
Abstract
Objective:
Mycobacterium leprae causes leprosy, an infectious disease that has persisted over centuries and is still an issue in public health in many nations. Diverse bioinformatics techniques have been effectively employed to annotate the functions of hypothetical proteins (HPs) originating from different pathogenic bacteria. The objective of the current research was to elucidate the functions of an HP obtained from M. leprae.
Methods:
A variety of in silico tools were utilized to make predictions regarding the structure and function of this protein. To identify homologous proteins, the BLASTp program was used to search for sequence similarity across the available biological databases. Additionally, using the proper bioinformatics methods, a number of properties were determined, including physicochemical characteristics, subcellular localization, phylogenetic analysis, functional annotation, pathway analysis, protein-protein interaction, secondary and tertiary structure determination, active site detection, quality assessment analysis, molecular docking, pharmacokinetic and toxicity profiling, and further molecular dynamics simulations.
Results:
The HP exhibited putative biological activity associated with a conserved functional domain, the CT_C_D superfamily domain. The allophanate hydrolase activity of the chosen HP was predicted by the functional annotation. Pathway analysis demonstrated the protein’s involvement in cellular and metabolic processes. Numerous functional partners that play a crucial role in bacterial survival were identified through the chosen HP’s protein-protein interactions. Furthermore, active site prediction and molecular docking analysis of the HP with ligands indicated that it could be a therapeutic target for M. leprae. ADMET analysis indicated that the selected compound has favorable bioavailability, drug-likeness, and safety. The stability of these complexes was verified by molecular dynamics simulations, which suggests they have therapeutic potential.
Conclusion:
This study emphasizes the effectiveness of in silico methods in predicting the biological functions of HP and generating hypotheses for potential therapeutic targets.
Introduction
Leprosy, or Hansen’s disease, is a severe neurological and infectious disease caused by Mycobacterium leprae (M. leprae), an intracellular, gram-positive, rod-shaped, slow-growing, and non-motile bacterium.1-3 M. leprae was first discovered in Norway in 1873 by G. H. Armauer Hansen, marking the first identification of a bacterium as the cause of a human disease.4,5 This pathogen exhibits a specific affinity for peripheral nerves, eyes, skin tissues, and mucosal surfaces of the upper respiratory tract, leading to nerve damage and disfigurement. In human leprosy, multiple organs, including the liver, spleen, bone marrow, lungs, lymph nodes, kidney, esophagus, and testes, can also be affected.3,6,7 Despite advancements in disease control, leprosy remains a significant global health concern. Over the past 2 decades, approximately 16 million leprosy cases have been recorded, with more than 200,000 new cases registered annually, according to 2020 data.7,8 Endemic regions such as Brazil, Congo, Madagascar, Mozambique, Nepal, and Tanzania continue to struggle with disease management due to a lack of adequate knowledge and resources. 9 As an obligate intracellular pathogen, M. leprae relies on host cells for survival and replication. It has the smallest genome (~3.3 Mb) among the Mycobacterium genus, with 1614 protein-coding genes and approximately 1300 pseudogenes.10-13 Notably, M. leprae primarily invades Schwann cells of the peripheral nervous system (PNS), which form myelin sheaths around axons and facilitate nerve function.14-17 In addition to humans, this pathogen can infect 9-banded armadillos and red squirrels, causing a disease similar to leprosy.18,19
With the advent of next-generation sequencing (NGS), the analysis of large genomic datasets has become more efficient, enabling the identification of hypothetical proteins (HPs) within various organisms. 20 HPs are proteins with unidentified functions in vivo, and their characterization can provide valuable insights into genomic and proteomic functions.21-23 Understanding the structural and functional properties of HPs facilitates the discovery of novel protein structures, pathways, and molecular interactions, enhancing our knowledge of disease mechanisms and potential therapeutic targets. 24 Computational approaches and in silico techniques have proven essential for annotating HPs, elucidating protein-protein interactions, and identifying novel drug targets. 25 HPs hold significant promise in drug discovery and vaccine development, offering advantages such as 3-dimensional (3D) structural predictions, novel domain identification, phylogenetic profiling, and functional annotation.26-30
“In this study, we conducted a comprehensive in silico analysis of the hypothetical protein (HP) with the accession number CAC32080.1 from the M. leprae TN strain. This HP was prioritized from the hypothetical proteome because of its high sequence conservation across M. leprae strains, absence of close human homologs, and predicted involvement in nitrogen metabolism pathways that are essential for bacterial survival.”
The M. leprae TN strain consists of 1605 proteins, approximately 595 of which are classified as hypothetical. 31 Given the potential role of HPs in disease pathogenesis and therapeutic intervention, their annotation is crucial for advancing research on leprosy. In this study, we conducted a comprehensive in silico analysis of the HP with the accession number CAC32080.1 from the M. leprae TN strain, which was selected because of its conservation across M. leprae strains and its predicted involvement in nitrogen metabolism pathways that are critical for bacterial survival. Our analysis aimed to characterize its physicochemical properties, secondary structure, subcellular localization, active sites, pathway analysis, and protein-protein interactions. Additionally, homology modeling techniques were employed to generate a high-quality structural model of CAC32080.1, providing deeper insights into its functional significance in M. leprae biology. Furthermore, molecular docking studies and molecular dynamics simulations were performed to investigate its interactions with potential ligands, along with pharmacokinetic and toxicity profiling to evaluate drug-likeness and safety. This study may facilitate the identification of novel pharmacological targets for screening, drug development, and development of treatments for leprosy disease.
Methods
HP Sequence Retrieval From NCBI
The protein database of the National Center for Biotechnology Information (NCBI) was used to identify the HP of M. leprae. The protein’s primary sequence was retrieved in FASTA format to facilitate further investigation. The steps involved in this study are depicted in Figure 1, and Table 1 contains the full list of the databases and software that were employed in this study.

Workflow for in silico functional annotation and molecular characterization of the targeted HP.
The List of the Databases Utilized for Functional Annotation and Molecular Characterization of HP From M. leprae.

