
Abstract
The inhibition of acetylcholinesterase plays a vital role in the treatment of Alzheimer disease. This study aimed to explore the acetylcholinesterase inhibition potential of Phyllanthus amarus and its phytoconstituents through an in vitro and in silico approach. The in vitro acetylcholinesterase inhibitory activity of P amarus was carried out, followed by the molecular docking studies of its phytoconstituents. The top-ranked molecules identified through molecular docking were subjected to molecular dynamics simulation (MDS) and density functional theory (DFT) studies. The results obtained revealed the methanolic extract of P amarus as a potent acetylcholinesterase inhibitor, while amarosterol A, hinokinin, β-sitosterol, stigmasterol and ellagic acid were identified as potential acetylcholinesterase inhibitors. The MDS and DFT results are in agreement with those obtained from the docking studies. Our findings suggest further studies on the hit molecules.
Introduction
Alzheimer disease (AD) is a progressive neurodegenerative disorder marked by cognitive and behavioural impairment that significantly interferes with social and occupational functioning. Although a disease of elderly individuals, evidence have shown that younger adults can be affected by AD as early as 40 years of age.1,2 Currently, more than 55 million people worldwide live with dementia and the number increases by 10 million every year, with AD contributing about 60% to 70%. Hence, AD is the leading cause of dementia. 3
The AD is a disease condition that comes with episodes of symptoms like memory loss, confusion about familiar location, compromised judgement, loss of spontaneity and mood and personality changes. 4 Although antidepressants, anxiolytics, antiparkinsonian agents, β-blockers and antiepileptic drugs, among others, have been used to treat secondary symptoms associated with AD, the most effective target for AD treatment is the cholinergic transmission. 5 An association has been shown between loss of cholinergic activity and the cognitive weakness in AD. Thus, the inhibition of AChE will result in reversal of deficiency of acetylcholine, which is among the major events in the pathology of AD expressed in the cholinergic hypothesis. 6 Furthermore, the promotion of rapid deposition of β-amyloid by AChE activities can also be prevented by AChE inhibitors, especially those with dual binding capacity. 7
The standard medical treatments for AD include cholinesterase inhibitors (AChEIs) and a partial N-methyl-D-aspartate (NMDA) antagonist. Donepezil, Galantamine and Rivastigamine (AChEIs) as well as Memantine (NMDA antagonist) are for symptomatic relief and do not treat the underlying cause, nor halt the progression of the disease.5,8,9 Furthermore, many side effects of these medications and questions on their sustainability as well as lack of response by some patients have led to a search for new drugs, of which herbal medicine is a potential source. In the treatment of diseases, plants have a long history in their use as medicines and as such are important sources of drugs that cannot but be explored in treatment of AD. 10
Phyllanthus amarus (Euphorbiaceae), also known as black catnip, windbreaker, or carry me seed, is a herb well known for its medicinal properties. 11 The plant belongs to the Phyllanthaceae family, which was previously part of the Euphorbiaceae family and is mostly found in the tropical and subtropical regions of the world, including Africa, Asia, South America and West Indies.11,12 The whole plant is traditionally used to treat diabetes, jaundice, flu, gonorrhoea, menstrual problem, skin diseases and memory enhancer and dropsy.11-15 Pharmacologically, its antiviral, anticancer, anti-inflammatory, antioxidant, hypolipidemic, antiplasmodial, antimicrobial and hepatoprotective activities have been reported from various studies.16-19
Molecular docking and molecular dynamics simulation have become useful methods of screening huge number of phytochemicals and identifying potential drug candidates.20-22 Also, density functional theory (DFT) is useful in probing the electronic properties in relation to the pharmacological potentials of phytochemicals. 23
Despite the evident efficacy of the whole part of P amarus as AChE inhibitor, 24 information on the identification of phytoconstituent responsible for its activity is scanty. Therefore, this study aims to validate the AChE inhibitory efficacy of P amarus through in vitro evaluation. It further performs the molecular docking studies of phytoconstituents previously isolated from the morphological parts of the plant. Also, molecular dynamics simulation and DFT were performed to elucidate the binding mode and quantum chemical properties of its hit molecules.
