
Abstract
A cold-adapted marine alkaline protease (MP, accession no. ACY25898) was produced by a marine bacterium strain, which was isolated from Yellow Sea sediment in China. Many previous researches showed that this protease had potential application as a detergent additive. It was therefore crucial to determine the tertiary structure of MP. In this study, a homology model of MP was constructed using the multiple templates alignment method. The tools PROCHECK, ERRAT, and Verify_3D were used to check the effectiveness of the model. The result showed that 94% of residues were found in the most favored allowed regions, 6% were in the additional allowed region, and 96.50% of the residues had average 3D-1D scores of no less than 0.2. Meanwhile, the overall quality factor (ERRAT) of our model was 80.657. In this study, we also focused on elucidating the molecular mechanism of the two “flap” motions. Based on the optimized model, molecular-dynamics simulations in explicit solvent environments were carried out by using the AMBER11 package, for the entire protein, in order to characterize the dynamical behavior of the two flaps. Our results showed an open motion of the two flaps in the water solvent. This research may facilitate inhibitor virtual screening for MP and may also lay the foundationknowledge of mechanism of the inhibitors.
Keywords
Introduction
Proteases execute a large variety of functions and have important biotechnological applications. 1 They represent one of the largest groups of industrial enzymes and are used the detergent, leather, food, and pharmaceutical industries, as well as in bioremediation processes. 2 They are currently classified into six broad groups: serine proteases, threonine proteases, cysteine proteases, aspartate proteases, metalloproteases, and glutamic acid proteases. The metzincin family of metalloproteinases is so named for the zinc binding motif HEXXHXXGXXH and a conserved methionine, which is located on a turn near the base of the metal binding pocket. 3 Among this family, some enzymes from a host of permanently cold habitats have been paid more and more attention. In recent years, there has been growing interest in cold-adapted enzymes as models in basic studies. Studies have attempted to investigate the thermal stability of these proteins and to understand the relationship between their stability and catalytic efficiency and their potential as candidates for industrial and biotechnological applications.4,5 The cold-adapted marine alkaline protease (MP, accession no. ACY25898) is one of these enzymes.
The MP molecule is a metallo endoproteases, which is made up of 480 amino acid residues. It is about 49 kD with an isoelectric point of about 4.5. It exhibits maximum activity at 30 °C, stability at pH values between 8 and 11, and insensitivity to phenylmethanesulfonyl fluoride. Sequence alignment and our previous study revealed that this protein belonged to the serralysin-type metalloproteases, 6 which in turn belong to the metzincine metalloprotease superfamily. 7 From the obvious study on the sequence of MP, we found the sequence segment HEIGHTLGLSH in the protein. This segment was equivalent to the zinc binding motif HEXXHXXGXXH. In addition, we also found that there are four continuous calcium-binding domains which increased the thermal stability of MP. This protein is homologous to Pseudomonas aeruginosa protease (AP, PDBid: 1H71_A), psychrophilic alkaline protease (PAP, PDBid: 1JIWP), and Serratia marcescens protease (SMP, PDBid: 1SAT_A), with strong similarity of more than 60%. Compared with mesophilic enzymes, cold adapted enzymes are usually more flexible. They usually have looser surface loops and weaker hydrogen bonding. Similar to PAP, MP is a cold adapted protease, but they have different living environments, namely temperature. Structural studies are important to elucidate the structure-environment adaptation of these proteases. In order to get some explanations about the differences in their biochemical properties, modeling the tertiary structure of MP became important. The interaction between enzyme and its ligands is often accompanied by changes in the conformation of the residues within the active site region. In AP, PAP, and SMP, there is a particular structural feature that two of the four loops surrounding the active-site region can flap. The two flaps corresponding to residues 126–129 and 182–189 in PAP play a role in restricting the access to the substrate binding cleft, and thus in controlling the substrate specificity.8,9 In MP, are there two flaps, just like PAP? To solve this problem, a twenty nanosecond-duration constrained molecular dynamics (MD) calculation in water was undertaken in an attempt to predict and understand the conformational changes of the two flaps. Molecular dynamics (MD) simulation was undertaken by using the AMBER11 software package. 10
In the present study, we focused on the investigation of MP homology modeling and explicit molecular dynamic simulation of two flaps. Our results show that the proper combination of homology modeling and MD simulation is powerful in predicting protein structure and obtaining a detailed description of the active site at the atomic level. The modeled structure will give us a better understanding of the relationship between protein structure and its function. Meanwhile, the modeled structure of MP will be of importance in the screening and designing of its inhibitor. This structure may also facilitate its industrial applications.