Physicochemical Properties Analysis
The ProtParam tool provided by Expasy, 32 a web tool was used for analyzing the physicochemical properties of the specific protein sequence. The tool was employed for the analysis of various properties encompassing molecular weight, atomic composition, estimation of half-life, theoretical isoelectric point (pl), amino acid composition, aliphatic index, stability index, count of positive and negative residues, and grand average of hydropathicity (GRAVY) of the HP.
Prediction of Subcellular Localization
The subcellular localization of proteins is essential to the efficient functioning of proteins and the accurate interpretation of genetic information. The CELLO 33 server was used to predict the subcellular location of the chosen HP. The PSORTb 34 and SOSUI 35 servers were also utilized to confirm the subcellular localization. Furthermore, the results were verified by employing the TMHMM, 36 and HMMTOP 37 tools.
Multiple Sequence Alignment and Phylogenetic Analysis
The BLASTp program at NCBI was used to do a similarity search against non-redundant protein sequences, 38 intending to locate homologous proteins from related animals that potentially share structural similarities with the chosen HP.
CLC Sequence Viewer version 8.0 was utilized to perform a multiple sequence alignment between the chosen HP and additional proteins acquired using the NCBI-BLASTp program. It was also used to conduct a phylogenetic analysis of the selected protein sequences.
Prediction of Function by Domain Analysis
An analysis of the HP domain was performed using NCBI Conserved Domain Search, 39 Pfam, 40 and InterProScan. 41 Conserved domain search was utilized to detect the presence of conserved domains in a given sequence of protein. It functions by performing comparison analysis via utilization of Reverse Position-Specific BLAST (RPS-BLAST) against position-specific score matrices resulting from alignments with the present conserved domains in the Conserved Domain Database (CDD). Pfam is a database of protein families that comprise multiple sequence alignments generated utilizing hidden Markov models (HMMs) and annotations. 40 This database is currently hosted by InterPro. The MOTIF server was employed to analyze the protein sequence motif. 42
Pathway Analysis
The KEGG server 43 was used to find the KEGG pathway data sets. The HP included in this investigation has the KEGG ID K14541. With the help of the KEGG pathway database, which is an extensive collection of expert-reviewed pathway maps, we can better comprehend the biochemical reactions, interactions, and relationship networks involved in a wide range of biological processes. These cover the development of drugs, human diseases, cellular processes, organismal systems, genetic information processing, and environmental information processing, as well as metabolism.
Protein-Protein Interaction Analysis
Prediction of the activity and function of a protein is frequently modulated by its interaction with other proteins. Thus, a thorough comprehension of the interactions among proteins may provide crucial information for predicting a protein’s function. In this study, the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING Version: 12.0), was used to perform an analysis of the HP’s interacting counterparts. 44 The interactions comprise functional (indirect) as well as physical (direct) associations, experimental or co-expression. The STRING server integrates the data of interactions from an ample number of organism sources quantitatively and performs information transfer between the organisms whenever appropriate.
Prediction of Secondary Structure
The secondary structure of the HP was predicted to assess the location of its amino acid residues. The SOPMA server 45 was used to conduct secondary structure predictions for the HP, and the PSIPRED server 46 was used to verify the precision of the SOPMA findings. The results of this study suggested the existence of random coils, alpha-helices, and beta-sheets.
Homology Identification and Quality Assessment of HP
Utilizing the Modeller 60 with the HHpred tool, 47 the 3D model of the HP was predicted. The YASARA 48 force field minimizer was employed to minimize the energy of the 3D model structure. After energy minimization, we refined the obtained model using GalaxyRefine-GalaxyWEB, an ab initio-based refinement server that improves protein structures based on input sequences. 49 The visualization of the 3D model structure was obtained using the PyMOL software.
A variety of assessment tools were employed to evaluate the accuracy of the predicted 3D structure. These are the PROCHECK, 50 ProSA-web, 51 Verify3D, 52 ERRAT, 53 and QMEAN. 54
Active Site Identification
The size and form of protein cavities and pockets are essential for active site evaluation and structure-based ligand design. In this study, the Computed Atlas of Surface Topography of Proteins (CASTp) was employed to identify potential binding sites, pockets, and cavities within the 3D structure of the target protein. 55
Molecular Docking and Normal Mode Analysis
Interactions between the chosen protein and 2 different ligand molecules were investigated through the use of molecular docking methods. One of the ligands acts as a substrate for the target protein, whereas the other acts as an inhibitor. Proper analysis through literature reviews was used to select ligands. Thus, allophanate and phenyl phosphorodiamidate were used as ligands in docking experiments with HP, where allophanate functions as a substrate and phenyl phosphorodiamidate as an inhibitor. 61 The 3D structure of the ligand was retrieved by using the PubChem database in the format of the Spatial Data File (SDF). 56 We utilized the AutoDock Vina using PyRx software to conduct blind docking analysis and predict the interactions between the HP and ligands. 62 In order to construct a grid box for the docking performance, the following parameters were set: exhaustiveness = 8, center x:y:z: = 34.5070:23.8889:63.3997, and dimension x:y:z = 53.9956:49.9286:37.6723. Our final analysis of the hydrophobic and hydrogen bond interactions was conducted using the Biovia Discovery Studio visualizer software. 63
Analysis of Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) Properties
To understand the drug-likeness of the selected inhibitor compound, the drug’s pharmacokinetics (PK) and ADME were analyzed. As a first step, the web-based SwissADME tool 57 was used and analyzed the regulatory parameters for the estimation of the oral bioavailability, such as molecular weight (MW), the number of hydrogen bond donors (HBD) and acceptors (HBA), topological polar surface area (TPSA) and the violations of Lipinski’s Rule of 5. For more refined ADME profiling, the QikProp module of the Schrödinger suite was used. This module predicted pharmacokinetic parameters which included predicted human oral absorption, Caco-2 permeability, brain/blood partition coefficient (QPlogBB), and the potential hERG channel inhibition (QPlogHERG). For predicting the toxicological potential of the target inhibitor compound, the ProTox-3.0 online server 58 was used. This server was used to assess parameters predicting possible organ- or cell-specific toxic risks related to cytotoxicity, immunotoxicity, mutagenicity, carcinogenicity, and to assess potential organ- or cell-specific toxic risks.
Normal Mode Analysis Using iMODS
Molecular dynamics (MD) simulation was conducted on the receptor-ligand complex that was derived from molecular docking assays to verify the stability and interaction mechanisms in a dynamic context. The mobility of the internal coordinates was determined by applying MD simulation to the HP-ligand complex by normal mode analysis (NMA) on the iMODS website. 59 In comparison to other MD simulation processes, this one is faster and less expensive. 64 This web server estimates eigenvalues, deformability, B-factors, and covariance. Eigenvalues are used to determine motif stiffness, and the deformity of the main chain is anticipated based on the efficacy measurement of the biological targets. 65
Molecular Dynamics Simulation
Using the GROningen MAchine for Chemical Simulations, also known as GROMACS (version 2022.3), 66 an MD simulation of the targeted HP-ligand complex was conducted for 100 ns. For the simulation, the CHARMM36 m force field was utilized. A water box was constructed using the TIP3P water model, with its edges spaced 10 nm apart from the protein’s surface. Using the necessary ions, the systems were neutralized. Following energy minimization, isothermal-isochoric (NVT) equilibration, and isobaric (NPT) equilibration of the system, a 100 ns MD simulation was performed with periodic boundary conditions and a time integration step of 2 fs. The trajectory data was analyzed at a snapshot interval of 100 ps. The root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), and solvent accessible surface area (SASA) analyses were performed using the rmsd, rmsf, gyrate, and sasa modules contained within the GROMACS software when the simulation was finished. The plots for these analyses were generated using RStudio’s ggplot2 tool.
Result and Discussion
Data Retrieval From the NCBI for HP
Functional relevance led to the selection of CAC32080.1, a 222-amino HP from M. leprae. It was identified as a conserved domain that is associated with vital biochemical pathways in nitrogen metabolism, which are essential for the survival of pathogens, among 595 HPs. Table 2 presents data obtained from the NCBI database.
Retrieval of HP Sequence of M. leprae.

Physicochemical Properties Analysis of the Chosen Protein Sequence
The ProtParam tool was utilized to evaluate several physicochemical properties of the HP (Table 3). The analysis of the physicochemical parameters indicated that the selected HP consists of 222 amino acids, with an estimated molecular weight of 23 992.32 Da. Its theoretical isoelectric point (pI) was estimated to be 8.66. The protein’s computed instability index of 21.43 classified it as stable, as an instability index value below 40 suggests stability, while a value above 40 implies instability. 67 The aliphatic index of the chosen protein (89.59) suggests that it is stable throughout a large temperature range. At the same time, the negative GRAVY value (−0.159) indicates that the HP is hydrophilic. 68
Physicochemical Properties of HP Protein as Predicted by the ExPASy Server.

