecular dynamics simulation
The dynamics of DNA polymerase III alpha (PolIIIα) subunit structure (PDB: 4jom, chain A) was investigated. From the crystal structure (X-ray structure), PDBFixer implemented in OpenMM, version 7.3, 25 on CPU platform was used to fix the protein. The fixed PDB file generated was loaded and OpenMM ForceField was instantiated using amber14/protein.ff14SB force field parameters for the protein and amber14/tip3p water model with constraints on the lengths of all bonds involving a hydrogen atom and TIP3P waters were added to a cubic box extending 10 Å beyond the outermost protein atoms with 300 mM NaCl. 26 The energy minimization was conducted until a tolerance of 50 kJ/mol using a Langevin integrator 27 with a time step of 2.0 fs, temperature of 300.0 K, and collision rate of 5.0 ps–1 using single precision. Nonbonded forces were modeled using the particle-mesh Ewald (PME) method 28 with a cutoff distance of 10 Å and a Monte Carlo Barostat with pressure of 1 atm, temperature of 300 K, and barostat update interval of 50 steps. The minimized protein was then subjected to fast simulation of structural flexibility using CAB-flex 2.0 server 29 with random number generation seed of 4956 while other parameters were at default settings. The contact map and root-mean-square fluctuations (RMSFs) of atoms in the server-analyzed protein was obtained.
Results and Discussion
The alterations of the TJs by infectious enteric agents often elicit inflammatory cascades and cause diarrhea. 9 Although the idea of safeguarding the TJ has been embraced (Table 2), the development of drugs with less side effect for treating microbial invasion of TJs with symptoms of diarrhea may not be feasible currently. Tight junctions are not designed by nature to be blocked. Blockage of TJs will affect intercellular passage of essential nanometer-sized molecules which could result to another pathogenesis. Previous in silico studies have reported several potential drug targets through inclusive stepwise subtractive process of comparative genomic analysis of E. coli O157:H7 (a typical EHEC) and C. jejuni.6,18,36
List of relevant antidiarrheal compounds.

About 60 antidiarrheal compounds were obtained from available literature (Table 2). Twenty-one of these compounds were found to possess relevant functional characteristics based on the nature of targets that were predicted for 5 reference ligands used in this study, as well as reported function related to diarrheal and absence of cases of liver injury or other acute toxicity. The result of predicted ADME parameter of these 21 compounds in Table 3 showed that 16 of the 26 selected ligands (such as racecadotril, alosetron, MMV676501, vibrepin, and N-coeleneterazine) have high gastrointestinal absorption (GA), 9 of the ligands (such as dicyclomine, diphenoxylate, loperamide, and MMV010576) were permeable through the blood-brain barrier (BBB), and 17 of the ligands (such as tetrodotoxin, d-galacturonic acid, amphotericin B, and toxtazin B) were substrates for P-glycoprotein (P-gp).
List of broad-spectrum potential antidiarrheal agents with similar pattern of targets in human and their predicted pharmacokinetics evaluated from SwissADME server.
