
Abstract
There is some evidence linking the mammalian paraoxonase-1 (PON1) loops (L1 and L2) to an increased flexibility and reactivity of its active site with potential substrates. The aim of this work is to study the structural, dynamical, and functional effects of the most flexible regions close to the active site and to determine the impact of mutations on the protein. For both models, wild-type (PON1wild) and PON1 mutant (PON1mut) models, the L1 loop and Q/R and L/M mutations were constructed using MODELLER software. Molecular dynamics simulations of 20 ns at 300 K on fully modeled PON1wild and PON1mut apoenzyme have been done. Detailed analyses of the root-mean-square deviation and fluctuations, H-bonding pattern, and torsion angles have been performed. The PON1wild results were then compared with those obtained for the PON1mut. Our results show that the active site in the wild-type structure is characterized by two distinct movements of opened and closed conformations of the L1 and L2 loops. The alternating and repetitive movement of loops at specific times is consistent with the presence of 11 defined hydrogen bonds. In the PON1mut, these open-closed movements are therefore totally influenced and repressed by the Q/R and L/M mutations. In fact, these mutations seem to impact the PON1mut active site by directly reducing the catalytic core flexibility, while maintaining a significant mobility of the switch regions delineated by the loops surrounding the active site. The impact of the studied mutations on structure and dynamics proprieties of the protein may subsequently contribute to the loss of both flexibility and activity of the PON1 enzyme.
Introduction
Serum paraoxonases (PONs) form a family of mammalian enzymes with three members including PON1, PON2, and PON3, which are clustered in tandem on the long arms of human chromosome 7(q21.22). 1 Among these enzymes, paraoxonase-1 (PON1) was the first identified and the most studied in the field of toxicology. Several studies have shown that PON1 exhibits a broad spectrum of activities and interacts with various substrates. It could hydrolyze the organophosphates such as paraoxon, diazinon, and nerve gases.2% PON1 is an HDL-associated enzyme involved in the pathogenesis of atherosclerosis by protecting lipoproteins against peroxidation. Studies on both mouse and human have demonstrated the antioxidative and atheroprotective effects of PON1. In 1991, Mackness et al. 3 have demonstrated the significant role of PON1 on cardiovascular diseases (CVD) by preventing the accumulation of oxidized lipids in low-density lipoprotein. At the genetic level, PON1 has two common polymorphisms in the coding region: Leucine (L)/Methi-onine (M) at position 55 and Glutamine (Q)/Arginine (R) at position 192. Both polymorphisms have been identified as genetic risk factors for CVDs in diabetes.4,5 Other studies showed that the Q192R polymorphism affects PON1 activity while the M and L alleles were associated, respectively, with lower and higher serum PON1 concentrations.6,7% These polymorphisms affect also the hydrolytic activity of the PON1 isoenzymes with respect to certain substrates, such as paraoxon and lipid peroxides.
PON1 is a 355 amino acid (43-kDa) glycosylated protein belonging to hydrolyze family and expressed mainly in the liver. The three-dimensional structure (3D) of a hybrid mammalian recombinant (rePON1) variant obtained by directed evolution has been determined, providing the first structural information of this protein. The 3D structure and directed evolution studies provide a detailed description of rePONl active site and an approach of the catalytic mechanism. 8 The refined 2.2 Å crystal structure contains one molecule per asymmetric unit along with two calcium atoms, a phosphate ion, and 115 water molecules. The structure shows all residues except for the N-terminal residues 1-15 and a surface loop called Ll (residues 72-79). Few studies have been focused on the importance of this loop in the rePON1 activity. In fact, the L1 loop may be a part of an active site lid and the mutation in the residue TYR74 led to changes in the substrate selectivity.8% The H3-extended loop called L2 (residues 292-300) is also important. This motion in the apoenzyme (apo) form is highly correlated with another small loop on the back of the active site and also stabilized with ligand binding. Both L1 and L2 loops are located at the front of the active site, forming a tunnel entrance to the binding pocket. Therefore, the high flexibility of these loops might play a selective determining role in substrate recognition. 9
In this work, we present a structural study at the atomic level of the rePON1 free of its substrate. In this work, we present a structural study at the atomic level of two polymorphisms of rePON1. The 3D structure of the rePON1 free of its substrate was first considered. This will help to better understand dynamics behavior and properties in the absence of the cofactor and will also identify the role of L1 loop within the active site. No structural study has ever demonstrated the impact of the rePON1 mutations on structure-activity relationship. Hence we perform a detailed computational analysis of wild-type and mutant rePON1 using molecular dynamics (MD) simulations. In the first part of our study, we investigate the behavior of the rePON1 wild-type regarding the mutant by performing MD simulations starting from X-ray crystallographic structure of the rePON1. In the second part of this work, we identify the regions responsible for catalytic movement based on the movement of L1 and L2 loops and also hydrogen bounds.
Methods
Model building
The 3D structural model of the rePON1 was built using MODELLER 9.11 package.10,11 MODELLER includes open-source software which predicts protein structures starting from their sequences. This prediction is based on domain boundary detection, sequence homology search, fold recognition, homology modeling, de novo design, and model evaluation. For the PON1 modeling, the crystal structure of the rePON1 (PDB identifier 1V04) 8 was used as the template to build both PON1wild-type (PON1wild) and PON1 mutant (PON1mut). The disordered L1 loop (Fig. 1) was reconstructed in the modeled structures of rePON1 by applying spatial restraints and energy minimization using MODELLER. In the same way, the mutant model loop was built following the same steps as the PON1wild and the mutations (Q/R 192 and L/M 55) were applied using the MODELLER script. After the geometry optimization of the PON1wild and PON1mut structures, models were analyzed and revealed that most of the structural features are observed and well constructed in both PON1wild and PON1mut (Fig. 2). This include H1 and H2 helices located in the PON1 surface, the six-bladed β propeller scaffold, the calcium-ligating residues centered in the tunnel, the three helices at the top of the propeller, and the phosphate ion bound to the catalytic calcium. All added mutations were also observed.