Materials and Methods
Homology Modeling
Homology modeling and molecular dynamic (MD) techniques were utilized to construct the three-dimensional structure of MP. The target sequence of MP (accession no. ACY25898) was downloaded from the National Center for Biotechnology Information (NCBI) protein database. Meanwhile, a sequence similarity search for this protease against sequences from the Protein Data Bank (PDB) database was performed on the BLAST online server (http://blast.ncbi.nlm.nih.gov). 11 We then found from the PDB that there were sixteen structures identified as homologous. They shared no less than 50% sequence identity with the query sequence. It was noteworthy that after excluding the redundant results, there were only three proteases (AP, PAP and SMP) left. Hence, they were selected as the template. The automated sequence alignment and analysis of the template and target was carried out using Espript2.2. 12 The Risler matrix was used to calculate the similarity score, with the similarity global score set to 0.7. The tertiary structure of MP was built using MODELLER 13 (version 9.9), which was used for homology modeling of protein three-dimensional structures. The predicted structures were saved in the PDB format and sorted according to scores calculated from discrete optimized protein energy (DOPE)14,15 scoring and GA341. 16 To assess the quality of the optimized models PROCHECK, 17 ERRAT, 18 and Verify_3D 19 analyses were also undertaken.
Molecular dynamics simulations
In this study, two separate sets of MD simulations were undertaken by using the Amber11 package. One was the model refinement, which was used to reduce steric clashes among residues. The other was focused on modeling the motion of the corresponding flaps.
The constructed model had to be refined because the changed side-chains could affect the backbone, which would have an effect on the predicted roamers. The constructed model was subjected to an energy minimization process by using the conjugate gradient method for about 10000 iterations and 2 ns isothermal, constant volume MD simulation, with AMBER FF03, all hydrogen amino acid parameters, and the TIP3P water box.
Using the refined model as the starting structure, another MD simulation was focused on modeling the conformation changes of two flaps. All bonds involving hydrogen atoms were constrained with the SHAKE 20 algorithm. Eleven Ca2+ ions were added to obtain charge neutrality. The systems were then solvated using the simple explicit water model. For explicit water simulation, a cubic box was set up and the molecules were positioned at the center of the box by defining a 10 Å distance between the protein and the box edge. The systems were minimized with the SANDER program using 2500 steps of the steepest descent algorithm, followed by 5000 steps of the conjugated gradient algorithm. The PMEMD program was used for molecular dynamics simulations.
Firstly, the restraining forces were applied to all atoms of the protein with 500 kcal/mol for 100 ps. Then, all constraints were removed and the whole system was minimized for 100 ps. Secondly, the whole system was heated from 0 K to 300 K in water over 500 ps, with constraints applied to all backbone atoms, followed by 20 ns of simulation where the proteins were free to move. The simulations were carried out for a total of 20 ns at constant temperature (303 K) and pressure (1 bar) conditions and the integration step was 2 fs. To avoid artifacts, five MD simulations were run twice, with different starting atomic velocities from the Maxwellian distribution. The resulting trajectories were analyzed using the Ptraj module of the AMBER 11 package. The RMSD was calculated for the protein backbone atoms using least-squares fitting. Distances between given residues were calculated with the position for Ca atoms.
Other Calculations
Ligplo 21 is a program used to plot schematic diagrams of protein-ligand interactions for a given ligand in a PDB file. In this study, this software was used to create a 2D diagram of hydrophobic and hydrogen bonding interactions for the Zn2+ ion and the coordinate residues.