Subcellular Localization Prediction
Predicting the features of a HP relies on determining its subcellular localization. A protein’s subcellular localization refers to its specific location within a cell, which can include the outer membrane, inner membrane, periplasm, extracellular space, or cytoplasm. 69 The results obtained from the CELLO server suggested that the HP is a membrane protein, whereas the PSORTb servers indicated it is a cytoplasmic protein. The SOSUI server identified the specified protein as a soluble protein. TMHMM and HMMTOP predicted no transmembrane helices. The results from the servers, namely PSORTb, SOSUI, TMHMM, and HMMTOP, are shown in Table 4.
Subcellular Localization of HP.
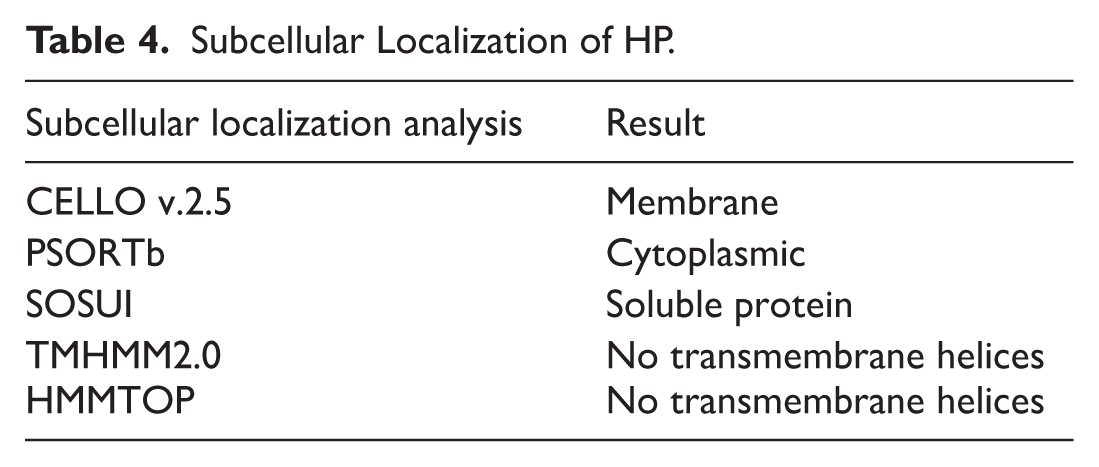
Considering that TMHMM and HMMTOP are specialized in identifying transmembrane helices and have predicted none, the protein is unlikely to be membrane-bound. The cytoplasmic and soluble predictions from PSORTb and SOSUI reinforce this hypothesis. The membrane localization suggested by CELLO may result from sequence characteristics similar to those of membrane proteins, yet without actual transmembrane domains. Consequently, synthesizing these findings, the protein is most likely cytoplasmic or soluble in nature.
Multiple Sequence Alignment and Phylogenetic Assessment
The BLASTp analysis against the non-redundant database indicated similarities with other allophanate hydrolase proteins, as indicated in Table 5.
Top Homologs of CAC32080.1 Identified Via BLASTp Search.

A limited number of proteins obtained from the BLASTp results were subjected to multiple sequence alignments to identify conserved and dissimilar residues among homologs (Figure 2). The alignment confirmed the high degree of conservation across homologs, as it indicated sequence identity ranging from 76.17% to 100%. According to BLASTp homology analysis, the query protein is structurally related to allophanate hydrolase proteins found in other species of Mycobacterium. The sequences chosen using BLASTp were aligned using CLC Sequence Viewer 8, a software which performs multiple sequence alignment. Using this same data, a phylogenetic tree was constructed (Figure 3). The target protein and another additional protein (WP_010908952.1.1-222) from M. leprae appear to share a common ancestor according to the phylogenetic tree analysis conducted using CLC Sequence Viewer 8. The scale bar is utilized to estimate sequence divergence, while the line segment bearing the numerical value (0.130) represents the level of genetic change.

Multiple sequence alignment of CAC32080.1 (row 4) and its top homologous proteins. M. lepromatosis is represented by rows 1 to 3, M. leprae by rows 4 to 5, M. haemophilum by rows 6 to 7, and different M. species by rows 8 to 11. The alignment was conducted via CLC Sequence Viewer 8.0.