Materials and Methods
Plant collection and extraction
P amarus was collected from Obafemi Awolowo University campus, Ile-Ife. Thereafter, it was identified and authenticated by Mr I. I. Ogunlowo of the Faculty of Pharmacy herbarium, Obafemi Awolowo University, Ile-Ife, Nigeria, and its voucher specimen (FPI 2353) was prepared and deposited. Later on, the whole plant was air-dried and pulverized. About 500 g of the powdered plant material was extracted with methanol with occasional shaking for 48 hours. The methanolic solution was filtered and concentrated in vacuo to afford the crude extract (5.2 g).
Receptor and ligand preparation
The 3-dimensional structure of AChE enzyme complexed with dihydrotanshinone I and coded with PDB ID: 4M0E was downloaded from the protein data bank (http://www.rcsb.org/structure/4M0E). The human acetylcholinesterase protein selected for this study consists of 4 chains. The protein’s crystal structure was prepared by removing water molecules, native ligand, co-factor, ions and 3 chains (B, C and D). Then, charges and hydrogen were added to the bare protein (chain A) using the MGLTools.
An literature review was conducted to collate the phytoconstituents previously isolated from P amarus (see supplementary material).25-36 The compounds were downloaded from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/) in SDF format, while others were built with Spartan 14 programme. Thereafter, the energy minimization of each ligand was performed under MMFF94x force field using the Open Babel interphase of the PyRx 0.8 software.
Validation of docking protocol and molecular docking studies
The docking protocol used for this study was validated before molecular docking studies were performed on AChE enzyme. In validating the docking protocol, amino acid residues resident with 5 Å in the binding site of the enzyme was chosen (Tyr72, Asp74, Tyr124, Trp286, Ser293, Val294, Phe295, Arg296, Phe297, Tyr337, Phe338 and Tyr341). Thereafter, water molecules, co-factor and native ligand were removed from the binding site of the enzymes. The native ligand removed was re-docked into the binding pocket of the bare protein. The best conformational pose of the re-docked ligand was selected and the root mean square deviation (RMSD) value was computed.
To perform molecular docking, the energy of the phytoconstituents from P amarus were minimized under MMFF94x force field using the steepest descent method for 200 steps with a step size of 0.02 and converted from PDB open babel programme in PyRx 0.8 software. Also, the bare proteins were prepared by adding polar hydrogens and merging non-polar hydrogens with MGLTools 1.5.2. Next, the grid box size was set at 403 and centre at X = –14.3536, Y = –42.8929, and Z = 24.9102 for the AChE enzyme. Molecular docking was performed to exhaustiveness of 50 using the AutoDock Vina tool in PyRx 0.8. 37 Results obtained with the lowest RMSD value and best binding energy. Later on, interactions like hydrogen bond, hydrophobic and pi were chosen and analysed using Discovery studio visualizer (2020).
Molecular dynamics simulation
The molecular dynamics simulation study was conducted in YASARA dynamics 38 with the aid of the AMBER14 force field. 39 A cubic simulation cell was created with a periodic boundary conditions and TIP3P water solvation model was used. 40 The docked complexes were initially cleaned, optimized and hydrogen bonds were oriented. The simulation cell was extended up to 20 Å at the each case of the complexes. The physiological conditions of the simulations cells were set as 298 K temperature, pH 7.4% and 0.9% NaCl. The long-range electrostatic interactions were calculated by the Particle Mesh Ewald algorithms by a cut-off radius of 8.0 Å.41-43 The initial energy minimizations of the complexes were done by the steepest gradient algorithms by simulated annealing methods (5000 cycles). The time step of the simulation cell was set as 2.0 fs. The simulation trajectories were saved after every 100 ps. 44 By following constant pressure and Berendsen thermostat, the simulations was extended to 50 ns. The simulations trajectories were used to calculate the RMSD, root mean square fluctuation (RMSF), hydrogen bond, solvent-accessible surface area (SASA) and radius of gyrations.45-49
Theoretical modelling and optimization studies
The DFT analysis of the 5 hit molecules from the molecular docking studies with the AChE enzyme was carried out using the Spartan 14 programme containing the functional B3LYP (Lee-Yang Parr exchange-correlation functional method) using a 6-31G basis set. 50 During the calculations, the values of the frontier orbital energies, energy gap, chemical potential, chemical hardness, softness and electrophilicity index were calculated.
