
Abstract
Prairie voles show strong pair bonding with their mating partners, and they demonstrate parental behavior toward their infants, indicating that the prairie vole is a unique animal model for analysis of molecular mechanisms of social behavior. Until a recent study, the signaling pathway of oxytocin was thought to be critical for the social behavior of prairie voles, but neuron-specific functional research may be necessary to identify the molecular mechanisms of social behavior. Prairie vole pluripotent stem cells of high quality are essential to elucidate the molecular mechanisms of social behaviors. Generation of high-quality induced pluripotent stem cells (iPSCs) would help to establish a genetically modified prairie vole, including knockout and knock-in models, based on the pluripotency of iPSCs. Thus, we attempted to establish high-quality prairie vole-derived iPSCs (pv-iPSCs) in this study. We constructed a polycistronic reprogramming vector, which included six reprograming factors (Oct3/4, Sox2, Klf4, c-myc, Lin28, and Nanog). Furthermore, we evaluated the effect of six reprogramming factors, which included Oct3/4 with the transactivation domain (TAD) of MyoD. Implantation of the pv-iPSCs into immunodeficient mice caused a teratoma with three germ layers. Furthermore, the established pv-iPSCs tested positive for stem cell markers, including alkaline phosphatase activity (ALP), stage-specific embryonic antigen (SSEA)-1, and dependence on leukemia inhibitory factor (LIF). Our data indicate that our newly established pv-iPSCs may be a useful tool for genetic analysis of social behavior.
Introduction
The prairie vole, a rodent, is a beneficial animal model for studies on molecular mechanisms of social behavior. Prairie voles show formation of enduring social attachments, including pair bonding between mating partners and parental care of pups (2,10,33). These social behavioral patterns are also observed in primates such as humans (6). These patterns are known to be regulated by intracerebral hormones, such as oxytocin and vasopressin (5,32). The latter is known to affect the ventral pallidum in males, and oxytocin affects the nucleus accumbens in females (5). The expression patterns of oxytocin receptors (OTRs) and vasopressin receptors (V1aRs) in the intracerebral area of prairie voles are similar to those of primates, but different from those of mice and mountain voles, the latter of which do not show any pair bonding in their behavior (10).
These observations suggest that the signaling pathways of oxytocin and vasopressin in the intracerebral region are crucial for regulation of various social behavioral patterns, including the pair bonds between mating partners or biparental care of infants (25). If we can successfully establish a neuron-specific knockout or a transgenic model of the prairie vole, the detailed mechanisms underlying social behavior can be elucidated.
For generation of genetically modified animals, there are two major methods: homologous recombination in stem cells and genome editing, such as the CRISPR/Cas system. The first option requires high-quality stem cells, which allow for germline transmission [e.g., high-quality embryonic stem cells (ESCs)]. In 2006, Yamanaka's research group showed that transduction of four reprogramming factors [octamer-binding transcription factor 3/4 (Oct3/4), Kruppel-like factor 4 (Klf4), sex-determining region Y-box 2 (Sox2), c-Myc] allows for cellular reprogramming from terminally differentiated cells to stem cells [induced pluripotent stem cells (iPSCs)], which have the capacity for differentiation toward all of the organs, including germ cells (26). Although we devoted significant efforts to establishing the ESCs from early prairie vole embryos, these experiments were not successful (unpublished data). Furthermore, recent advances in genome editing [e.g., zinc finger nucleases (ZFN), TALEN, and CRISPR/Cas] seem to be powerful tools for creation of a simple deletion (1,3,17,29) or a small (less than 100 bp) insertion, but a replacement in the genome involving a relatively large region (more than several kb of DNA) is still problematic. Accordingly, establishment of iPSCs from the prairie vole is expected to overcome the limitations for the neuron-specific genetic modification of these animals. In previous reports, prairie vole iPSCs (pv-iPSCs) were established by using a monocistronic retroviral vector (18). However, in this study, we developed a polycistronic transposon-based vector, which expresses six reprogramming factors. We found that expression of six reprogramming factors is more advantageous for obtaining high-quality porcine iPSC lines with both activated X chromosomes, when compared with four reprogramming factors (under submission). Therefore, there is a possibility that expression of six reprogramming factors allows us to efficiently obtain high-quality pv-iPSCs. Furthermore, the piggyBac transposon system allows us to precisely excise out the expression cassette from the genome after the expression of the transposase. The excision of the expression cassette would facilitate the full reprogramming of iPSCs (7,30,34).
