
Abstract
Horse health has become a major concern with the expansion of horse-related industries and sports; the importance of healthy muscles for horse performance and daily activities is undisputed. Here we generated equine-induced pluripotent stem cells (E-iPSCs) by reprogramming equine adipose-derived stem cells (E-ADSCs) into iPSCs using a polycistronic lentiviral vector encoding four transcription factors (i.e., Oct4, Sox2, Klf4, and c-Myc) and then examined their pluripotent characteristics. Subsequently, established E-iPSCs were transplanted into muscle-injured Rag/mdx mice. The histopathology results showed that E-iPSC-transplanted mice exhibited enhanced muscle regeneration compared to controls. In addition, E-iPSC-derived myofibers were observed in the injured muscles. In conclusion, we show that E-iPSCs could be successfully generated from equine ADSCs and transplanted into injured muscles and that E-iPSCs have the capacity to induce regeneration of injured muscles.
Introduction
Musculoskeletal injuries or disorders are common among athletic animals such as horses (5,6,11). Horses offer a unique opportunity to explore treatment strategies for musculoskeletal disorders under conditions that resemble the pathophysiology of human patients (28); therefore, they constitute an ideal animal model for studying new therapies, particularly in the field of regenerative medicine. Stem cells have been gaining attention in human and veterinary biomedical research because of their potential as therapeutic agents for musculoskeletal injuries and disorders. Mesenchymal stem cells (MSCs), embryonic stem cells (ESCs), and induced pluripotent stem cells (iPSCs) have been actively studied as a source for stem cell therapy in horses. In addition to the advantages afforded by equine iPSCs (E-iPSCs) in the field of veterinary medicine, E-iPSCs also open up the possibility to use horses for the validation of stem cell-based therapies prior to moving into the human clinical setting.
Skeletal muscles comprise approximately 40% of the human body mass and are important for normal body function and homeostasis. Skeletal muscles produce the force for locomotion, breathing, and posture support and provide heat during cold stress. Moreover, skeletal muscles are a major regulator of blood glucose levels; impaired insulin signaling in skeletal muscles can lead to type 2 diabetes (9,22,29).
Skeletal muscles possess remarkable regenerative capacity; acute injury to healthy muscles produces an inflammatory reaction that removes damaged myofibers and promotes their replacement. The main source for muscle regeneration and growth is a population of muscle stem cells called satellite cells. However, this regenerative capacity decreases with age and can be dramatically affected by multiple types of muscle-related pathological conditions. These conditions include chronic inflammatory myopathies, muscular dystrophies, neuromuscular disorders, sarcopenia, cachexia, and metabolic disorders.
Most of the skeletal muscle disorders are characterized by progressive muscle weakness and wasting. More severe volumetric skeletal muscle loss and injury can also result from mild lacerations (22). Under such conditions, the satellite cell pool is depleted, thus decreasing the regenerative capacity of the skeletal muscles. Therefore, many researchers in the field of muscle regenerative medicine have focused on the maintenance and replenishment of the satellite cell pool. Other sources of progenitor and stem cells with myogenic capacity exist, which may similarly enable skeletal muscle repair. Specifically, recent studies have shown the active myogenic role of human and mouse iPSCs during muscle regeneration in preclinical mouse models (7,8,10,13,23,31,35,36).
In this study, we report the generation of iPSCs from equine adipose-derived stem cells (E-ADSCs) using a TetO lentiviral vector to induce the expression of octamer-binding transcription factor 4 (Oct4), (sex-determining region Y)-box 2 (Sox2), Kruppel-like factor 4 (Klf4), and c-Myc. In addition, we show that E-iPSCs transplanted into injured muscles of Rag/mdx mice exhibit therapeutic effects.
Materials and Methods
Preparation of Equine ADSCs
Adipose tissue from a male 8-month-old horse was used for the isolation of E-ADSCs. The adipose tissue was harvested and washed with Dulbecco's phosphate-buffered saline (DPBS; GeneDEPOT, Barker, TX, USA) and 70% ethanol (Duksan Pure Chemicals, Ansan, Republic of Korea). The adipose tissue was minced and digested with 0.2% type I collagenase (Worthington Biochemical, Lakewood, NJ, USA) in DPBS in a shaking incubator at 37°C for 10 min. The digested tissue was passed through a 70-μm nylon cell strainer (SPL Life Sciences, Pocheon, Republic of Korea) to remove the debris and then centrifuged at 200 × g for 3 min to obtain the cell pellet. The pellet was resuspended and washed twice with DPBS.