In vitro acetylcholinesterase inhibitory assay
The AChE inhibitory activity of the methanolic crude extract of P amarus was evaluated by Ellman method 51 as described by Obuotor. 52 The 96-well plates were added with 240 µl of buffer (50 mM Tris-HCl, pH 8.0), 20 µl of varying concentrations (10, 5, 2.5 and 1.25 mg/ml) of the extract dissolved in 5% dimethyl sulphoxide and 20 µl of the enzyme preparation. The reaction mixture was then incubated for 30 min at 37°C, followed by the addition of 20 µl of 10 mM DTNB (5,5ʹ-dithiobis-(2-nitrobenzoic acid)). The reaction was initiated by addition of 20 µl of 25 mM ATChI. The rate of hydrolysis of ATChI was then determined spectrophotometrically by measuring the change in the absorbance per minute (∆A/min) due to the formation of the yellow 5-thio-2-nitrobenzoate anion at 412 nm over a period of 4 minutes at 30 seconds interval. The buffer solution was used as a negative control. The percentage inhibition (%I) of each sample and the positive control (eserine) were obtained using the formula

where: I (%) = percentage inhibition, Vi = enzyme activity in the presence of the extract and positive control (eserine) and Vo = enzyme activity in the absence of the extract and positive control (eserine).
Results and Discussion
Analysis of molecular docking studies with acetylcholinesterase enzyme
In validating the docking protocol, co-crystallized and re-docked ligands were aligned and the RMSD value between them was calculated (Figure 1). The RMSD value obtained at 0.01 Å was low and within the acceptable range.

Superimposed image of co-crystallized ligand (green) and re-docked ligand (yellow).
Molecular docking has become a useful method of predicting the bioactive potential of chemical compounds against a target receptor implicated in a disease condition.53-55 In this study, 5 top-ranked molecules were selected based on their binding energy with the AChE enzyme compared with eserine (–8.2 kcal/mol) (Table 1).
Detailed interaction analysis of top-ranked molecules with AChE enzyme.

Abbreviation: AChE, acetylcholinesterase.
Amarosterol A elicited the best binding energy of –10.0 kcal/mol towards the AChE enzyme. It further established 16 hydrophobic interactions with Tyr124, Phe297, Tyr337, Tyr341 and His441. Also, both pi-sigma and pi-alkyl interactions were formed with Tyr124, Phe297, Tyr337, Tyr341 and His441. However, no hydrogen bonding and electrostatic interactions were established between the AChE enzyme and amarosterol A moiety (Figure 2A).

Interaction between amino acid residues in the binding site of AChE and amarosterol A (A), hinokinin (B), β-sitosterol (C), stigmasterol (D) and ellagic acid (E).
Hinokinin gave a considerably high binding affinity towards the AChE enzyme at –9.8 kcal/mol. Also, the oxygen atom on the hinokinin moiety participated in 2 hydrogen bond interactions with Tyr124 at 2.3133 Å and Phe295 at 2.4255. The phytochemical further established 3 hydrophobic interactions with Tyr286 andTyr341. Furthermore, 2 pi-alkyl interactions were observed between the hinokinin moiety and Tyr286 and Tyr341 (Figure 2B).