The Phylogenetic tree was constructed using the CLC Sequence Viewer version 8. The scale bar estimates sequence divergence, while the line segment bearing the value (0.130) represents the quantity of genetic change.
Functional Annotation Analysis
Functional annotation of the chosen HP was conducted using NCBI Conserved Domain Search, Pfam, and InterProScan to identify conserved domains and predict its function. These analyses suggested the presence of a carboxyltransferase domain, subdomains C and D (CT_C_D), which are essential for the enzyme urea carboxylase’s function.
The NCBI-CDD server identified a specific hit with allophanate hydrolase subunit 1 (COG2049) in the target HP, with the CT_C_D domain spanning amino acid residues 25-221 and an E-value of 7.78E-84 (Figure 4). Similarly, Pfam detected the carboxyltransferase domain (PF02682) between residues 21-211, with an E-value of 3.30E-57. InterProScan further confirmed the presence of this domain (InterPro Entry: IPR003833) at positions 19-211, with an E-value of 3.7E-87. Additionally, the MOTIF database identified the same domain at residues 21-211, with an E-value of 1.1E-60.
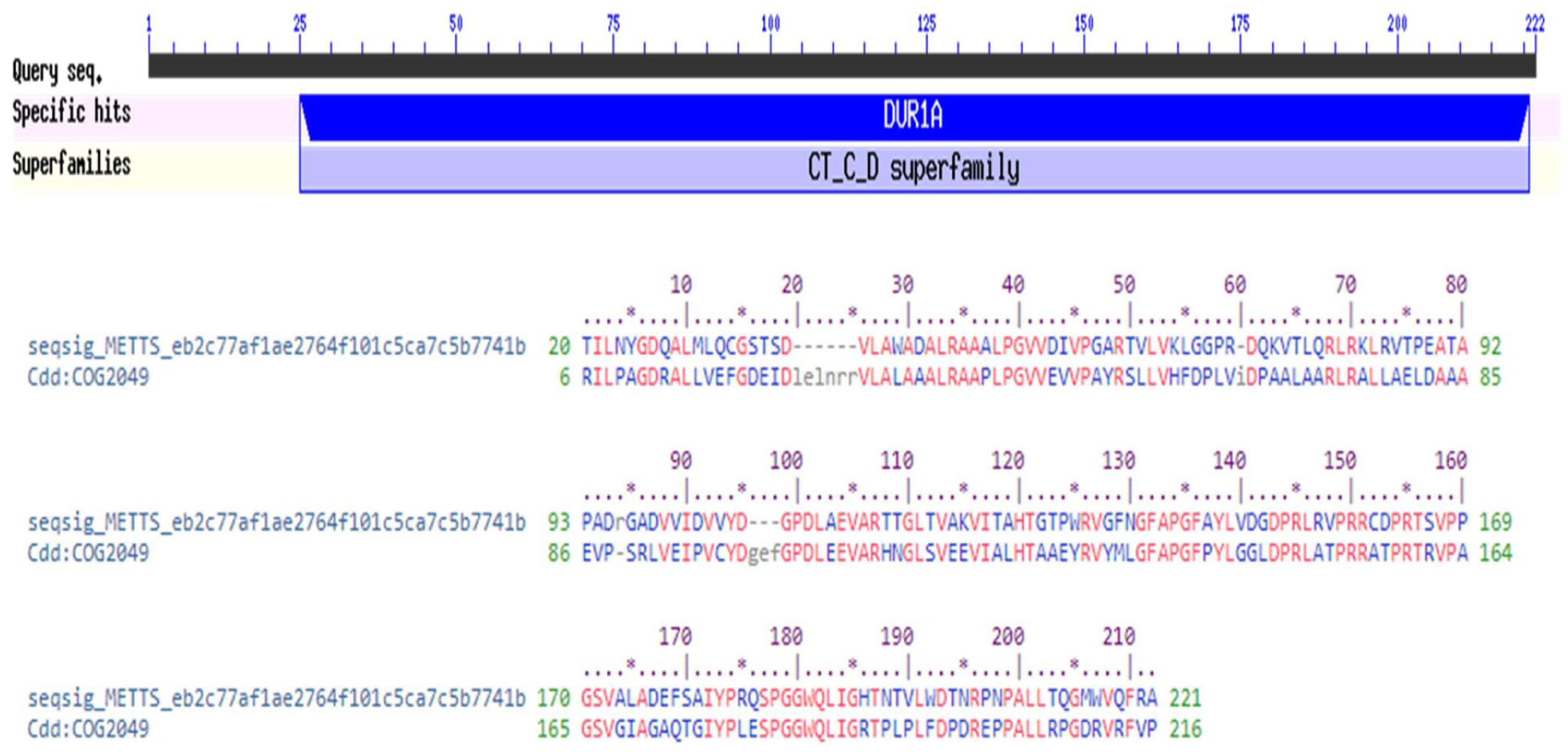
Functional annotation of the HP by identifying conserved domain.
The identification of conserved domains is crucial for functional annotation, as they represent functional units within proteins that contribute to molecular evolution. These domains often recombine in different ways to generate proteins with distinct functions. The presence of the CT_C_D domain suggests that the HP belongs to a specific protein family characterized by this domain architecture. The data were then utilized for functional annotation of the HP based on hits to particular superfamilies, resulting in the identification of similar domain-containing proteins. 70
Urea carboxylase is an ATP- and biotin-dependent enzyme that catalyzes a 2-step carboxylation reaction of urea. It consists of 3 key domains: carboxyltransferase (CT), biotin carboxyl carrier protein (BCCP), and biotin carboxylase (BC). The carboxyltransferase domain is further divided into 4 subdomains: A, B, C, and D. The CT_C_D superfamily domain specifically covers the C and D subdomains of this enzyme. This domain has been identified in Bacillus subtilis, where it spans the entire length of the kinase A inhibitor (kipI). 71 Furthermore, in Saccharomyces cerevisiae, the multifunctional biotin-dependent enzyme Dur1,2, which exhibits both urea carboxylase and allophanate (urea carboxylate) hydrolase activities, also contains the CT_C_D superfamily domain. 72
The results of this analysis provide a strong functional annotation for the HP, linking it to known biochemical pathways involving urea metabolism. The presence of conserved domains across different databases reinforces the reliability of these findings and suggests that the HP may play a role in carboxylation reactions similar to those observed in urea carboxylase and related enzymes.
KEGG Pathway
The HP is potentially associated with significant metabolic processes in M. leprae, primarily within the arginine biosynthesis pathway, according to KEGG pathway analysis. The mapping emphasizes its participation in enzymatic processes associated with the urea cycle, amino acid metabolism, and nitrogen metabolism (Supplemental Figure 1). These findings suggest that the HP may play a role in regulating arginine production as well as cellular nitrogen handling and disposal via urea formation.
Protein-Protein Interaction Analysis
Protein-protein interaction (PPI) databases play a crucial role in identifying biological networks and pathways in cells. 73 PPIs are fundamental to almost all cellular processes, as proteins often function through interactions with other biomolecules. Identifying interacting partners of a protein provides valuable insights into its potential role and biological significance.
The PPI analysis of the HP predicted interactions with 3 proteins at high confidence (0.7 or more; Supplemental Figure 2). These interacting proteins include: a conserved HP analogous to carboxyltransferase domain-containing protein (UniProt ID: Q9CD25), which shares similarity with numerous proteins of undefined function; a conserved HP analogous to 5-oxoprolinase subunit A (UniProt ID: Q9CCW2), an enzyme involved in the ATP-dependent cleavage of 5-oxoproline to form L-glutamate; and aminoglycoside 2′-N-acetyltransferase (UniProt ID: Q9CD24), which exhibits high similarity to mycobacterial aminoglycoside-resistance determinants.
The local network cluster identified in this analysis belongs to the cyclophilin-like domain superfamily and the acetyltransferase (GNAT) family. These findings further support the functional annotation of the HP by linking it to well-characterized protein families with defined enzymatic activities and structural features. The data obtained from PPI analysis were utilized for the functional classification of the HP based on specific domain similarities and evolutionary relationships. 70
The results from both conserved domain identification and PPI analysis provide a comprehensive functional annotation of the HP, suggesting its involvement in enzymatic processes related to carboxylation and acetylation. These insights contribute to understanding the broader biological context of the HP and its potential role within cellular pathways.
Prediction of Secondary Structure, Homology Modeling, and Quality Evaluation of the HP
The target protein’s secondary structure was analyzed using a server called SOPMA, which estimated the proportions of the extended strands (22.52%), alpha helices (28.83%), beta turns (7.66%), and random coils (40.99%; Supplemental Figure 3). PSIPRED analysis also yielded similar results, as seen in Figure 5. The secondary structural elements, including helix, sheet, coil, and turn, strongly correlate with protein functioning, architecture, and interaction.74-77

The PSIPRED server predicted the secondary structure of the HP.
The HHpred is a tool that is effective for remote homology determination and structure prediction. It was initially developed as HMMs and was subsequently popularized by the first pairwise comparison study of homologous protein patterns. A number of repositories are supported by it, including PDB, CDD, Pfam, SMART, SCOP, and COG. 78 HHpred is capable of generating a variety of 3-dimensional representations, various model alignments with a variety of schemes, and multiple inquiry prototypes from these combinations using the Modeler program. 79 The template with the highest probability (100%), E-value (5e-61), and target length of 228 was chosen. The template’s HHpred ID is 3 MML_F and its PDB ID is 3MML.
The 3D structure of the HP became more stable after the YASARA energy minimization process, with a value of −0.21, and the energy decreased from −59 286.9 to −113 439.1 KJ/mol. Using the GalaxyRefine server, the target protein model was refined to produce 5 top models based on RMSD, Clash score, Rama favorability, and MolProbity (Supplemental Table 1). We selected the model that achieved the highest ranking from these for further analysis. PyMOL was used to visualize the structure (Figure 6A).