The modeled structure of rePON1 Phosphate ion (red and orange balls) bound in the active site as well as two calcium atoms are shown in the
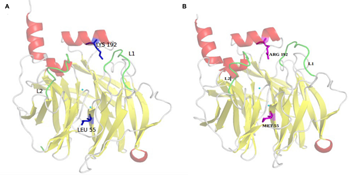
PON1wild and PON1mut models illustrated using PyMol. (
PON1 topology
Before applying MD simulations, we have generated the PON1wild and PON1mut topologies with bonded and nonbonded interaction parameters using the GROMACS package 4.5.4. 12 Missing hydrogen atoms were added with the pdbmgx module. For the neutralization of the systems, 16 Na counter-ions were added in PON1-wild and 18 in PON1mut, respectively. For the solvation, explicit TIP3P water 13 was used using a cutoff distance of 10 Å. The parameters of PO4 molecule was generated using PRODRG program. 14
MD simulations
Twenty nanoseconds of MD simulations of each variant were performed with explicit solvent using the GROMACS package. Coordinate files were created with the FF99SB force field of AMBER.
15
The time step for all MD simulations was set to 2 fs. A nonbonded cutoff of 10 Å was used. Periodic boundary conditions were applied to simulate a continuous system. The Particle Mesh Ewald method was used to calculate the long-range electrostatic interactions.
16
Before starting MD simulations, energy minimization was performed. Thousand steps using the steepest descent algorithm were used to remove close contacts and to relax the system. The systems simulations were conducted in two phases. The first phase was conducted under an NVT ensemble (constant Number of particles, Volume, and Temperature). This ensemble is also referred to as
The resulting trajectories were analyzed using a set of GROMACS modules as g_rms, g_rmsf, g_hbond, g_traj, and g_dihedals from the GROMACS package. The root-mean-square deviations (RMSDs) of the backbone were calculated from the trajectories at 1-ps interval, using the initial structure as a reference. The MD simulations of the wild-type system were carried out in Moroccan Grid in Centre National de la Recherche Scientifique et Technique, Rabat, while the MD simulations of mutant form were performed at the Biology and Health Laboratory (Laboratoire de Biologie et Santέ) in our faculty.
Results
Structural model and dynamics of PON1wild
RMSD, root-mean-square fluctuation, hydrogen bonds, and dihedral analysis
We analyzed 20 ns of MD for PON1wild model built using MODELLER. To assess the stability of each MD trajectory, the RMSDs of the Cα coordinates were calculated for PON1wild system using g_rms module with respect to the first trajectory frame. In this simulation, drifts of the whole system were observed successively at 5, 8, and then at 13 ns. Drifts reached the stability plateau around 2 Å after 13 ns. Variations of internal movements seem to be detected in the 5- to 13-ns interval (Fig. 3).