Beta-sitosterol elicited a binding energy of –9.7 kcal/mol at the active site of the AChE enzyme. Six amino acid residues (Tyr124, Trp286, Phe297, Tyr337, Phe338 and Tyr341) participated in 10 hydrophobic interactions with the β-sitosterol moiety. In addition, pi-sigma and pi-alkyl interactions were observed with Tyr124, Trp286, Phe297, Tyr337, Phe338 and Tyr341, while no hydrogen bonding and electrostatic interactions were established between the β-sitosterol moiety and the active site of the AChE enzyme (Figure 2C).
Stigmasterol inhibited the AChE enzyme with a binding energy of –9.5 kcal/mol. It further established 14 hydrophobic interactions with Tyr124, Trp286, Leu289, Val294, Tyr337, Phe338 and Tyr341. Also, both pi-sigma and pi-alkyl interactions were formed with Tyr124, Trp286, Tyr337, Phe338 and Tyr341. However, no hydrogen bonding and electrostatic interactions were established between the AChE enzyme and amarosterol A moiety (Figure 2D).
Ellagic acid gave binding energy of –9.2 kcal/mol with the AChE enzyme’s active site. Three hydrogen bonding interactions (Tyr124 at 2.1989 Å, Phe295 at 2.2097 Å and Tyr74 at 1.9720 Å) were established between 2 oxygen and 1 hydrogen atoms. Furthermore, 9 hydrophobic interactions were formed between Trp286 and Phe341, while pi-pi stacked interactions were established with Trp286 and Phe341. However, no electrostatic interaction was formed with the AChE enzyme (Figure 2E).
The top-ranked phytochemicals and eserine established common hydrophobic and pi interactions with Tyr286. However, only the phytochemicals formed common hydrophobic and pi interactions with Tyr341. Generally, the hydrogen bond, hydrophobic and pi interactions formed between the each phytochemical and the active site of the AChE enzyme account for the unique stability and high binding affinity exhibited by each molecule in the receptor’s binding pocket. Hence, the binding energies of the top-ranked phytochemicals are in the order of amarosterol (10.0 kcal/mol) > hinokinin (–9.8 kcal/mol) > β-sitosterol (–9.7 kcal/mol) > stigmasterol (–9.5 kcal/mol) > ellagic acid (–9.2 kcal/mol) > eserine (–8.2 kcal/mol). The results obtained showed that amarosterol A, hinokinin, β-sitosterol, stigmasterol and ellagic acid played a significant role in the acetylcholinesterase activity observed from the methanolic extract of P amarus.
Molecular dynamics simulation of top-ranked molecules with AChE enzyme
The molecular dynamics simulation study was conducted to understand the structural variations among the docked complexes across the simulation trajectories. The RMSDs from the C-alpha atoms were assessed to understand the stable nature of the complexes. The RMSD value from the C-alpha atoms from the simulations study demonstrates the degree of flexibility and the mobile nature of the complexes. The higher RMSD related to the more flexible nature, whereas the lower RMSD defines more stable nature of the complexes. Figure 3A indicates that the hinokinin, amarosterol A, β-itosterol, stigmasterol and ellagic acid had initial upper trend and this might be responsible for the flexible nature of the complexes. Therefore, all of the complexes tend to be reached in the steady state after 15 ns times. The β-sitosterol and stigmasterol had comparatively higher trend in RMSD which indicates the relatively flexible nature of this complexes, whereas the hinokinin and amarosterol A had lower RMSD than other complexes. However, all complexes exhibit RMSD lower than 2.5 Å which demonstrates the docked complexes’ rigid nature in atomistic simulations.

Amarosterol A, hinokinin, β-sitosterol, stigmasterol and ellagic acid with 4M0E; RMSD (A), SASA (B), radius of gyration (C), hydrogen bond (D) and RMSF (E).
Moreover, the SASA of the complexes was analysed to understand the changes in the protein surface area where the higher SASA related to the expansion of the surface area and the lower SASA defines the truncated nature of the complexes. Figure 3B indicates that the ellagic acid exhibits relatively higher SASA which demonstrates the extended surface area of the protein complexes upon ligand bindings. The other 4 complexes exhibited a similar trend in SASA and maintained lower degrees of deviations in simulations. The SASA results from the simulations trajectories indicate that the upon binding with the ligand molecules the complexes were stable except ellagic acid.