Until now, two kinds of iPSCs have been reported: naive iPSCs and primed iPSCs (19). Naive iPSCs show three-dimensional colonies in a culture dish (19) and are positive for stage-specific embryonic antigen-1 (SSEA-1) (8); their growth is leukemia inhibitory factor (LIF) dependent (20). Furthermore, naive iPSCs form a rapidly growing teratoma (24) and can contribute to embryonic tissues, which is characteristic of inner cell mass-derived cells from an early stage embryo (11,19). In the case of primed state iPSCs, they form flattened colonies in a cell culture dish (e.g., human ESCs or iPSCs) (19). Although there are no significant findings due to the limitations on clinical experiments with humans and primates, primed iPSCs are believed to contribute only to the extra embryonic tissues, and this characteristic is close to that of epiblast stem cells (EpiSCs). Therefore, to achieve our goal of establishing a genetically recombinant prairie vole, we need to establish the prairie vole-derived iPSCs that are close to the naive state.
In an early study on iPSCs, the expression of Lin28 and Nanog with Oct3/4, Sox2, Klf4, and c-Myc was reported to be effective for the establishment of iPSCs. The first two genes are also key regulators of the maintenance of pluripotency (21,28). On the basis of this idea, we introduced six reprogramming factors into prairie vole-derived fibroblasts, trying to obtain high-performance stem cells.
Materials and Methods
Primary Cell Culture From a Prairie Vole
Prairie vole handling and maintenance were approved by the animal committee of Tohoku University (approval number 2013-Nodo-049). Prairie vole-derived fibroblasts were collected from an embryo (hereafter prairie vole embryonic fibroblasts: pv-EFs). These fibroblasts were cultured in Dulbecco's modified Eagle medium/F12 (DMEM/F12, 048-29785; Wako Pure Chemical, Osaka, Japan), containing 10% fetal bovine serum (FBS, 35-010-CVR; Corning Inc., Corning, NY, USA), and a 1% antibiotic–antimycotic mixed solution (02892-54; Nacalai Tesque, Kyoto, Japan). The fibroblasts were incubated at 37°C in a humidified atmosphere containing 5% CO2.
Reprogramming Vectors
We chemically synthesized the expression cassette that included the six reprogramming factors (Oct3/4, Sox2, Klf4, c-Myc, Lin28, and Nanog, all derived from mice). At the junction of the coding region, the self-cleaving 2A peptide was inserted (Fig. 1A). The cDNA insert was transferred from a shuttle vector to the piggyBac transposon vector (PJ547-17; DNA 2.0, Menlo Park, CA, USA). In the original transposon vector, the elongation factor-1 (EF1) promoter was used to drive expression of the cDNA cassette. We replaced the original EF1 promoter with the CAG promoter [C: the cytomegalovirus (CMV) early enhancer element; A: the promoter, the first exon, and the first intron of chicken β-actin gene; G: the splice acceptor of the rabbit β-globin gene] by means of restriction digestion and ligation; then we named it the PB-CAG vector. We inserted the cDNA fragment encoding six reprogramming factors into the multiple cloning sites of PB-CAG, with restriction enzymatic digestion and ligation, and named the resulting plasmid PB-CAG-6F. Furthermore, we chemically synthesized the six modified reprogramming factors. According to Hirai et al., the fusion of the protein Oct3/4 with the transactivation domain (TAD) of MyoD dramatically improves the efficiency of reprogramming (9). According to this idea, we also created the six reprogramming constructs in a tandem fashion with Oct3/4-TAD. We hereafter refer to the reprogramming factor including Oct3/4-TAD as redesigned 6F (R6F). We also introduced an expression cassette of R6F into the multiple cloning sites of PB-CAG-6F. This plasmid is called PB-CAG-R6F (Fig. 1B).

Structure of reprograming vectors. (A) Structure of the reprograming vector PB-6F. (B) Structure of the R6F vector.
Establishment of iPSCs
According to the protocol shown in Figure 2A, prairie vole-derived iPSCs (pv-iPSCs) were established in this study. The culture conditions were as follows: pv-EFs were seeded in a six-well plate at 105 cells per cell culture dish (3.5 cm diameter). The reprograming vector was transfected into the cells by means of the Lipofectamine 2000 transfection reagent (11668019; Life Technologies, Carlsbad, CA, USA). After hygromycin selection (085-06153; Wako Pure Chemical Industries), the transfected pv-EFs were reseeded on a mouse embryonic fibroblast (MEF) feeder layer. The protocol for MEF feeder cells was described previously (4). After day 12 from the genetic transduction, the serum-based medium was replaced with a serum replacement-based medium (hereafter: iPSC medium). iPSC medium composition was as follows: DMEM/F12 including 15% StemSure Serum Replacement (197-16775; Wako Pure Chemical Industries), a 1% antibiotic–antimycotic mixed solution, 1% nonessential amino acids (139-15651; Wako Pure Chemical Industries), 2 mM glutamic acid (16948-004; Nacalai Tesque), and 0.1 mM β-mercaptoethanol (21438-82; Nacalai Tesque). After appearance of iPSC-like colonies, they were picked and seeded on the MEF feeder in a 48-well plate. The iPSCs were digested with Accutase (A11105-01; Life Technologies) at every passage and maintained on an MEF feeder. We used 1,000× human leukemia inhibitory factor (LIF; 125-05603; Wako Pure Chemical Industries), 4.0 ng/ml fibroblast growth factor (FGF; KHFGF001; DS Pharma Biomedical, Osaka, Japan), 0.25 μM thiazovivin (04-0017; Stemgent, Cambridge, MA, USA), 0.75 μM CHIR99021 (Axon1386; Axon Medchem, Groningen, Netherlands), and 0.25 μM PD0325901 (163-24001; Wako Pure Chemical Industries) as the supplements in the iPSC medium.