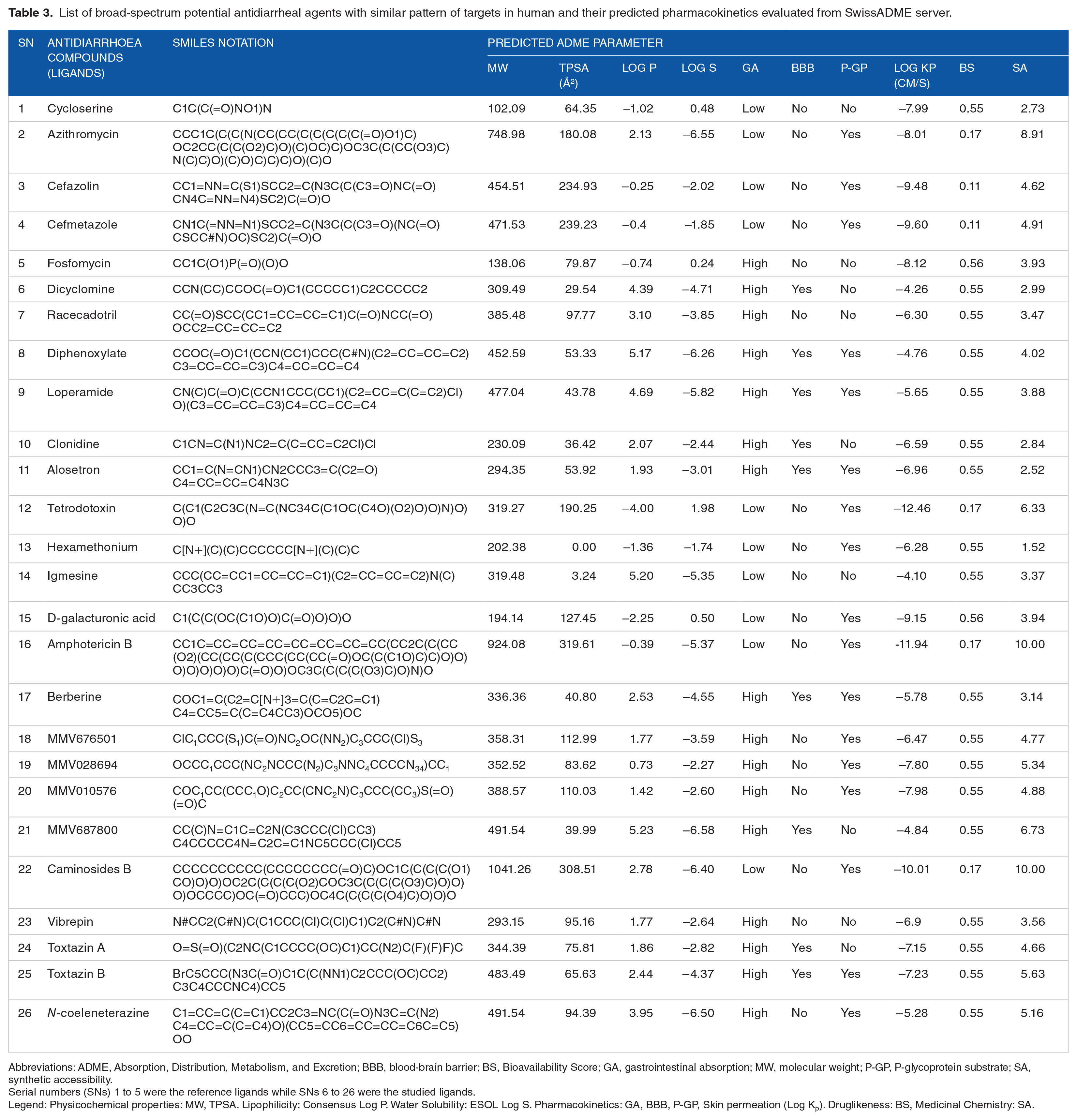
Abbreviations: ADME, Absorption, Distribution, Metabolism, and Excretion; BBB, blood-brain barrier; BS, Bioavailability Score; GA, gastrointestinal absorption; MW, molecular weight; P-GP, P-glycoprotein substrate; SA, synthetic accessibility.
Serial numbers (SNs) 1 to 5 were the reference ligands while SNs 6 to 26 were the studied ligands.
Legend: Physicochemical properties: MW, TPSA. Lipophilicity: Consensus Log P. Water Solubility: ESOL Log S. Pharmacokinetics: GA, BBB, P-GP, Skin permeation (Log Kp). Druglikeness: BS, Medicinal Chemistry: SA.