The radius of gyrations of the complexes was also assessed to understand the mobile nature of the complexes where higher Rg related to the more flexibility while lower Rg related to the strict nature of the complexes. Figure 3C indicates the hinokinin had the lower Rg than other complexes and stigmasterol had the higher Rg; this trend correlates with the RMSD trend of these complexes in simulations. These results demonstrate that the stigmasterol complexes exhibit more flexibility, whereas the hinokinin had more rigid nature.
The hydrogen bond plays an important role in determining the ligand protein interactions in the macromolecular systems. Figure 3D indicates that all complexes had stable hydrogen bond patterning in simulations. Therefore, the RMSFs of the complexes were assessed to explore the flexibility across the amino acid residues of the protein. Figure 3E indicates that maximum residues had RMSF of less than 2.5 Å which defines the stability of the docked complexes. These results indicate that maximum residues from the all complexes exhibit more stable nature.
Frontier molecular orbital and quantum chemical calculation
The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of molecules are vital parameters that give detailed understanding on the chemical reactivity and stability of chemical constituents. They play important roles in the interactions of the compounds with different enzymes. 56 The HOMO helps to explain the electron-donating potential of the molecule while the LUMO is associated with the electron-accepting ability of the molecule. Therefore, a higher EHOMO value corresponds to a high-donating ability while a low ELUMO value corresponds to a better accepting ability. 57