Establishment of prairie vole-derived induced pluripotent stem cells (pv-iPSCs) from primary fibroblasts. (A) The protocol for the establishment of pv-iPSCs. (B, C) Morphological features of pv-iPSC colonies on a feeder layer. Scale bar: 100 μm. (D) Colony morphology of pv-iPSCs (generated with the PB-6F vector) at an early passage. Scale bar: 50 μm. (E) Colony morphology of pv-iPSCs (generated with PB-6F) at a late passage. Scale bar: 100 μm. (F) Colony morphology of pv-iPSCs (generated with PB-R6F) at an early passage. Scale bar: 50 μm. (G) Colony morphology of pv-iPSCs (generated with PB-R6F) at a late passage. Scale bar: 100 μm.
Alkaline Phosphatase Staining
The pv-iPSCs were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) (09154-85; Nacalai Tesque) for 3 min. The alkaline phosphatase (ALP) enzymatic activity was detected based on the protocol described in our previous study (4). In brief, the staining solution, consisting of 0.6 mg/ml Fast Red TR Salt [hemi(zinc chloride) salt, F8764; Sigma-Aldrich, St. Louis, MO, USA], 0.1 mg/ml naphthol phosphate (23821-24; Nacalai Tesque), 0.7 mM N, N-dimethylformamide (13016-65; Nacalai Tesque), 0.2 mM MgCl2 (20908-65; Nacalai Tesque), and 0.1 mM Tris-HCl (pH 8.5), was mixed with the fixed cells, and incubated at 37°C for 10 min. After the staining reaction had run its course, it was terminated with a 4% paraformaldehyde solution.
Immunostaining of the iPSCs
pv-iPSC-like cells were fixed with 4% paraformaldehyde in PBS for 3 min. Permeabilization treatment was carried out with 0.5% Triton X-100 (35501-15; Nacalai Tesque) for 60 min. After three washes with PBS, pv-iPSCs were blocked by 1% bovine serum albumin (BSA, 01863-06; Nacalai Tesque) for 45 min. The iPSCs were incubated with a primary antibody overnight, then exposed to the corresponding secondary antibodies with a florescent label. The counterstaining was performed with a 4′,6-diamidino-2-phenylindole (DAPI) solution (340-07971; Wako Pure Chemical Industries). The immunostained images were obtained under an Olympus FSX100 (Tokyo, Japan) fluorescence microscope. The following antibodies were used in this study: mouse anti-human/mouse SSEA-1 (1:100 dilution, NL2155R; R&D Systems, Minneapolis, MN, USA), rat anti-mouse/human SSEA-3 (1:100 dilution, 09-0014; Stemgent), mouse anti-human/mouse SSEA-4 (1:100 dilution, MAB1435; R&D Systems), and rabbit anti-human Oct3/4 (1:100 dilution, SC-9081; Santa Cruz Biotechnology, Dallas, TX, USA). Alexa Fluor 488-conjugated goat anti-rabbit IgG (A11070; Life Technologies) was used for detecting Oct3/4, Alexa Fluor 568-conjugated goat anti-mouse IgG (A11019; Life Technologies) was used for detecting SSEA-1/SSEA-4, and Alexa Fluor 488-conjugated donkey anti-rat IgG (1:300 dilution, A21208; Life Technologies) was used for the detection of SSEA-3.
Genomic PCR
Genomic DNA was extracted by means of the NucleoSpin Tissue Kit (740952.50; Macherey-Nagel, Düren, Germany). PCR was carried out with 100 ng of template DNA, PCR buffer of KOD-FX neo (KFX-201; Toyobo, Osaka, Japan), 0.4 mM dNTPs (KFX-201; Toyobo), 0.5 U KOD-FX neo (KFX-201; Toyobo), and 0.3 μM of each primer. In genomic PCR analysis, the PB-6F vector, the R6F vector, and TSC2, which was the internal control, were detected in pv-iPSCs and pv-EFs. The sequences of primers are listed in Supplemental Table 1 (available at: http://www.agri.tohoku.ac.jp/iden2/iden2/Protocols_files/Supplemental%20Table%20final.pdf). The PCR was conducted under the following conditions: predenaturation at 94°C for 2 min, denaturation at 98°C for 10 s, and extension at 68°C for 30 s. There were 40 cycles (denaturation and extension). The PCR products were analyzed by electrophoresis on 1.0% agarose/Tris–acetate–EDTA (ethylenediaminetetraacetic acid) gels and stained with ethidium bromide (14603-64; Nacalai Tesque).