Evaluation of tertiary structure. (A) HHpred’s predicted 3D structure. (B) Statistical evaluation of 3D structure using Ramachandran plot. (C) The Z-score from ProSA-web.
The quality of the model was then evaluated using various tools such as PROCHECK, ProSA-web, Verify 3D, ERRAT, and QMEAN. According to PROCHECK, 94.8% of the amino acid residues were found in the most preferred regions of the Ramachandran plot, which is considered a reliable measure of model quality (Supplemental Table 2; Figure 6B). The Z-score of the HP was −7.21, which was retrieved from ProSA-web (Figure 6C).
Based on the analysis of the 3D plot, we determined that 85.78% of the residues had an average 3D to 1D score greater than or equal to 0.1, indicating that the model’s environmental profile is favorable (Figure 7A). The projected protein’s total quality factor, as determined by the ERRAT program, was 95.3608, indicating that the model is satisfactory (Figure 7B). The QMEAN score was used to assess the reliability of the projected model by comparing its structure to an experimentally established structure of similar size. The QMEAN global score of our target protein was 0.19, which signifies a high-quality model (Figure 7C). Table 6 shows the quality parameters of the predicted HP structure both before and after energy minimization with refinement.
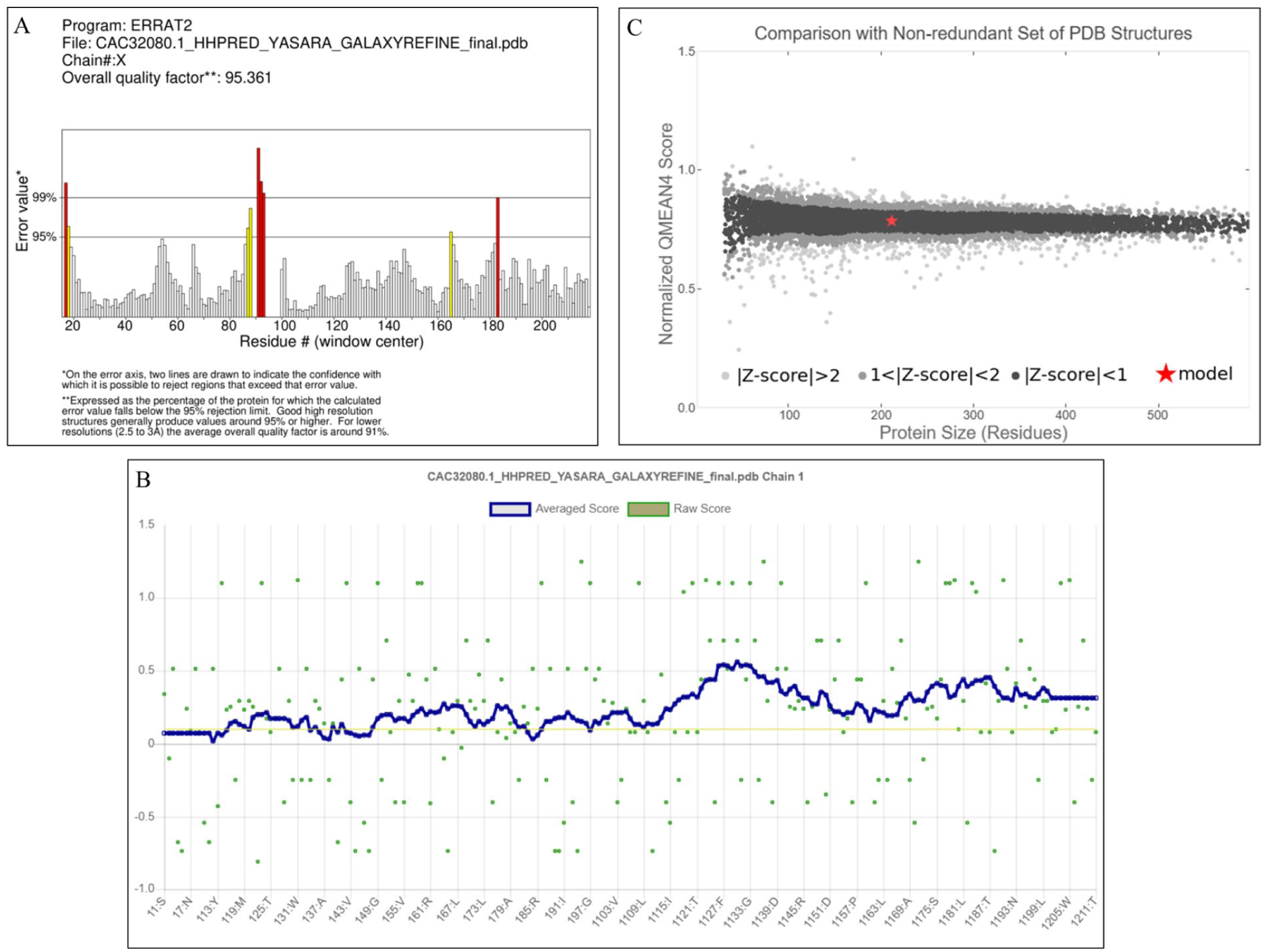
(A) Graphical representation of the tertiary structure’s averaged 3D-1D scores for the amino acid residues, as calculated by the VERIFY3D server. (B) Graphical representation of the estimated overall quality factor (ERRAT) value, which is 95.3608. (C) Graphical representation of the QMEAN estimation of the model’s absolute quality.
Analysis of the Protein’s Quality Before and After Energy Minimization With Refinement.

Active Site Identification of the HP
The chosen HP exhibited 31 active pockets, as determined by the CASTp calculation. The optimal active site was identified with a volume (SA) of 337.877 Å 3 and an area (SA) of 217.277 Å 2 (Figure 8). The CASTp server accurately quantified the volumes, areas, and dimensions of the mouth openings of the active pockets. The metrics were computed analytically, utilizing 2 different surface models: Lee and Richards’ solvent-accessible surface model 80 and Connolly’s molecular surface model. 81