Gastrointestinal absorption and BBB permeation was predicted based on permeability through the white and yolk of a boiled egg, respectively. 21 The best bioavailability score and synthetic accessibility score is 1.0 which is an indication of the amount of the compound that could reach the active site and extent of ease of synthesis of the compound, respectively. 21 In addition, the ADME parameter showed similarity that exists between the reference and study ligands. For example, fosfomycin has similar GA, BBB, and P-gp properties and close bioavailability score with racecadotril and vibrepin. The overall ADME parameter of the reference ligands shows that favorable antidiarrheal drug should have low GA, are not permeable through BBB, and should not be greatly affected by P-gp.
The range value of a drug-like compound has been reported as 5 ⩽lipophilicity ⩾0 ⩽hydrophilicity ⩾–5 and that drug-like compound may possibly violate not more than one of the Lipinski’s rule. 38 Based on the available published literatures on relevant studies of diarrheal infection and antidiarrhea agents, 17 potential molecular targets were manually extracted based on the following criteria: (1) their absence in human metabolic pathway while present in broad-spectrum bacterial essential pathways, (2) nature of the functional group of their inhibitory compounds, and (3) availability of experimental or approved drug (Table 4).
List of selected potential target proteins obtained from available literatures.

Three molecular target genes, murA, dxr, and DnaE, which code for UDP-N-acetylglucosamine-1-carboxyvinyltransferase, 1-deoxy-D-xylulose-5-phosphate reductoisomerase and PolIIIα subunit, respectively, were found to be highly conserved in diarrhea-causing microbes. In all the 3 gene set which covered 7 diarrhea-causing microbes that were selected in this study, E. coli and S. flexneri were found to be the ancestor of diarrhea-causing microbes while H. pylori and C. jejuni were the most recently evolved (Figure 1) and showed similar phylogenetic tree pattern.

The phylogenetic trees (A) DnaE gene (B) dxr gene, and (C) murA gene; from 7 microorganisms causing diarrhea visualized from phylo.io.
The exclusive result of the screening of potential drug targets and antidiarrhoea compounds in this study showed that currently available therapeutics for diarrhea were antibacterial in mechanism. However, an experimental study has shown that auranofin (MMV688978) has limited activity against Cryptosporidium in vitro. 40 The lack of drug targets that do not have human homologues is one of the key difficulties faced when developing antiparasitic treatments. 34
As shown in Table 5, the active site amino acid residues were obtained from curated information on UniProt and from available literatures.41-45 At free energy cutoff of −8.0 kcal/mol, the highest effective molecular target was PolIIIα subunit (PDB ID: 4JOM) followed by UDP-N-acetylglucosamine-1-carboxyvinyltransferase (PDB ID: 5UJS), and 1-deoxy-D-xylulose-5-phosphate reductoisomerase (PDB ID: 1ONN) while the highest effective lead compound was N-coeleneterazine followed by amphotericin B, MMV010576, MMV687800, MMV028694, azithromycin, and diphenoxylate (Table 6). The docking pose of N-coeleneterazine on PolIIIα subunit (−10.2 kcal/mol) is shown in Figure 2. The compound N-coeleneterazine (DB04118) has been reported as a potential inhibitor of novel broad-spectrum antibiotic targets, specifically the PolIIIα subunit (DnaE) of E. coli O157:H7. 36
Selected broad-spectrum targets and docking parameters generated in AutoDock Tools.

Free energy score of the binding interaction between selected broad-spectrum targets and ligands obtained from AutoDock Vina.


Docking pose of N-coeleneterazine on DNA polymerase III alpha subunit (−10.2 kcal/mol) on ADT (upper) and PyMol (down). The PyMol view shows distance (Å) between the ligand and protein backbone.