Mode Analysis of Karyotype Analysis About pv-iPSCs

Normal karyotype chromosome number is 54.
Karyotype Analysis
Karyotype analysis was performed on two pv-iPSC clones, which were derived from pv-EFs (PB-6F and PB-R6F). Those iPSCs were treated with 0.02 μg/ml colcemid (Sigma-Aldrich) overnight. After trypsinization, iPSCs were exposed to a hypotonic solution (Sigma-Aldrich) and fixed with Carnoy's fluid (Sigma-Aldrich). Fixed iPSCs were placed on a glass slide to be viewed under a microscope (Sigma-Aldrich) and stained by G-band staining. In this study, the chromosome number of 50 cells was counted, and G banding of 20 cells was examined.
Teratoma Formation
Teratoma formation experiments were approved by the animal committee of Tohoku University (approval number 2013-Nou-Dou-003). In this experiment, 106 pv-iPSCs were injected into the testicular tissue of mice with severe combined immunodeficiency (SCID; CLEA Japan, Tokyo, Japan). After several weeks, we tested the mice for the presence of teratomas by abdominal palpitation. After euthanasia of the mice and excision of the tumors, the tumor tissues were fixed with a 4% paraformaldehyde (PFA) solution. The fixed tissues were processed by means of the standard histological analysis, and the slices were stained with hematoxylin and eosin (H&E) (Sigma-Aldrich).
Copy Number Detection of the Expression Cassette
Genomic DNA was extracted with a NucleoSpin Tissue Kit (740952.50; Macherey-Nagel). Real-time PCR was performed in a 12.5-μl volume containing 2× THUNDERBIRS SYBR qPCR Mix (QPS-201, Toyobo), 10 ng of cDNA solution, and 0.3 μM of each primer. The sequences of the primers are available in Supplemental Table 2 (available at: http://www.agri.tohoku.ac.jp/iden2/iden2/Protocols_files/Supplemental%20Table%20final.pdf). Quantitative PCR was performed in triplicate using the Thermal Cycler Dice Real Time System Single (TP850; Takara Bio, Otsu, Japan). The copy number of the reprograming vector was obtained with the computer software for the relative quantitation, Multiplate RQ (Takara Bio). As the control target, we used the tuberous sclerosis type 2 gene (Tsc2), since there is no redundancy of the Tsc2 gene in humans, rats, or mice (12–14). Furthermore, in these species, there is no pseudogene in the genome, indicating that the Tsc2 gene only has two copies within the genome. Based on the relative quantitation between Tsc2 and the expression cassette, the copy number of the inserted transposon vector was estimated.
Microscopic Examination of pv-iPSCs

Normal karyotype is 54, XY.
Embryo Body Formation
To form the embryonic bodies, pv-iPSCs were seeded in Petri dishes for bacteria, to prevent the cell attachment to the dish. After 4 to 5 days, floating embryo bodies (EBs) were picked up and transferred to 0.1% gelatin-coated six-well plates and cultured for 4 additional days. The composition of the EB-forming medium was as follows: DMEM/F12 including 15% FBS, a 1% antibiotic–antimycotic mixed solution, 1% nonessential amino acids, 2 mM glutamic acid, and 0.1 mM β-mercaptoethanol.
Detection of Gene Expression with Real-Time PCR
Total RNA was isolated from pv-iPSCs, EBs (derived from pv-iPSCs), and pv-EFs in six-well plates using NucleoSpin RNA II kits (740955; Takara Bio). Then complementary DNA (cDNA) was synthesized with the PrimeScript reverse transcription (RT) reagent kit (Perfect Real Time, RR047A; Takara Bio) from total RNA. The detailed method for the synthesis of cDNA was provided by the manufacturer. Real-time PCR was performed in a 12.5-μl volume containing 2× THUNDERBIRS SYBR qPCR Mix (QPS-201; Toyobo), 10 ng of cDNA solution, and 0.3 μM of each primer. The sequences of the primers are available in Supplemental Table 3 (available at: http://www.agri.tohoku.ac.jp/iden2/iden2/Protocols_files/Supplemental%20Table%20final.pdf). To detect the linearity of the gene expression, a cDNA solution of pv-iPSC PB-R6F clone 1 was diluted, 2×, 4×, 8×, and 16× dilutions. The reaction was carried out with a duplicate reaction. The cycling was programmed as 95°C for 60 s (initial denature), 95°C for 15 s (denature), and 60°C for 20 s (annealing and extension) for 40 cycles. The expression level of target genes was normalized with that of GAPDH. The expression level was obtained with the computer software, Multiplate RQ (Takara Bio), for relative quantitation.