Determination of active sites. The red sphere represents the target protein’s active sites.
Molecular Docking Analysis
Multiple intermolecular interactions between the ligands (allophanate and phenyl phosphorodiamidate) and HP were identified by the molecular docking analysis. Molecular docking is a method that elucidates the binding interactions between small molecules and receptors, including the strength of their interactions. 82 Autodock Vina was used to conduct the docking analysis, which included the HP and ligands. In order to evaluate the substrate’s specificity, the blind docking technique was carried out. Consequently, software such as PyMOL and Discovery Studio were utilized to conduct a more in-depth analysis of the interactions following the docking study. The binding affinity for allophanate was found to be −4.5 (kcal/mol) after docking (Figure 9A). The docking score of −4.8 (kcal/mol) indicates that phenyl phosphorodiamidate exhibited stronger binding affinity than allophanate to the chosen HP (Figure 9C). The HP-phenyl phosphorodiamidate complex contains 4 interacting amino acid residues and the HP-allophanate complex contains 3 interacting amino acid residues, as predicted by blind docking. Both HP-phenyl phosphorodiamidate and HP-allophanate docked complexes have 2 conventional hydrogen bonds in total. There were 2 conventional hydrogen bonds and 1 carbon-hydrogen bond in the HP-allophanate complex, while 2 conventional hydrogen bonds and 2 Pi alkyl bonds were found in the HP-phenyl phosphorodiamidate docked complex. The allophanate ligand interacts with the residues Asp152, Asp176, and Glu177 (Figure 9B), whereas the phenyl phosphorodiamidate ligand interacts with the residues Asp150, Pro153, Arg156, and Glu177 (Figure 9D). Similar interaction residue such as Glu177 appeared in the structures of both ligands (Table 7).
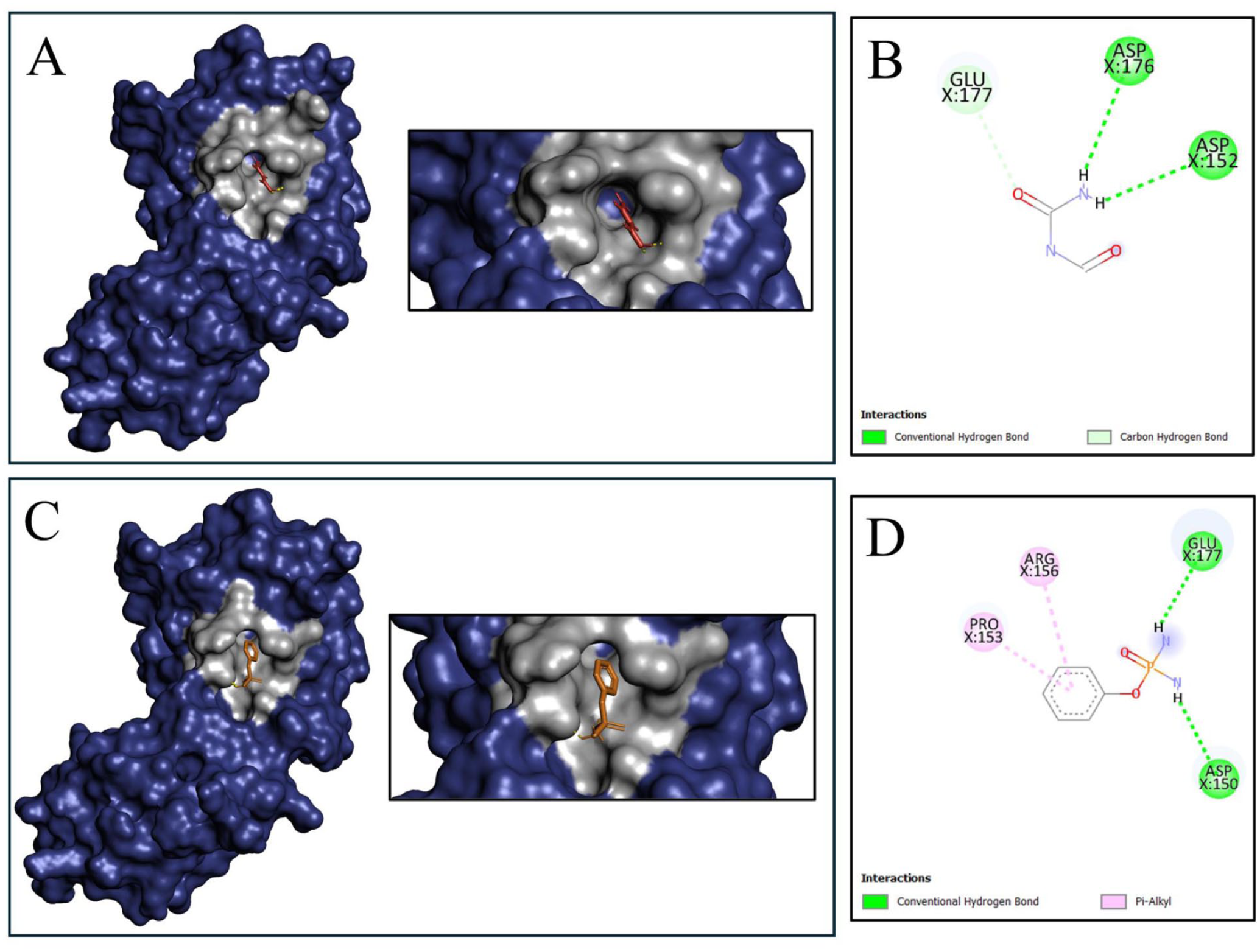
Analysis of molecular docking and amino acid interaction of the HP with the allophanate and phenyl phosphorodiamidate. (A) Around the allophanate ligand (red), the gray color represents the binding residues of HP. (B) 2D interaction between the allophanate ligand and the targeted protein; green dashed lines represent conventional hydrogen bonds, and light green dashed lines indicate carbon hydrogen bonds. (C) The gray color around the phenyl phosphorodiamidate ligand (orange) indicates the binding residues of HP. (D) 2D interaction between the phenyl phosphorodiamidate ligand and the targeted protein; green dashed lines represent conventional hydrogen bonds, and pink dashed lines indicate Pi-alkyl interactions.
Licist of Ligands.

Indeed, the binding affinities between the HP and ligands were insufficient, as indicated by the docking analysis results. However, to address this limitation of the HP-ligand complexes, we conducted additional analyses, such as MD simulations.
ADMET and Toxicity Analysis of the Selected Compound
The SwissADME web tool was used to assess the pharmacokinetic potential and drug-likeness of the inhibitor compound, phenyl phosphorodiamidate (Table 8). The results indicated a very promising profile for oral medication development. There are no violation of Lipinski’s Rule of 5 in this molecule, which has a balanced lipophilicity (Consensus Log Po/w:0.24), a low molecular weight (172.12 g/mol), and an ideal number of hydrogen bond acceptors (4) and donors (2). In addition to its beneficial lipophilicity, it has outstanding aqueous solubility; the Ali and ESOL models agree that it is “very soluble.” In addition, the molecule has a low number of rotatable bonds (2), which indicates good structural rigidity This is generally linked to enhanced oral bioavailability. Importantly, these characteristics come together to suggest that it has the potential to be taken orally due to its high gastrointestinal absorption. With a synthetic accessibility score of 3.07, which is considered “Easy,” it appears that the molecule is easily produced, which opens the door to more research. Phenyl phosphorodiamidate is a drug-like candidate with good chances of oral bioavailability, according to the SwissADME study.
SwissADME Predicted Physicochemical and Pharmacokinetic Properties of Phenyl Phosphorodiamidate.