This study identified d-galacturonic acid as one of the potential antidiarrheal compounds. This may be due to the importance of galactose in the formation of neutral glycolipids; globotriosylceramide (Gb3; Galα(1–4)-Galβ(1–4)-Glcβ1-ceramide) and globotetraosylceramide (Gb4; GalNAcβ(1–3)-Galα(1–4)-Galβ(1–4)-Glcβ1-ceramide), in the cellular receptors of the Stxs by Stx-producing EHEC.6,46-48 Development of galacturonate-containing compounds could be another game changer because prebiotic (nondigestible food ingredients that promote beneficial bacterial growth in the gut) in combination with probiotics (such as Saccharomyces cerevisiae), aid intestinal cleansing by causing displacement of adhesive bacteria and parasites from the intestinal mucosa lining.49,50
For example, rhamnogalacturonan (RGal) isolated from Acmella oleracea (L.) leaves has been reported to ameliorate intestinal barrier function in vivo and in vitro and shown to be a promising molecule for the therapeutic management of ulcerative colitis which is a chronic relapsing and idiopathic disease that affects the colonic mucosa with bloody diarrhea. 51 The compounds, toxtazin A, toxtazin B, and toxtazin B′, were reported as toxT transcription inhibitors that reduce production of cholera toxin in V. cholerae. 10 However, the efficacy of antivirulence drugs is subject to query in diarrheal treatment despite their present adoption in combating diseases due to their molecular targets which are unrelated to the mechanisms of diarrhea pathology. It could be noted that the antivirulence mechanism at the level of DNA polymerization and transcription will be suitable for diarrhea therapy.
N-coeleneterazine, and not amphotericin, was selected for further investigation in this study due to its overall pharmacological properties from the previous steps (Tables 3 and 6). The original indication for amphotericin has been as an antifungal agent; however, drug repurposing study has revealed its new indication as an effective therapeutic agent against leishmaniasis. 52 Thus, it will not be a mystery to see amphotericin serve as a potent antidiarrhoea agent.
DNA polymerase III (PolIII) is a multisubunit enzyme responsible for the replication of bacterial genome with actual DNA synthesis carried out by PolIIIα subunit which is also known as DnaE1 and belongs to the class of C-family of DNA polymerases.41,45 Positively, for therapeutic purposes, studies have shown that PolIIIα subunits are both structurally and evolutionary distinct from eukaryotic and archaeal replicative DNA polymerases that belong to the B-family.45,53 DNA polymerase III alpha subunit was selected for molecular dynamic (MD) flexibility simulation due to the fact that it was a target for 6 ligands with binding free energies that were below −8.0 kcal/mol.
The superimposition of the structure and contact map of 10 models of flexibility simulation of PolIIIα was shown in Figures 3 and 4, respectively. The extrapolation of the active site amino acid residue from the RMSF (Figure 5) showed that Asp593 could be unstable in PolIIIα during catalysis than others. Previous study has shown that the acidic amino acid at position 593 is not conserved in most of the bacteria. 41 This study also showed that residues at position 40 to 65, 125 to 150, 320 to 345, 545 to 570, and 755 to 780 were relatively stable, and the indication is that the residues at the metal-binding site of the enzyme may not be stable during catalysis. Study has shown that the DnaE1 sequence of proteobacteria (α, β, and γ), bacteroidetes, and fusobacteria, all have significant substitutions in the polymerase and histidinol phosphatase (PHP) metal-binding site. 45

Superimposition of top 10 simulated structures of DNA polymerase III alpha subunit obtained from CAB-flex 2.0 server.