Introduction of Fluorescence Marker Gene into Prairie Vole-Derived iPSCs
Due to the internal ribosome entry site (IRES)-enhanced green fluorescent protein (EGFP) at the 3′ downstream of the expression cassette (Fig. 1A), the established pv-iPSCs should show the fluorescence, although its expression level is not so high. To efficiently address the contribution of obtained pv-iPSCs in the chimeric experiment, pv-iPSCs were introduced with EGFP expressing markers. Having iPSCs with high expression levels of the EGFP marker protein would allow us to identify the contribution of iPSCs to embryonic tissue. To achieve this, we introduced the marker expression cassette via a lentiviral vector. PGK-EGFP-IRES-Puro (pLenti PGK GFP Puro) (w509-5) (plasmid #19070; Addgene, Cambridge, MA, USA) was used. After transfection of PGK-EGFP-IRES-Puro in pv-iPSCs, transfected iPSCs were selected by puromycine (1 μg/ml) on DR-4 feeder (DS Pharma Biomedical). Subsequently, pv-iPSC colonies that showed a high level of EGFP expression were picked out and expanded.
Results
Generation of pv-iPSCs From Primary pv-EFs
The finalized protocol for establishing pv-iPSCs is detailed in Figure 2A. Key points of the protocol are the hygromycin selection step on day 5 and the use of a growth factor (LIF). Furthermore, we used 2i (CHIR99021 and PD0325901) and thiazovivin. For the genetic modification, we used two kinds of expression vectors, PB-6F (piggyBac with six factors) and PB-R6F (piggyBac with redesigned six factors). PB-R6F contains an Oct3/4 fusion protein with the TAD of MyoD (M3O). According to the study by Hirai et al., the Oct3/4 with TAD of MyoD dramatically increases the number of murine iPSC colonies (9). We therefore used the two kinds of reprogramming vectors to obtain the pv-iPSCs. Around day 19 or 20, the pv-iPSC colonies that showed three-dimensional cell growth appeared in the MEF feeder layer (Fig. 2B, C). We picked more than 20 colonies from the primary dishes, and two representative clones were maintained on the feeder layers with sequential passaging. Although the colony morphology of cells generated with PB-6F and PB-R6F did not show any difference from the early passage (up to passage 5) (Fig. 2D and F), pv-iPSCs that were generated with PB-6F showed flattened morphology at a late passage (more than passage 10). The pv-iPSCs with PB-R6F did not show major morphological changes even at the late passage (Fig. 2G).
Detection of Pluripotent Makers in pv-iPSCs
We successfully detected the expression of pluripotency markers in our established pv-iPSCs. At first, we detected the ALP activity with chromogenic staining. Our established pv-iPSCs showed strong ALP activity (Fig. 3A). Fluorescence immunostaining for SSEA-1 and SSEA-4 yielded positive results, but SSEA-3 staining did not (Fig. 3B). Although we cannot distinguish whether the positive signal came from the endogenous Oct3/4 or exogenous mouse-derived Oct3/4 due to the high homology, we confirmed that the anti-Oct3/4 antibody showed reactivity to the pv-iPSCs (Fig. 3B). From these data, we concluded that our established pv-iPSCs have pluripotent properties, judging by the marker expression.

Detection of pluripotent markers in pv-iPSCs. (A) Alkaline phosphatase activity was detected by chromogenic staining. Scale bar: 50 μm. (B) SSEA-1, SSEA-3, SSEA-4, and Oct3/4 staining of pv-iPSCs. Right side is pluripotent markers. Left side is nuclear counterstaining. Scale bar: 100 μm.
Detection of Inserted Vectors and Karyotypic Analysis
We detected the insertion of the reprogramming vectors in genomic PCR analysis. We tested two representative clones for PB-6F and PB-R6F. After PCR amplification, all of the pv-iPSCs tested positive for the corresponding expression cassette (Fig. 4A). Furthermore, we analyzed the karyotype in both PB-6F- and PB-R6F-generated iPSCs. More than 70% of cells had a normal karyotype in the pv-iPSC clone that was generated with PB-R6F, whereas only 30% of the cells showed the normal karyotype in pv-iPSCs with PB-6F (Fig. 4B, Tables 1 and 2). Furthermore, copy numbers of the reprograming vector in pv-iPSCs were detected by real-time PCR analysis. All R2 values of the calibration were more the 0.9 (Fig. 4C), indicating that the quantitation is quite reliable. We used the Tsc2 gene for comparative control gene, because Tsc2 exists in only two copies (derived from paternal and maternal), and there is no pseudogene in the genome of humans, mice, or rats. As a result of analysis, the estimated copy number of reprograming cassette copy number in all iPSC clones (both PB-6F and PB-R6F) is close to identical to Tsc2 (Fig. 4D). From these results, we concluded that pv-iPSCs have around two copies of the expression cassette, which was introduced with a transposon.