Coordinates RMSD of MD-simulated system (PON1wild) in comparison with the starting structure. The RMSD is calculated on Cα atoms of the protein.
Other studies have established the movement of PON1-wild apo form with atomic fluctuations in the interval of 5-10 ns. 18 Our work is consistent with these results but shows a second region with greater flexibility beyond 10 ns. The ability of the PON1wild to be flexible among different protein regions is clear from the inspection of the root-mean-square fluctuation (RMSF) calculated using the g_rmsf module of GROMACS and shown in Figure 4.

Atomic fluctuations (RMSF) calculated from the backbone of the PON1wild during MD simulations.
The overall profile of internal fluctuations is quite stable for all systems except for three regions that display large fluctuations. The first region corresponds to the N-terminal tail of H1 (residues 16-25) and the second region, which is the most flexible, corresponds to the L1 loop (residues 72-79). In addition, the H3-extended loop L2 (residues 290-300) shows significant fluctuations. We can notice that both loops L1 and L2 are at the front of the active site, forming a tunnel entrance to the catalytic pocket. Therefore, the large RMSD observed for PON1wild is mostly induced by the internal motion of these two loops. Hydrogen bonds, which were noticed during MD simulations and identified using the g_hbond module of GROMACS, show various fluctuations. Figure 5 displays the mean of hydrogen bonds at each time step of MD. The overall movement of PON1wild is thus supported by an increasing H-bond number at three distinct regions of HB drift: 5- to 6-ns interval, 8- to 10-ns interval, and 13 ns. These three oscillations are perfectly correlated to the regions defined previously by the RMSD drifts (Fig. 3).

Mean of the hydrogen bond movements for PON1wild system.
Analysis of the trajectories
Full-length trajectories were analyzed using g_traj module of GROMACS and then visualized using PyMol software. 19 A detailed trajectory analysis of 20-ns MD simulations revealed that the active site undergoes conformational changes according to the L1 and L2 loop movements. Visualization of trajectories (Fig. 6) showed an alternative movement between a closed- and an open-loop conformation.

Conformations of catalytic loops L1 and L2 of PON1wild. (
Special attention was given to the movement of residues 71-79 and 292-300 of flexible loops L1 and L2, respectively. These loops play an important role in the enzyme stability and may act as a
Recurrence of the

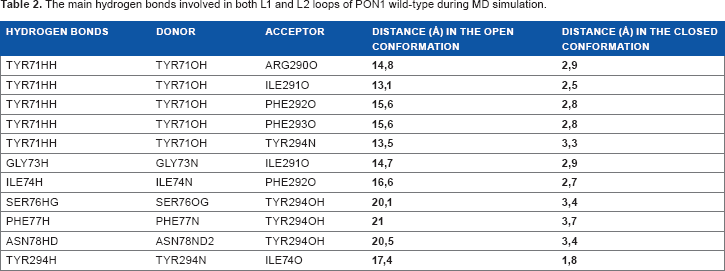
To further understand the loop movements, we have focused on hydrogen bonds between L1 and L2 especially during three time intervals where the conformational changes have occurred. Thus, 11 hydrogen bonds were identified for the first time as being responsible for the consistency of the catalytic movement (Table 2). Among them, one important hydrogen bond stands out and involves the hydroxyl group of TYR71 of the loop L1, which interacts with ARG290, ILE291, PHE292, and PHE293 of the loop L2 on the one hand and with (N) of TYR294 on the other. In this way, TYR71 of L1 loop forms five pairs connected to specific amino acids in the second loop L2. Another set of hydrogen bonds involved residues of L1 loop ILE74N, SER76OG, PHE77N, ASN78ND2, and TYR294N, which may influence the residues PHE292O, TYR294OH, TYR294OH, TYR294OH, and ILE74O from loop L2.
The main hydrogen bonds involved in both L1 and L2 loops of PON1 wild-type during MD simulation.
Dihedral angles
To monitor the local movement of L1 and L2 residues, we calculated the dihedral angles using the