The energy gap is a parameter obtained from the EHOMO and ELUMO values and has found wide usefulness in predicting the chemical reactivity of chemical compounds. 58 It is the difference between the ELUMO and EHOMO. Hence, a molecule has high stability and lower reactivity when it has a higher energy gap, while a lower stability and high reactivity corresponds to a low energy gap. 57 Sitosterol elicited the highest energy gap while ellagic acid has the lowest energy gap, thereby suggesting that sitosterol is the most stable molecule and least reactive while ellagic acid is the least stable and most reactive phytochemical. Hence, the calculated energy gap value is in the order of sitosterol > stigmasterol > amarosterol A > hinokinin > ellagic acid (Figures 4 and 5).
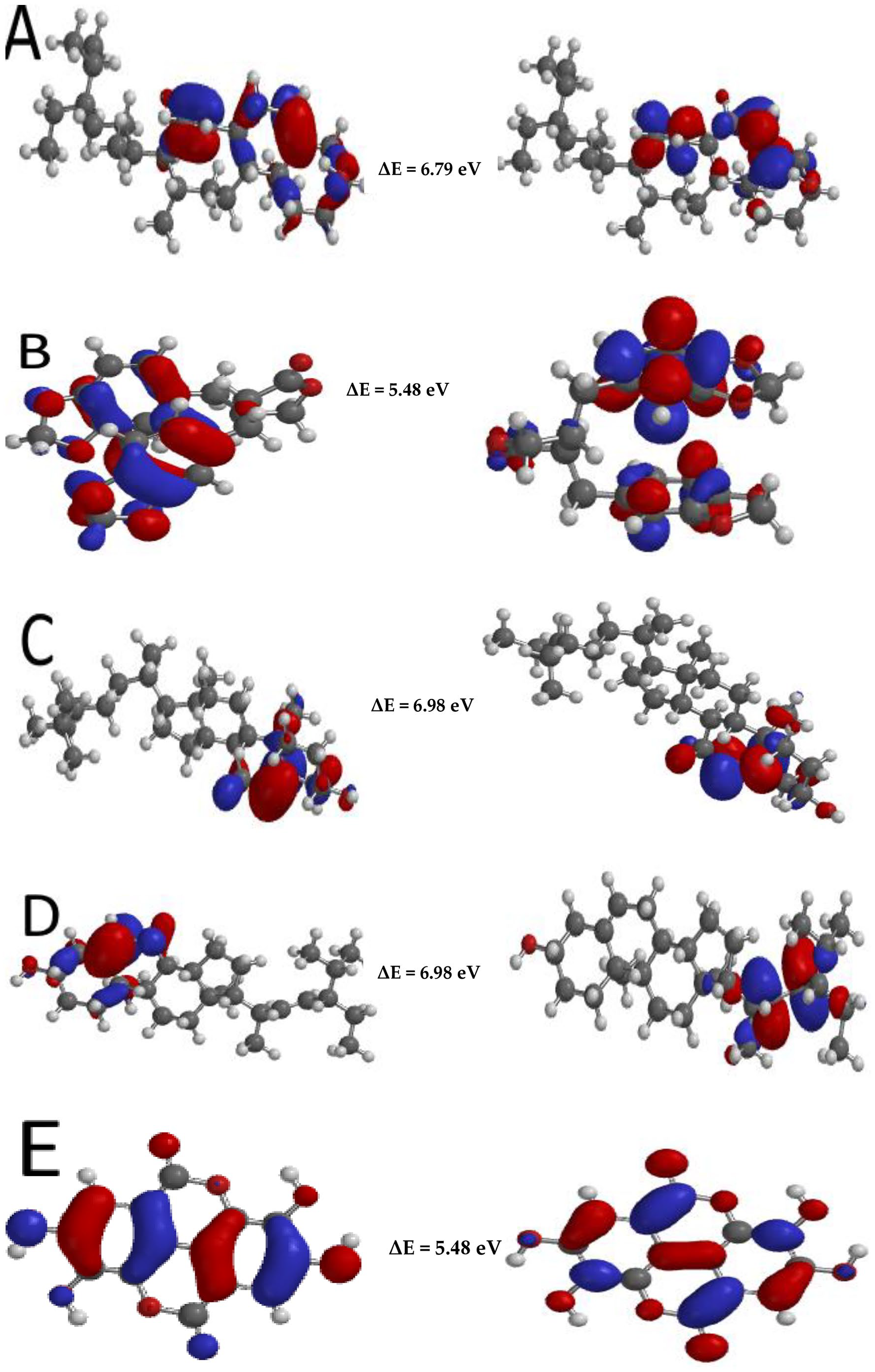
Illustration of the distribution map of the HOMO (A-E), LUMO (unlettered) and the gap energies of amarosterol A (A), hinokinin (B), β-sitosterol (C), stigmasterol (D) and ellagic acid (E).
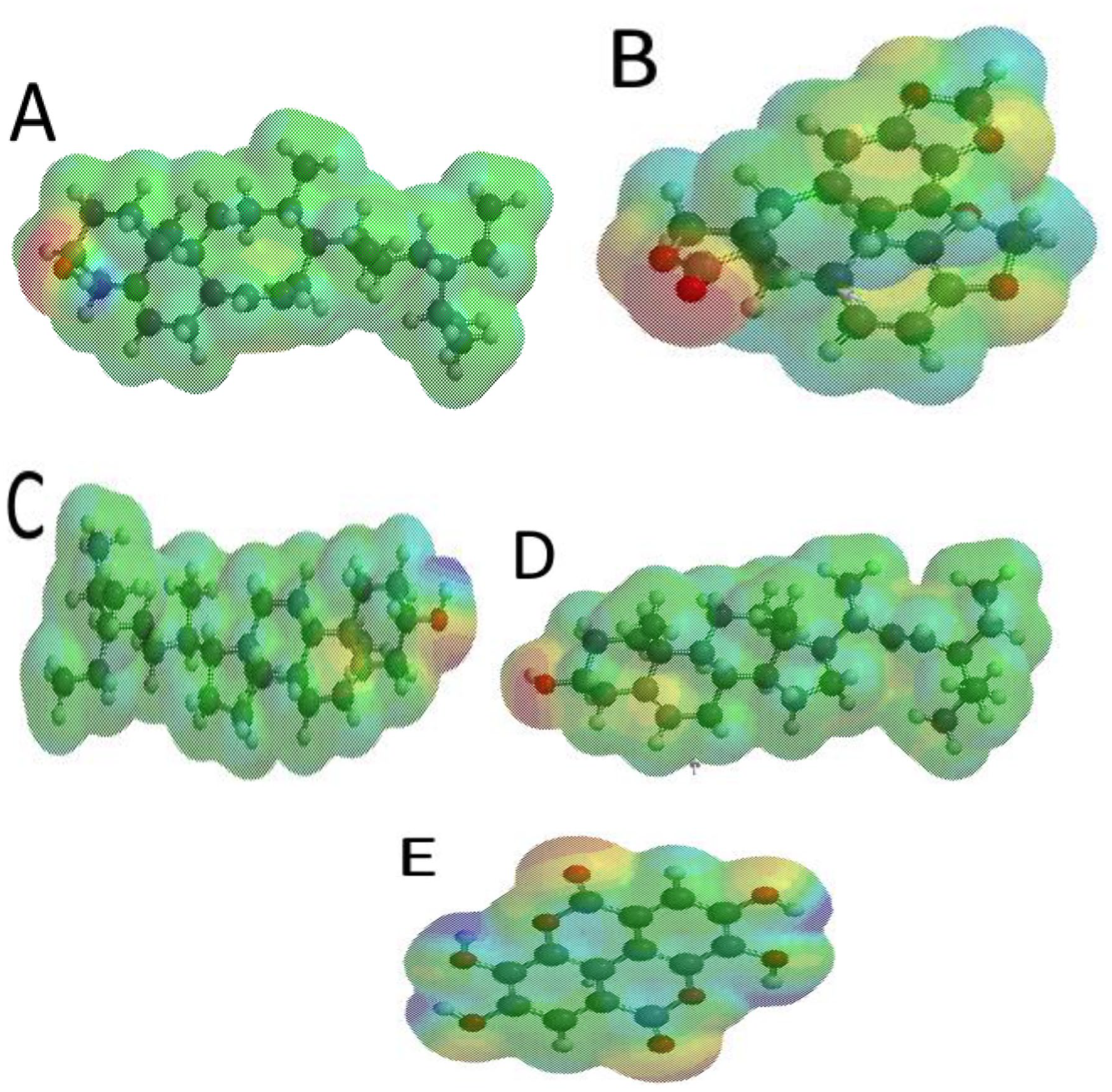
Molecular potential surface of amarosterol A (A), hinokinin (B), β-sitosterol (C), stigmasterol (D) and ellagic acid (E).
To have a vivid understanding of the electronic properties of the phytochemicals, important electronic parameters like chemical potential (μ), chemical hardness (η), softness (S) and electrophilicity index (ω) were calculated (Table 2).
EHOMO, ELUMO and electronic properties of the chemical compounds.