Detection of the reprogramming cassette by genomic PCR in the established iPSCs and karyotype analysis. (A) Detection of reprogramming cassettes by genomic PCR. Lane 1: prairie vole embryonic fibroblasts (pv-EFs). Lane 2: pv-iPSCs (generated with the PB-6F vector; clone 1). Lane 3: pv-iPSCs (generated with PB-6F; clone 2). Lane 4: pv-iPSCs (generated with PB-R6F; clone 1). Lane 5: pvi-PSCs (generated with PB-R6F; clone 2). Lane 6: no-template control. As the amplification control, the tuberous sclerosis type 2 gene (Tsc2) was used. (B) Karyotypic analysis of pv-iPSCs. The chromosomal pattern at metaphase is shown. The chromosomal set of the prairie vole is 2n = 52 + XY. (C, D) Copy number detection of reprogramming cassette by real-time PCR. Calibration curve of PB-6F, PB-R6F, and Tsc2 (C). Relative copy number quantitation of reprograming cassette (D).
Effects of Growth Factors and Small-Molecule Inhibitors
In this study, we used two kinds of growth factors: LIF and FGF. Furthermore, three kinds of low molecular weight inhibitors, PD0325901, CHIR99021 (2i), and thiazovivin were used. PD0325901 is known to inhibit MEK signaling, and CHIR99021 is known to inhibit glycogen synthase kinase-3β (GSK-3β) signaling (16,22,31). Thiazovivin targets Rho-associated kinase (ROCK) (16). A combination of these three compounds was reported to dramatically enhance the establishment of iPSCs (16,31). To elucidate which component is most essential for maintenance of the pv-iPSCs, we conducted the experiments with ALP staining. We found that FGF was not required to maintain iPSCs (Fig. 5, upper left). When we removed LIF from the medium, the cell growth of the colonies slowed down, and ALP activity decreased (Fig. 5, upper right). When we removed 2i and thiazovivin, cell growth and ALP activity also decreased (Fig. 5, lower left). Furthermore, removal of all supplements from the medium (LIF, FGF, 2i, and thiazovivin) reduced cell growth (Fig. 5, lower right) and partially caused differentiation into the epithelial cell lineage (data not shown). From these results, we concluded that our established pv-iPSCs require the presence of LIF and low molecular weight inhibitors.

Growth dependence of pv-iPSCs. (A) Growth dependency of the newly established pv-iPSCs, generated with the PB-6F vector. (B) Growth dependency of the established pv-iPSCs generated with PB-R6F. Scale bars: 100 μm. Control, LIF + 2i (CHIR99021 and PD0325901) + thiazovivin; LIF(-), 2i + thiazovivin; 2i thiazovivin (-), only LIF; No supplements, the basal medium only.
Teratoma Formation in SCID Mice
We evaluated teratoma formation and the differentiation ability of pv-iPSCs cells in vivo. The pv-iPSCs cells were injected into the testicular tissues of SCID mice. At 2–3 weeks after the injection, we already detected formation of teratomas by abdominal palpitation (Fig. 6A, B). Our research group works with iPSCs from various animals; formation of a teratoma within 2–3 weeks is quite rare, and this finding is suggestive of the strong pluripotent properties of these pv-iPSCs. We injected pv-iPSCs into bilateral testes of four SCID mice and detected teratoma formation in all injected areas (the teratoma formation ratio was 100% in five animals).

Teratoma formation by pv-iPSCs and its histological analysis. (A) Teratoma formation by pv-iPSCs generated with the PB-6F vector. (a) Gross appearance of the teratoma tissues, (b) the gastrointestinal tract (endoderm), (c) cartilage (mesoderm), (d) fat and smooth muscle (mesoderm), (e) skin (ectoderm), (f) neural tube (ectoderm). Staining with H&E. Scale bars: 25 μm. (B) Teratoma formation by pv-iPSCs generated with PB-R6F. (a) Gross appearance of teratoma tissues, (b) gastrointestinal tract (endoderm), (c) cartilage (mesoderm), (d) fat and cartilage (mesoderm), (e) hair root and skin tissues (ectoderm), (f) neural tube (ectoderm). Staining with H&E. Scale bars: 50 μm (b, f); 500 μm (c). 25 μm (d, e).