Dihedral angles of the amino acid TYR71 of PON1wild.
The closed TYR71 conformation in the PON1wild was observed first in the early stage of the simulation with a TYR71 pointing into the active site. The increased fluctuations of loop L1 in the PON1wild structure caused a serious conformational change of TYR71 with the side chain pointing away from the active site, which lead to an open conformation (Fig. 8). A short period of transition from
Important variations of the TYR71 side chain were observed at 5 and 8 ns. TYR71 is known by its reactivity with polar aromatic amino acids and by the great role of its phenolic hydroxyl group. It has an ability to form hydrogen bonds and, hence, it has an important role in the PON1wild flexibility. Our results show clearly that the movement of the TYR71 was mainly responsible for the fluctuations observed in RMSD and HB drifts.
By analyzing MD trajectory, we followed the movement of TYR71 more precisely at 5 and 8-9 ns of simulation. In Figure 8, we demonstrate that the TYR71 is very consistent with the movement of opening and closing of the two catalytic loops within a specified time. In the closed conformation, TYR71 adopts a spatial orientation and points inside the active site, while in the open conformation, it takes another direction pointing outside the active site.

The position of TYR71 in the PON1wild active site. (
Structural model and dynamics of PoN1mut: comparison with the wild-type protein

RMSDs of PON1wild (black line) and PON1mut proteins (red line).
The RMSD plots for both mutant and “wild-type have relatively the same shape in the entire simulation except for the region 3-12 ns as shown in Figure 9. The RMSD values for the mutant seem to be slightly higher compared to those for the wild-type. At the end of the simulation, RMSD remains stable. The weaker of the fluctuations together with very small difference between the average RMSD values indicate that simulation produced stable trajectories providing a suitable basis for further analyses.
The RMSF values of mutant backbone residues were also calculated and compared with the wild-type system. The analysis of fluctuation scores showed that the flexibility of amino acids in the mutant system has been altered. The mutations Q/R associated to L/M seem to induce constrains in the flexibility of protein structure except for the regions designated as switches. For both mutant and wild-type structures, all the regions concerned by this flexibility were reported with RMSF plots in Figure 10.

RMSF of Cα backbone atoms of wild-type and mutant PON1 protein versus time. PON1wild is shown with a black line and PON1mut with a red line.
The primary and secondary structures of the various regions are given in the following list. In order to be clear, we have given the same beta sheet number to the switch region. Expect the N-terminal helix H1 at the top of the propeller, we have localized six switch regions. Switch region 1: (aa 30-50) corresponding to the l1 loops connecting two antiparallel strands in the same β1 sheet; Switch region 2: (aa 59-60) corresponding to a small loop l2 connecting two antiparallel strands in the same β2 sheet; Switch region 3: (aa 123-124) and (aa 149-150) corresponding to l3 loop connecting the first two antiparallel strands of the β3 sheet and l3’ loop connecting the second two antiparallel strands in the same sheet, respectively. Switch region 4: (aa 174-177) and (aa 209-211) corresponding to l4 loop connecting two antiparallel strands of the β4 sheet and l4’ loop connecting the second antiparallel strands in the same sheet, respectively. Switch region 5: (aa 231-223) and (aa 251254) corresponding to l5 loop connecting the first two antiparallel strands of the β5 sheet and l5’ loop connecting the second antiparallel strands in the same sheet, respectively. Switch region 6: (aa 276-277) and (aa 310-311) corresponding to l6 loop linking the first two antiparallel strands of the β6 sheet and l6’ connecting the second antiparallel strands in the same sheet. In order to localize the position of the six small loops switching strands in beta propeller surrounding the active site, we analyzed PONlmut trajectories of MD simulations using PyMol viewer (Fig. 11).

Representation of PON1mut switch regions named in this work as (I1, I2, I3-I3’, I4-I4’, I5-I5’, and I6-I6). (
Additional information on the structural flexibility of PON1mut protein was obtained by the analysis of time-dependent HB formation. During MD simulations of 20 ns, a rapid decrease of the number of HB occurs at 3 ns (Fig. 12) and the number of hydrogen bonds of PONlmut remains stable.
Based on the HB average calculation, the wild-type protein appears to form an average number of HB in the range of 260, whereas the mutant appears to form fewer, around 255. This analysis reflects that electrostatic interactions between donors and acceptors of the HB could be slightly repressed in the active site of PONlmut. This may cause the loss of the functional activity of the PONlmut protein compared to PONlwild.