Attrition at later stages of drug development due to lack of efficacy and potential adverse effects, mainly associated with ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) problems, is common. The analysis of ADMET and drug-likeness properties of Phenyl Phosphorodiamidate using QikProp extended the prediction confidence of initial SwissADME profile (Supplemental Table 3). For any compound to be a potent drug candidate, it should be readily absorbed by the body. The hit compound showed a 68.053% Percent Human Oral Absorption, which is classified as medium and surpasses the minimum absorption threshold of 30%. 83 The predicted Caco-2 cell permeability (QPPCaco) of 305.762 nm/s suggests moderate permeability, which is between <25 (poor) and >500 (great). The predicted value of QPlogBB (−0.96) is within an acceptable range of (−3 to 1.2) which means that the compound has low potential for CNS penetration which is good to prevent possible neurotoxicity. 84 This is also supported by the predicted CNS activity value (−1) which is indicative of low diffusion potential to the CNS. The compound’s lipophilicity was predicted to be QPlogPo/w = −0.577 which also falls within an acceptable range (−2 to 6.5). This value indicates the compound has a moderate hydrophilic-lipophilic character which should enhance absorption and distribution within the body. The PSA of 78.702 Å 2 along with 4 hydrogen bond donors and 6 acceptors is within the recommended range for oral bioavailability which is also in accordance with and as stated by Veber’s rule. 85 In addition, the compound showed 0 violations of the Lipinski’s Rule of Five and the Rule of Three, which signals strong probabilities of the compound having the drug-like qualities. 86 The stars parameter which is a number of property descriptors falling outside of optimum range for 95% of known drugs was only 1 for Phenyl phosphorodiamidate, which highly indicates that the compound possesses drug-like attributes. With regards to safety, the predicted value of −3.972 for QPlogHERG indicates a low risk of cardiotoxicity and is also a non-concerning value as it doesn’t fall below the −5 threshold. The prediction for binding to human serum albumin (QPlogKhsa) was −0.836, which belongs to the ideal range (−1.5 to 1.5) and indicated there is a balanced distribution profile which does not exhibit excessive plasma protein binding. The predictive model also formulated low likely metabolic reactions (0) which indicate simplicity in the metabolic profile. The SASA and molecular volume was also within the recommended ranges which confirms the appropriate molecular size and shape in relation to drug likeness.
To ascertain the probable adverse effects of the inhibitor compound, an in silico assessment on toxicity using the ProTox-3.0 server was conducted following the actionability of the ADMET properties and drug-likeness estimations. The assessment predicted the compound to be inactive on cytotoxicity (Probability: .73), immunotoxicity (Probability: .99), and mutagenicity (Probability: .62), although a potential concern was the predicted active carcinogenicity of the compound, albeit with low probability (Probability: .52; Supplemental Table 4). Even though there is a potential risk, the low probability indicates a need for experimental assessment to accurately characterize the compound’s carcinogenic potential.
The in silico profiling taken together demonstrates that phenyl phosphorodiamidate has drug-like potential. The analysis on SwissADME showed a high likelihood of oral availability and low-cost synthetic accessibility. This was corroborated by the QikProp assessment which outlined predicted absorption as satisfactory, the compound’s lipophilicity was acceptable, and there was a favorable ADMET profile with low risk of cardiotoxicity. The single most important issue was potential carcinogenicity which arose from ProTox-3.0. The compound’s overall profile with respect to no violations of the drug-likeness rules and favorable physicochemical properties, as outlined by both tools, has positioned it with high likelihood for further development in experimental validation for safety and efficacy.
Normal Mode Analysis
The iMODS server was used for NMA to assess the complex’s stability and effectiveness. Although both HP-ligand complexes showed weaker binding affinity, the HP-phenyl phosphorodiamidate was chosen for the MD simulations study due to its comparatively stronger binding affinity than the HP-allophanate. The mobility of the docked complex is reflected in the NMA B-factor values, which are proportional to the averaged root mean square (RMS) value. A molecule’s main-chain deformability indicates how much it can bend at each of its residues. High deformability regions can be used to determine the location of the chain hinges (Figure 10A). A RMS is provided by the B-factor column (Figure 10D). According to Figure 10B, the complex appears to have a reasonably low stiffness along the appropriate mode of motion, as indicated by the eigenvalue of 9.814225e-05 for the docked complex. This indicates that the complex is more likely to undergo conformational modifications or deformation along this particular mode in comparison to complexes that have higher eigenvalues. An association between eigenvalue and variance existed for every normal mode. 87 Figure 10C shows that eigenvalues are inversely related to variances, indicating that lower eigenvalues are associated with larger variances and higher eigenvalues are associated with lower variances. The graph depicts the relative contribution of variance to equilibrium movements for each normal mode, with green and violet bars representing group and single variance.

The following parameters were analyzed during a normal mode analysis of HP- phenyl phosphorodiamidate complex: (A) deformability, (B) eigenvalue, (C) variance, (D) B factor, (E) elastic network, and (F) covariance matrix.
The elastic network model specifies the atom pairs that are connected by springs. When the electron-nuclear magnetic resonance (ENM) is seen graphically, each dot on the graph stands for a spring that links 2 atoms. The spring stiffness is shown by the color of the dots. Lighter shades of gray suggest less rigid connections, while darker shades typically indicate more rigid springs (Figure 10E). The correlation matrix is used by the iMODS server to find patterns of coordinated atomic fluctuations, which indicate protein regions that move in a correlated manner. The covariance diagram shows how different parts of a molecule move relative to 1 another; red, blue, and white colors indicate related, unrelated, and anti-related motions, respectively (Figure 10F).
Molecular Dynamics Simulation Analysis of the HP
The MD simulation of the HP-phenyl phosphorodiamidate complex provided information about the system’s structural stability and flexibility. The stability of the systems was assessed through the use of RMSD calculations. Protein conformational changes are correlated with variations in the RMSD value. In Figure 11A, the complex’s RMSD profile is depicted. The RMSD initially rose and appeared to stabilize after approximately 25 ns, but clear fluctuations and an increase after about 60 ns indicated further conformational changes rather than a strictly stable plateau. Thus, the complex showed a relatively stable binding mode over 100 ns while still undergoing noticeable conformational rearrangements.