Abbreviations: HOMO, highest occupied molecular orbital; LUMO, lowest unoccupied molecular orbital.
The chemical hardness of a molecule is referred to as the opposition of a molecule to exchange electron density with the environment. 56 A hard molecule is characterized by a large energy gap while soft molecules possess a low energy gap. 57 The energy gap and the hardness of the compounds followed the order ellagic acid < hinokinin < amarosterol A < stigmasterol < sitosterol. Therefore, sitosterol is the hardest while ellagic acid is the softest (Table 2).
Chemical potential are changes in the energy of a molecule with respect to the electron number at a fixed potential. It elucidates the potential of a chemical compound to exchange electron density with its environment at the ground state and also linked to the electrophilicity index of the molecules.57,58 Electrophilicity index is a measure of the energy stabilization of a chemical compound as it acquires an extra amount of electronic density from the environment. A strong electrophile possesses a low chemical potential and high chemical hardness, while a weak electrophile possesses high chemical potential and low chemical hardness. Hence, strong electrophiles exhibit high electrophilicity index values while weak electrophiles possess higher lower electrophilicity values.58-60 Ellagic acid had the highest electrophilicity index while stigmasterol had the lowest. Since the calculated values are more than 0.8, it can be inferred that all the hit molecules had high electrophilicity index values and are likely to act as electrophiles (Table 2).