Mean of the hydrogen bond movements for PON1mut in relation to PON1wild system.
PONlmut trajectory was inspected to check both L1 and L2 movement and TYR71 direction inside the active site. Table 3 shows the recurrence of the active site
Recurrence of the

In conjunction with this movement and to further characterize the dynamic behavior of TYR71 side chain in PON1mut, we analyzed this movement over the time course of the simulation. This was displayed with representative snapshots extracted from the trajectories (Fig. 8). The open conformation is characterized by the TYR71 outside the active site. This shape was predominantly observed in the presence of mutations. However, the conformation of the TYR71 pointing outside the active side seems to be absent. The PON1mut structure seems to be more stable with the open conformation. This is very consistent with the impact of mutations on both flexibility and reactivity of PON1 protein (Fig. 13).

The position of TYR71 in the PON1mut active site. (
Discussion
PON1 is an important protein, which plays many crucial roles in the body and partakes in the prevention of cardiovascular damage. Moreover, PON1 gene polymorphisms promote pronounced systemic antioxidant effects in humans, and these effects translate into reductions in the prevalence of coronary artery disease and reductions in the risks of major adverse cardiac events. This original work aimed at studying the fundamental structural underpinnings affecting flexibility and reactivity of PON1 enzyme. Twenty nanoseconds of MD were simulated on both PON1wild and PON1mut (Q/R and L/M mutations) with an explicit solvent. This comparative study started from crystal structure of the rePON1 (1V04. pdb) completed with the missing loop L1 (72-79) and also with the integration of the rePO4 molecule parameters. For both systems, we have chosen to work with the apo form in order to understand the molecular bases of PON1 behavior free of substrate.
To gain a deeper insight into the stability of the simulation, the RMSD was calculated. Then, RMSF, hydrogen bonds, and dihedral angles were analyzed and gave an overview of the most flexible regions of PON1wild. The overall stability of the PON1wild structure is maintained, but large displacements around the active site are observed including L1 and L2 loops, which perform a repeated movement of opening and closing during the simulation. It is important to note that the loops L1 and L2 are at the front of the active site, forming an inlet of the catalytic pocket. Our results are in complete harmony with those of Xin Hu and collaborators.18% In addition, the higher fluctuations described in this work are consistent with a previous study, which shows that the flexibility of L1 and L2 catalytic loops could play a decisive role in PON1 mechanism of action and then in the selectivity and the recognition of substrates. 9 These conformational changes reveal an overall structural plasticity of the active site that allows the enzyme to optimize interaction of the hydrogen-bonding network. This is correlated with the exchanges of hydrogen bonds between the pairs of amino acids belonging to L1 and L2 loops.
The opening and closing movement of the two loops are correlated with l1 of HB and involve several amino acids, in particular TYR71 in the L1 loop. This key residue forms hydrogen bonds successively with ARG290, ILE291, PHE292, and PHE293 belonging to L2 loop. TYR71 plays apparently a donor role with different amino acid acceptors leading to the successive formation and breaking of HB. Our results have identified the following acceptors/donors pairs of amino acids SER76 (O)/TRY294 (OH), PHE77 (N)/TYR294 (OH), ASN78 (N)/TYR294 (OH), and TYR294 (N)/ILE74 (O) involved in L1 and L2 interactions.
A detailed trajectory analysis revealed that the active site in the wild-type structure adopts a
To give further details about catalytic conformations, we have studied the torsion angle of the TYR71 side chain over the time of dynamics simulations. Indeed, fluctuations of the dihedral angles of the TYR71 are consistent with the variation of the RMSD and HB profiles. The transition zones from the torsion angle profiles appear to be in perfect harmony with the opening and closing transition movement of catalytic loops. The closed conformation characterized by the TYR71 pointing into the active site is mainly observed at 5 and 8 ns during MD. This is in perfect agreement with another study showing that the closed conformation is characterized by the TYR71 pointing to the active site of the substrate-bound complex. 9 In our case, the stability of this conformation is because of the interaction between the OH group of TYR71 and phosphate molecule in the catalytic pocket. Contrastingly, the increase of L1 loop fluctuations pushes TYR71 pointing outside the active site and leads to an open conformation. TYR71 seems to be involved in the changes of conformational catalytic loop thus facilitating the recognition and routing substrate through the tunnel of active site. These results confirm the important influence of TYR71 on the PON1wild active site. 20