Analysis of HP-ligand complex using MD simulation. (A) RMSD profile of the HP-phenyl phosphorodiamidate complex. (B) RMSF profile of the HP-phenyl phosphorodiamidate complex. (C) Rg profile of the HP-phenyl phosphorodiamidate complex. (D) SASA profile of the HP-phenyl phosphorodiamidate complex. (E) H-bond profile of the HP-phenyl phosphorodiamidate complex.
RMSF was employed to evaluate the protein’s regional flexibility. The flexibility of a specific amino acid position increases as the RMSF increases. The RMSF profile of the complex is illustrated in Figure 11B. The most flexible region was observed to be the region between the 200th and 215th residues. These residues are located in close proximity to the predicted binding pocket; however, they do not establish direct contact with the ligand during the MD frames that were examined. This implies that their impact on pocket dynamics may be indirect or allosteric. This suggests that the flexible region could still have a substantial impact on the protein’s function, particularly in terms of allosteric regulation and dynamics, despite the fact that it does not directly interact with the ligand. Protein regions, particularly those at the C-terminus, showed enhanced flexibility according to RMSF analysis. The inherent flexibility of terminal regions is expected to increase the mobility of the C-terminus. Several peaks on the RMSF plot indicated potential flexible loops or domains that could be critical to the protein’s function. Additional research into these flexible regions may shed light on the structural changes of the protein.
The Rg was used to determine the degree of compactness. A protein’s folding is stable when the Rg is relatively steady. The fluctuation in the Rg indicates the protein’s unfolding. Figure 11C illustrates the complex’s Rg profile. The complex’s Rg value increased after 75 ns but was relatively constant at roughly 1.875 nm throughout the simulation. The protein in this case generally maintained its overall compactness.
SASA was employed in MD simulation to forecast the hydrophobic core of protein’s exposure to solvents. Higher SASA values suggest that much of the protein is accessible to water, whereas lower values indicate that much of the protein is buried within the hydrophobic core. Figure 11D shows the complex’s SASA profile. Throughout the simulation, the SASA values remained greater than 115 ns. The hydrophobic core of the protein complex was folded securely with limited water exposure, as indicated by the steady state of the SASA values. This provided more evidence that the complex structure was securely folded. The protein-ligand complex maintained a stable hydrogen bond interaction with minor fluctuations throughout the simulation, as indicated by H-bond analysis (Figure 11E).
Overall, the MD simulation demonstrated that the docked HP-phenyl phosphorodiamidate complex structure was very stable. Presumably, the complex was folded correctly as a result of its low solvent exposure and retained compactness. The HP and the phenyl phosphorodiamidate ligand also appeared to interact stably through hydrogen bonding. Further research could delve into the dynamics of HP’s structure, with a particular emphasis on its flexible loops and domains, to uncover its functional mechanisms.
The present study has certain limitations that must be acknowledged. The docking interaction of both ligands with the hypothetical protein demonstrates that their binding affinities are poor, falling below the threshold considered favorable for strong ligand-protein interactions. This limitation underscores that these findings are preliminary in silico observations, rather than definitive evidence of inhibitory potential. This work relies solely on in silico analysis and lacks support from in vitro or in vivo experiments. The structural stability of the docked complexes was maintained during the 100 ns MD simulation; however, this stability is also based on its computational environment and does not necessarily correspond to biological efficacy. The findings are indicative and offer preliminary insights into the HP’s structure and function, demanding experimental validation to assess its viability as a therapeutic target.
Conclusion
Understanding the functions of HPs plays a crucial role in comprehending biological processes. Systematic investigation of these proteins may contribute to a broader comprehension of the organism’s fundamental biology and pathogenesis. Hence, this study aimed to use an in silico approach to determine the biological and structural functions of an HP from M. leprae. The study employed several in silico tools to identify the roles and characteristics of the HP, whose structure and functions are yet to be determined. The findings of this HP will strengthen the basic knowledge of M. leprae and suggest this protein as a putative therapeutic target that requires further experimental validation, particularly given the relatively weak docking affinity observed. However, further in vitro research must be conducted to validate the experimental possibilities shown here and determine the target protein’s roles in biological research.
Supplemental Material
sj-docx-1-evb-10.1177_11769343261438525 – Supplemental material for Functional Annotation and Molecular Characterization of a Hypothetical Protein From Mycobacterium leprae Through In Silico Approaches: A Potential Therapeutic Target
Supplemental material, sj-docx-1-evb-10.1177_11769343261438525 for Functional Annotation and Molecular Characterization of a Hypothetical Protein From Mycobacterium leprae Through In Silico Approaches: A Potential Therapeutic Target by Tasnim Hosen Tanha, Shoaib Saikat, Rabiul Hasan, Md Mehedi Hasan Yeamin, Rehana Parvin, Md Imtiaz and Md Hasib in Evolutionary Bioinformatics
Supplemental Material
sj-docx-2-evb-10.1177_11769343261438525 – Supplemental material for Functional Annotation and Molecular Characterization of a Hypothetical Protein From Mycobacterium leprae Through In Silico Approaches: A Potential Therapeutic Target
Supplemental material, sj-docx-2-evb-10.1177_11769343261438525 for Functional Annotation and Molecular Characterization of a Hypothetical Protein From Mycobacterium leprae Through In Silico Approaches: A Potential Therapeutic Target by Tasnim Hosen Tanha, Shoaib Saikat, Rabiul Hasan, Md Mehedi Hasan Yeamin, Rehana Parvin, Md Imtiaz and Md Hasib in Evolutionary Bioinformatics
Supplemental Material
sj-docx-3-evb-10.1177_11769343261438525 – Supplemental material for Functional Annotation and Molecular Characterization of a Hypothetical Protein From Mycobacterium leprae Through In Silico Approaches: A Potential Therapeutic Target
Supplemental material, sj-docx-3-evb-10.1177_11769343261438525 for Functional Annotation and Molecular Characterization of a Hypothetical Protein From Mycobacterium leprae Through In Silico Approaches: A Potential Therapeutic Target by Tasnim Hosen Tanha, Shoaib Saikat, Rabiul Hasan, Md Mehedi Hasan Yeamin, Rehana Parvin, Md Imtiaz and Md Hasib in Evolutionary Bioinformatics
Supplemental Material
sj-docx-4-evb-10.1177_11769343261438525 – Supplemental material for Functional Annotation and Molecular Characterization of a Hypothetical Protein From Mycobacterium leprae Through In Silico Approaches: A Potential Therapeutic Target
Supplemental material, sj-docx-4-evb-10.1177_11769343261438525 for Functional Annotation and Molecular Characterization of a Hypothetical Protein From Mycobacterium leprae Through In Silico Approaches: A Potential Therapeutic Target by Tasnim Hosen Tanha, Shoaib Saikat, Rabiul Hasan, Md Mehedi Hasan Yeamin, Rehana Parvin, Md Imtiaz and Md Hasib in Evolutionary Bioinformatics
Supplemental Material
sj-docx-8-evb-10.1177_11769343261438525 – Supplemental material for Functional Annotation and Molecular Characterization of a Hypothetical Protein From Mycobacterium leprae Through In Silico Approaches: A Potential Therapeutic Target
Supplemental material, sj-docx-8-evb-10.1177_11769343261438525 for Functional Annotation and Molecular Characterization of a Hypothetical Protein From Mycobacterium leprae Through In Silico Approaches: A Potential Therapeutic Target by Tasnim Hosen Tanha, Shoaib Saikat, Rabiul Hasan, Md Mehedi Hasan Yeamin, Rehana Parvin, Md Imtiaz and Md Hasib in Evolutionary Bioinformatics
Supplemental Material
sj-pdf-5-evb-10.1177_11769343261438525 – Supplemental material for Functional Annotation and Molecular Characterization of a Hypothetical Protein From Mycobacterium leprae Through In Silico Approaches: A Potential Therapeutic Target
Supplemental material, sj-pdf-5-evb-10.1177_11769343261438525 for Functional Annotation and Molecular Characterization of a Hypothetical Protein From Mycobacterium leprae Through In Silico Approaches: A Potential Therapeutic Target by Tasnim Hosen Tanha, Shoaib Saikat, Rabiul Hasan, Md Mehedi Hasan Yeamin, Rehana Parvin, Md Imtiaz and Md Hasib in Evolutionary Bioinformatics
Supplemental Material
sj-pdf-6-evb-10.1177_11769343261438525 – Supplemental material for Functional Annotation and Molecular Characterization of a Hypothetical Protein From Mycobacterium leprae Through In Silico Approaches: A Potential Therapeutic Target
Supplemental material, sj-pdf-6-evb-10.1177_11769343261438525 for Functional Annotation and Molecular Characterization of a Hypothetical Protein From Mycobacterium leprae Through In Silico Approaches: A Potential Therapeutic Target by Tasnim Hosen Tanha, Shoaib Saikat, Rabiul Hasan, Md Mehedi Hasan Yeamin, Rehana Parvin, Md Imtiaz and Md Hasib in Evolutionary Bioinformatics
Supplemental Material
sj-pdf-7-evb-10.1177_11769343261438525 – Supplemental material for Functional Annotation and Molecular Characterization of a Hypothetical Protein From Mycobacterium leprae Through In Silico Approaches: A Potential Therapeutic Target
Supplemental material, sj-pdf-7-evb-10.1177_11769343261438525 for Functional Annotation and Molecular Characterization of a Hypothetical Protein From Mycobacterium leprae Through In Silico Approaches: A Potential Therapeutic Target by Tasnim Hosen Tanha, Shoaib Saikat, Rabiul Hasan, Md Mehedi Hasan Yeamin, Rehana Parvin, Md Imtiaz and Md Hasib in Evolutionary Bioinformatics
Footnotes
Acknowledgements
The authors express their appreciation to the Bioinformatics Division of the National Institute of Biotechnology, Bangladesh, for their assistance with MD simulation.
Ethical Considerations
No ethical approval was required for this manuscript.
Author Contributions
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.