In histological analysis, teratomas from PB-6F and PB-R6F iPSCs showed differentiated tissue from the three germ layers, such as the gastrointestinal tract (endoderm), cartilage (mesoderm), fat (mesoderm), and neural tube (ectoderm) (Fig. 6A, B). Therefore, we concluded that pv-iPSCs from PB-6F and PB-R6F cells have the properties of stem cells. It should be noted that R6F-derived iPSCs showed cellular differentiation into keratinocytes and skin hair follicle cells (Fig. 6Bb–e). In the histological analysis, the teratoma tissue from PB-R6F cells showed differentiation into a greater number of lineages when we compared these cells with those derived from PB-6F (data not shown).
Expression of Pluripotent-Related Genes
In this study, expression of pluripotent-related genes was evaluated with real-time PCR between established iPSCs and pv-EFs (Fig. 7B). Furthermore, we also evaluated the gene expression pattern after the embryoid body (EB) formation (differentiation state) (Fig. 7A). With regard to gene expression, Tbx3 was well expressed in pv-iPSCs, and Klf2 was more highly expressed in pv-EFs. Esrrb was highly expressed in pv-iPSCs, and Sox2 and Oct3/4 expression levels were a bit higher, when compared with that of pv-EFs (Fig. 7B). Interestingly, although there is some variation among the multiple clones, the expression levels of these pluripotent genes in EBs closely resembled that of pv-EFs after differentiation (Fig. 7C). For example, expression levels of Oct3/4, Sox2, and Klf2 became lower after EB formation. The levels of Klf4 and Tbx3 were higher in EBs when compared to iPSCs.

Pluripotent-related gene expression in pv-iPSCss and EBs. (A) Cytomorphology of pv-iPSC-derived embryoid bodies. Scale bar: 100 μm. (B) Pluripotency-related gene expression in iPSCs and pv-EFs. (C) Pluripotency-related gene expression in pv-iPSCs and EBs (derived from pv-iPSCs).
Introduction of Fluorescent Marker Gene into pv-iPSCs
In this study, we established pv-iPSCs with expression cassettes with IRES-EGFP at the 3′ end. Interestingly, we observed a relatively strong expression of EGFP in the early passage, and the intensity of EGFP became much lower in later passages (around passage 20) (Fig. 8, left side). To address the chimeric contribution of iPSCs, we need to introduce an efficiently expressed marker in iPSCs, regardless of the differentiation status. To achieve this, we introduced the recombinant lentivirus, which expresses EGFP and a puromycin-resistant gene, that is driven by a PGK promoter (pLenti PGK GFP Puro). The introduced iPSCs were selected with puromycin. As a consequence, strong green fluorescence was confirmed in EGFP-transfected pv-iPSCs (Fig. 8, right side). The established pv-iPSCs could be a powerful tool to address the contribution of iPSCs to various developmental stages.
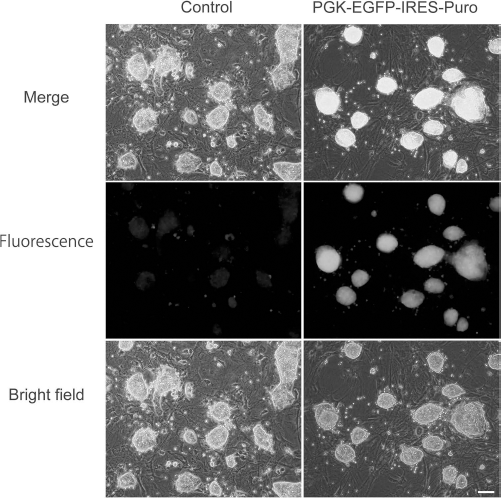
Establishment of pv-iPSCs, which efficiently expresses the fluorescent marker. Left side (three panels): fluorescence, bright field, and merged image of control pv-iPSCs. Right side (three panels): fluorescence, bright field, and merged image of PGK-EGFP-IRES-Puro pv-iPSCs after the selection. Scale bar: 100 μm.
Discussion
In this study, we established pv-iPSCs from fibroblasts. In our preliminary studies, we first tried to create a lentiviral-based monocistronic vector: STEMCCA-loxP (23). STEMCCA-loxP is a powerful vector for establishment of iPSCs from human and mouse cells. Our first attempt to establish pv-iPSCs with the STEMCCA lentivirus was not successful. Then we started to use immortalized cells for optimization of the experimental conditions.