Molecular electrostatic potential (MESP) is useful in investigating the chemical reactivity of molecules.60,61 It is a plot of electrostatic potential over constant electron density of molecular systems. The MESP provides a visual representation of the molecular size, shape and potential (positive, negative and neutral) of molecules through a colour grading representation. 23 The molecular electrostatic potential can help in the identification of the reactive sites of electrophilic and nucleophilic attack in bonding interactions and in the field of biological recognition. 62
In vitro acetylcholinesterase inhibitory activity of extract of P amarus
The methanolic extract of P amarus was tested for its AChE inhibitory potential and compared with eserine (standard drug). The results showed that the extract elicited a considerably high AChE inhibitory activity at IC50 = 0.09 ± 0.02 mg/ml compared with eserine at 1.93 ± 0.04 µg/ml. The methanolic extract of P amarus showed better AChE inhibitory activity compared with the extracts of Ipomoea aquatica, Terminalia bellirica and Nelumbo nucifera.62-64 Polar and non-polar chemical constituents have been identified as potent AChE inhibitors. 65 In our study, the AChE activity elicited by the methanolic extract of P amarus can be attributed to the presence of phytochemicals like tannins and terpenoids that are resident in the extract. This can be further substantiated from the binding energy elicited by the terpenoids (amarosterol A, β-sitosterol, stigmasterol and hinokinin) and tannin (ellagic acid).
Conclusions
This study validated the ethnomedicinal usage of P amarus as a memory-enhancing agent. The binding affinity obtained from the molecular docking studies identified compounds in terpenoids and tannin classes as therapeutic agents responsible for the inhibition of AChE. The RMSD and RMSF plots obtained for the top-ranked molecules in the binding pockets of the AChE enzyme showed their stability throughout the entire simulation period. The EHOMO, ELUMO, energy gap and other parameters in the quantum chemical calculations revealed amarosterol A, hinokinin, β-sitosterol, stigmasterol and ellagic acid as promising AChE inhibitors. Hence, the in silico AChE inhibitory activity of the phytochemicals is in the order of amarosterol A > hinokinin > β-sitosterol > stigmasterol > ellagic acid. The results obtained need to be further validated through in vitro studies to attest to the activity of the compounds.
Supplemental Material
sj-docx-1-bbi-10.1177_11779322221118330 – Supplemental material for Revealing the Acetylcholinesterase Inhibitory Potential of Phyllanthus amarus and Its Phytoconstituents: In Vitro and in Silico Approach
Supplemental material, sj-docx-1-bbi-10.1177_11779322221118330 for Revealing the Acetylcholinesterase Inhibitory Potential of Phyllanthus amarus and Its Phytoconstituents: In Vitro and in Silico Approach by Kolade O Faloye, Shafi Mahmud, Emmanuel G Fakola, Yemisi M Oyetunde, Sunday J Fajobi, Jeremiah P Ugwo, Ayobami J Olusola, Samson O Famuyiwa, Oluwabukunmi G Olajubutu, Temitope I Oguntade and Ahmad J Obaidullah in Bioinformatics and Biology Insights
Footnotes
Funding:
The author(s) received no financial support for the research, authorship and/or publication of this article.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
Conceptualization, K.O.F and S.O.F; methodology K.O.F, S.M, O.G.O, S.J.F and E.G.F; data analysis, O.G.O, S.J.F and J.P.U; writing – original draft preparation, K.O.F, S.M, Y.M.O and E.G.F; writing – review and editing, S.O.F, K.O.F, S.M, and E.G.F; supervision, K.O.F, S.O.F, Y.M.O and E.G.F; funding acquisition, S.M. All authors have read and agreed to the published version of the manuscript.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.