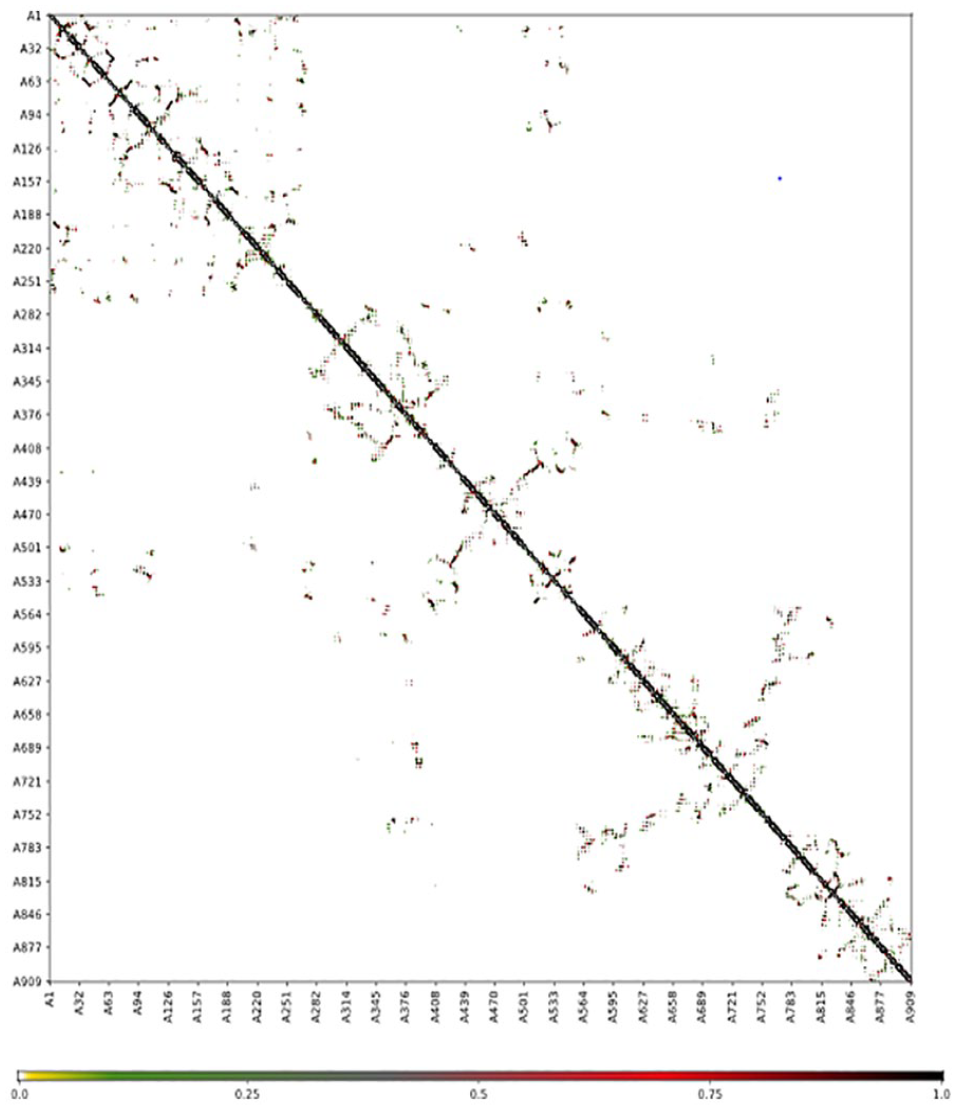
Contact map of superimposition of top 10 simulated structures of DNA polymerase III alpha subunit obtained from CAB-flex 2.0 server.

Root-mean-square fluctuations (RMSF) of atoms in the DNA polymerase III alpha subunit obtained from CAB-flex 2.0 server.
Conclusion
This in silico study has identified bacteria DnaE gene or its protein as a highly promising molecular target for the next generation of antidiarrheal and antibacterial drugs of the class of N-coeleneterazine. The clinical significance of this study will require further research to validate the predicted data obtained. The next research will focus on experimental investigation of inhibitory kinetics, pharmacokinetics, and pharmacodynamics of N-coeleneterazine in respect to bacteria PolIIIα in an in vitro study and in vivo study in model organisms such Drosophila melanogaster and zebrafish.
Footnotes
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
Authors HUU, OCN and SUO designed the study, while HUU and THF conducted the analyses. All authors were involved in the interpretation of the results, preparation and revision of the manuscript, and approved the final version of the manuscript.