In this study, we used two kinds of reprogramming vectors: PB-6F (piggyBac with six reprogramming factors) and PB-R6F (piggyBac with redesigned reprogramming factors). The difference between these two reprogramming vectors is the structure of the Oct3/4 gene. Oct3/4 was reported to be the most critical reprogramming factor to obtain iPSCs. Hirai et al. showed that the fusion protein of Oct3/4 with the transcriptional activation domain (TAD) of MyoD (M3O) dramatically increases the efficiency of iPSC reprogramming of murine and human cells (9). They stated that the increased efficiency of TAD of MyoD changes the epigenetic status of the chromosome, and the effects of Oct3/4 on the downstream genes are expected to be enhanced. In this study, we did not observe a major morphological difference between pv-iPSCs generated with the PB-6F vector and those with PB-R6F at the early stage of sequential passages. Nonetheless, at a late passage, pv-iPSCs with PB-R6F were more advantageous than were PB-6F-generated cells. This difference in morphology might be explained by the differences in transcriptional activity between original Oct3/4 and Oct3/4 with TAD. Furthermore, we found out that the expression level of vole-derived Oct3/4 is still highly maintained in differentiated EBs in the case of PB-R6F, when it was compared with that of PB-6F (Fig. 7C). The fusion protein of mouse Oct3/4 with transactivation domain of MyoD was reported to change the epigenetic status of the promoter region. There is a possibility that the expression of modified Oct3/4 (M3O) keeps their endogenous transcriptional activity through the enhanced transcriptional activity, even after EB formation. Detection of these differences in pluripotency-related genes caused by normal Oct3/4 and M3O, after EB formation, is the first data in prairie vole.
Furthermore, in this study, we conducted a karyotypic analysis. In both pv-iPSC clones, some of the cells showed normal diploidy at a frequency of ~50%. Although our established pv-iPSCs showed an abnormal number of chromosomes, previously established pv-iPSCs also showed diploidy at a frequency of ~70% (18). Although there is a difference between our data and previous data, the present pv-iPSCs may be sensitive to genomic instability. In our data, swine iPSCs with six reprogramming factors showed completely normal diploidy in the 50 cells examined (unpublished data). Therefore, there is a species difference in terms of genomic stability during the process of reprogramming.
In this study, we used a polycistronic vector for reprogramming. The polycistronic vectors (a mixture of monocistronic expression vectors, as in the original method of iPSC induction) are useful for induction of iPSCs; however, the introduction of polycistronic vectors requires high efficiency of the DNA transfection method, and achievement of high efficiency is sometimes difficult in the case of primary cells. Accordingly, we created six reprogramming polycistronic expression vectors. We successfully obtained 26 lines out of 35 primary colonies, indicating that the successful ratio is around 74.28%. However, in a previous report using a monocistronic expression vector, only 11 iPSC lines were established from 2,800 primary colonies (18). From this result, we established polycistronic vectors (PB-6F or PB-R6F), which are advantageous tools for establishment of iPSCs in prairie vole. In most of the expression vectors derived from piggyBac, because of the structure of the transposon and expression vector, two different vectors (transposon vector and transposase expressing vector) have to be introduced into the target cells. In contrast, our piggyBac expression vector here contained both the transposon cassette and the expression cassette of transposase. Therefore, our established vector system combines several features. Our vector may be a useful tool for the creation of iPSCs from various types of tissues and cells.
In a previous report, pv-iPSCs were established by using a retroviral vector (18). Retroviral vectors cannot be removed from the host's genome. In this study, pv-iPSCs were established with a piggyBac transposon system. The piggyBac system is a beneficial system, because introduced genes can be removed by the expression of transposase (7,30,34). In mice and humans, iPSCs have established with a piggyBac system (27,30).
Our pv-iPSCs showed growth dependence on the presence of LIF and low molecular weight inhibitors. Because the prairie vole is a rodent, it is reasonable that vole-derived stem cells show a dependence on LIF for growth. The presence of low molecular weight inhibitors is also required for the growth of rat iPSCs (15). The growth dependence of pv-iPSCs on these small-molecule inhibitors might be a common feature of rat cells.
Our newly established pv-iPSCs showed positive expression of SSEA-1 and rapid tumor growth after implantation into the testicular tissues of SCID mice. This biological characteristic closely resembles that of mouse iPSCs (8,24,26). Therefore, we are now analyzing the usefulness of our pv-iPSCs for creation of chimeras. Our newly established pv-iPSCs are expected to be a useful tool for analysis of the molecular mechanisms underlying social behavioral patterns.
Footnotes
Acknowledgments
We thank Dr. Kenji Sakimura (Niigata University) and Dr. Shizu Hidema (Tohoku University) for helpful discussions. We thank Dr. Larry J. Young (Yerkes National Primate Research Center, Department of Psychiatry, Emory University) for providing prairie voles. This work was supported by JSPS KAKENHI, grant No. 13J04781. The authors declare no conflicts of interest.