Furthermore, MD simulations on the PON1mut (Q192R and L55M) were performed for the first time in order to explore the structural impact of the two mutations. Experiments have shown that PON1 activity is sensitive to amino acid substitutions in the active site. 21 The 20 ns of MD trajectory analysis revealed that the mutant structure remains stable during the whole simulation runs. A detailed analysis of the RMSD pattern shows that the two mutations do not affect greatly the global shape of the PON1mut structure. However, the analysis of the RMSF has shown that the mutations affect the dynamical fluctuations around the active site. The higher mobility concerns especially residues near the switch regions, which can cause loss of the activity.
Hence, it was very noteworthy to observe that the amino acid residues present in the switch region of PON1mut protein showed distinct RMSF changes. These regions seem to play the role of hinge zones which flows the dynamical movement of the catalytic site in the case of wild-type protein. Nevertheless, the two mutations in the active site may influence the motion of the catalytic loops without affecting the movement of the most flexible switch regions in the PON1mut. All these results concluded that the mutations seem to play an important role in decreasing the structural and dynamical flexibility of the catalytic core of the mutant protein. The identified hydrogen bonds firmly support the hierarchy of activity suppression. They in fact decreased clearly in the case of PON1mut since mutations appear to suppress the movement of the two loops responsible for catalytic activation. This result provides more evidence on the involvement of L1 and L2 loops in the overall flexibility of the PON1mut and brings insights to the loops disorders additional to the ones described in the studies by Peterson et al.22% and Sanan et al.
23
In line with these analyses, useful information about the PON1mut conformations have been gained from examining the trajectories. The PON1mut adopts a
To better understand the movement sequence in PON1mut, it seems that mutations act in two ways. First, there is a direct effect on the movement of the active site, which seems to be reduced and could cause loss of the activity of PON1mut. Second, mutations have an indirect effect on the free regions qualified by switch regions. In fact, while the movement of the active site was blocked by mutations, the switch regions seem not to be influenced by the decrease of this movement, which is because of their localization outside the active site and then they showed high mobility. This is discussed for the first time as it is the first MD study showing the impact of common polymorphisms on PON1 movement at the atomic level. The structure-activity relationship was the primordial notion upon which the authors’ studies are based. Furthermore, in the case of mutated structure, experimental studies which most often associate the two mutations (Q192R and L55M) found a decrease in the PON1 activity and concentration. 7
Conclusion
This paper describes for the first time a computational study of the MD simulations of both PON1wild and PON1mut apo forms. The analysis of MD trajectories provides information about the role of L1 and L2 loops in maintaining the flexibility of the PON1wild along with their role as an essential element for catalytic activity. Residues (especially TYR71) and HB highlighted in this work display a fundamental role in this flexibility. In conjunction with this, a series of simulations have given insight into the overall dynamics and conformational changes of mutant PON1. The mutant protein simulation allows the structural impact of PON1 mutation on the active site flexibility and switch regions. The global movement of these regions seems to be not influenced by the negative effect of mutations on the catalytic site. Finally, further studies can be performed using docking simulations for future experimental and theoretical investigations of PON1 enzyme, such as more complete considerations of protein-ligand interactions and structure-function relationships in both systems.
Author Contributions
Conceived and designed the experiments: KA, AK, AEK, AN. Analyzed the data: KA, AK, LM. Wrote the first draft of the manuscript: KA. Contributed to the writing of the manuscript: AM, LM. Agreed with manuscript results and conclusions: RS, AK. Jointly developed the structure and arguments for the paper: AK. Made critical revisions and approved the final version: AM. All the authors reviewed and approved the final manuscript
Footnotes
Acknowledgments
The authors are grateful to Professor Ibnou Zahir from Hassan II University of Casablanca for English language correction.