Results and Discussion
Sequence Alignment
An optimal sequence alignment is essential to the success of homology modeling. We performed the sequence alignment on the clustalw 22 server at the EBI 23 using the default parameters. A consensus sequence was generated using criteria from MULTALIN 24 and the result was shown in the bottom of Figure 1. The relative accessibility of each residue was extracted from DSSP 25 and PHD 26 files. The alignment results showed that the MP displayed 91% sequence identity with PAP and 63% with AP. Meanwhile, the flap loops of MP and PAP were strictly conserved and the conserved flaps were 140–147 and 237–243 in corresponding positions of MP.

Sequence alignment for MP, PAP, AP and SMP.
Model Construction and Refinement
Because of the importance of metallo-endoproteases in many diseases (eg, cancer and arthritis) and to in order to get a better understanding of the experimental findings, especially to predict substrate specificity of MP, it was critical to know the three-dimensional structure of MP. In this study, homology modeling technique was used to construct models of MP based on the strong similarity among the structures of marine alkaline protease (MP), P. aeruginosa (AP), psychrophilic alkaline protease (PAP) and S. marcescens (SMP).
The homology modeling approach was used to construct the structure of MP using MODELLER9v9. In Table 1, the five resulting three-dimensional models of MP are listed. They were sorted according to DOPE and GA341 score. From the table, we can see that the GA341 score all equal 1.00000 and their DOPE scores are all lower than—48000. By comprehensive analysis of these three scores, the B1.pdb was selected as the initial model. PROCHECK was used to evaluate the Stereochemical properties of the model. A Ramchandran plot was built and it showed that 92.9% of residues were in the most favored regions and 7.1% were in the additional allowed regions. After the MD refinement, the check process was also undertaken. The Ramachandran plot of our model showed that 94% of residues were found in the most favored allowed regions and 6% were in the additional allowed region. Verify-3D graphs showed that 96.50% of the residues had average 3D-1D scores of no less than 0.2. Thus, side chain environments are acceptable. Meanwhile, the overall quality factor (ERRAT) of our model was 80.657. Thus the model was acceptable and was chosen for subsequent molecular dynamical simulation.
The topic 5 results of modeled structures.

The refined model is shown in Figure 2, as drawn by the Pymol 27 program. In this model, the MP protein had two domains and might have one Zn2+ in its active site. Its tertiary structure was comparable to those of the available homologues PAP with the N-terminal domain comprising residues 1–264 and the C-terminal domain comprising residues 265–480.

General overview of the structure of MP.
Catalytic Domain and Active Site
From the above analysis we knew that MP was one of the Metzincins which had an essential Zn2+ for the catalytic activity. A feature that characterized this subfamily was the sequence motif HEXXHXXGXXHZ, wherein the histidine residues were ligands to the Zn2+ and Z was characteristic in the different subfamilies. This conserved segment corresponded to H186-P197 in MP. Moreover, a conserved methionine residue that was likewise characteristic of the metzincin superfamily was also present in MP (Met224). From the LigPlus result (Fig. 3), we could easily find that Zn2+ was bound to the side-chains of HIS186, HIS190, HIS196, TYR 226 and a water molecule. Meanwhile, the water molecule was also bound to the side-chain of Glu187 and TYR226, which was thought to be important in activating the water molecule during catalysis. 28 The hydroxyl group of this tyrosine served as the fifth ligand for the Zn2+. However, upon substrate binding, the tyrosine hydrogen bonded to the main chain of the bound peptide. Just as shown in Figure 4, the distance of the residues His 169, 173, 179, and Tyr209 to the zinc ion at the active site in PAP (1H71_A) are 2.29, 2.24, 2.26, and 2.96 respectively. However, in MP, the distance of residues His 186, 190, 196, and Tyr226 to the zinc ion are 2.20, 2.22, 2.0,3 and 2.44 respectively (as shown in Fig. 3). In order to inspect whether the active zone is associated with histidine, we studied the relationship between the histidine residue and enzyme activity. Through the sequence analysis we know that there is no mercapto amino acid in this protein; therefore we used acetate bromine (BrAc) to modify this enzyme directly. We determined that the leaving enzyme activity after BrAc and the enzyme reacted 30 minutes in the NaAc-HAc buffer. The pH of the solution was 6.0 and the reaction temperature 30 °C. From the result, we found that, with the increase of BrAc's concentration, the relative activity decreased gradually. The binding site defined in DS client showed that this cavity could include 216 detector molecules and its volume was 27 Å 3 (with the threshold set at 2.5 Å). The position of the cavity center was –14.866, 100.425, –5.154, and the cavity radius was 5 Å. In the active site, Zn2+ was coordinated with His 186, 190, 196, and Tyr226. Similar to PAP, they formed a coordinate in trigonal bipyramidal geometry to four or five ligands depending on the position of TYR226. Docking might occur within this range, which gave us something to inspect in a further study.

The molecular interaction of the active sites of MP.
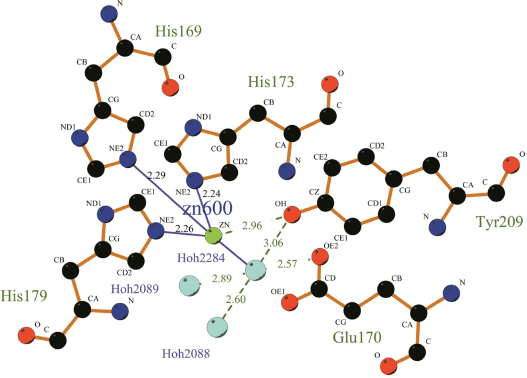
The molecular interaction of the active sites of PAP.
MD Simulation
In order to investigate the motion of the two loops, the modeled structure was simulated by the explicit water MD and a 20 ns MD simulation was performed. The starting model was built from the homology modeling of model1 described above. The MD simulation was run twice using different starting velocities to avoid artifacts. Because the movements of flaps in two MD simulations were the same, the analysis of only one MD simulation is described below.
As expected, a conformational change was observed during the MD simulation. The backbone RMSD of the conformation after 20 ns when compared to the initial structure was 3.025 Å for the global structure (Fig. 4C). As far as flap1 (S140-A147) was concerned, there was a displacement of 2.6 Å in the conformation after 20 ns and a displacement in the initial structure for Ca atoms of the G144 (Fig. 5A). This was different from the approximately 6 Å in PAP. For flap2 (T237-A244), there was an evident conformation change. The distance between the Ca atoms of Thr239 was 16.3 Å (Fig. 4B). This was in agreement with the generally accepted concept that cold adapted proteins were more flexible.

The trajectory of the MP was analyzed by calculating RMSD.
Conclusion
We presented the optimized tertiary structure of MP by homology modeling method. The modeled structure of MP in this study allowed us to better understand the binding of Zn2+. Because the substrate specificity of alkaline metalloproteases was controlled by the structural rearrangement of the two mobile loops, we performed large scale MD simulation that revealed the movement of two flaps. These results might further the understanding of the molecular design of inhibitors and facilitate the virtual screening of inhibitors for the cold-adapted marine alkaline protease MP.
Author Contributions
Conceived and designed the experiments: XJ, MS. Analysed the data: XJ, YZ, JH. Wrote the first draft of the manuscript: XJ. Contributed to the writing of the manuscript: XJ, WW. Agree with manuscript results and conclusions: XJ, WW, YZ, JH, MS. Jointly developed the structure and arguments for the paper: MS. Made critical revisions and approved in all version: XJ. All authors reviewed and approved of the in all manuscript.
Funding
This work is supported by the Specific International Cooperation and Exchanges in Science and Technology (2011DFB30250) and the National High Technology Research and Development Program (“863” Program) of China (2011AA090703).
Competing Interests
Author(s) disclose no potential conflicts of interest.
Disclosures and Ethics
As a requirement of publication author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.