Following isolation, the cells were plated with growth media [low-glucose Dulbecco's modified Eagle's medium (DMEM); Gibco, Carlsbad, CA, USA] containing 10% fetal bovine serum (FBS; Atlas Biologicals, Fort Collins, CO, USA) and 1% penicillin/streptomycin solution (Welgene, Daegu, Republic of Korea) and incubated at 37°C in a humidified environment with 5% CO2. Passage 2 ADSCs were used for reprogramming. Cell handling and experimental procedures were approved by the Kyungpook National University Institutional Animal Care and Use Committee (IACUC; Approval No. 2013-0082).
To demonstrate the phenotype of E-ADSCs, a fluorescence-activated cell sorting (FACS) analysis was performed using cluster of differentiation (CD) markers. Passage 3 ADSCs were harvested from the culture dish, and the 5 × 105 cells per 200 μl of PBS were suspended. The cells were incubated with fluorescein-conjugated monoclonal antibodies: anti-human CD31-fluorescein isothiocyanate (FITC; BD Pharmingen, San Jose, CA, USA), anti-human CD34-phycoerythrin (PE; eBioscience, San Diego, CA, USA), anti-human CD44-PE (eBioscience), anti-human CD45-FITC (BD Pharmingen), and anti-human CD90-PE (BD Pharmingen). For detection of CD29, E-ADSCs were stained with anti-human/equine integrin β1/CD29 monoclonal antibody (R&D Systems, Minneapolis, MN, USA), followed by FITC-conjugated anti-mouse IgG secondary antibody (Invitrogen, Carlsbad, CA, USA).
Generation of Equine Induced Pluripotent Stem Cells (E-iPSCs)
E-iPSCs were generated as previously reported (4). Plasmids TetO-FUW-OSKM (containing Oct-4, Sox2, Klf4, and Myc genes) and FUW-M2rtTA (containing the M2rtTA gene) were obtained from Addgene (Cambridge, MA, USA). Lentiviral vectors (OSKM and M2rtTA) were produced using the ViraPower™ Lentiviral Packaging Mix (Invitrogen) according to the manufacturer's instructions.
E-ADSCs were plated 1 day prior to transduction at 1 × 105 cells per 100-mm culture dish. The cells were incubated overnight with equal volumes of lentiviral supernatants (OSKM and M2rtTA) and 10 ng/ml polybrene (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Transduced E-ADSCs were cultured in ESC medium [high-glucose DMEM (Gibco) containing 1× GlutaMAX (Gibco), 1× minimum essential medium nonessential amino acids solution (MEM-NEAA; Gibco), 0.1% mercaptoethanol (Gibco), 1 U of leukemia inhibitory factor (LIF; Millipore, North Ryde, Australia), 1% penicillin/streptomycin solution, 20% FBS, and 2 μg/ml doxycycline hyclate (Dox; Sigma-Aldrich, St. Louis, MO, USA)]. The medium was changed every other day. Colonies were picked 18 to 30 days after viral transduction and manually plated onto culture plates seeded with 10 μg/ml mitomycin C (Sigma-Aldrich)-treated mouse embryonic fibroblasts (MEFs); 10 mg/ml type IV collagenase (Gibco) in DPBS was used for passaging.
Alkaline Phosphatase (AP) Staining
E-iPSCs were fixed with 4% paraformaldehyde (PFA; Yukari Pure Chemicals, Tokyo, Japan) for 1–2 min and then stained with the AP detection kit (Millipore) according to the manufacturer's instructions.
Immunofluorescence (IF) Staining of E-iPSCs
E-iPSCs were fixed in 4% PFA at room temperature (RT) for 15 min and then immunostained with the following primary antibodies: anti-mouse Oct3/4 (1:200 dilution; Santa Cruz Biotechnology), anti-rabbit Sox2 (1:200 dilution; Abcam, Cambridge, MA, USA), anti-mouse stage-specific embryonic antigen 1 (SSEA-1) (1:200 dilution; Santa Cruz Biotechnology), and anti-rabbit Nanog (1:200 dilution; Abcam). FITC-conjugated anti-mouse IgG and 5/6-tetramethyl-rhodamine isothiocyanate (TRITC)-conjugated anti-rabbit IgG were used as secondary antibodies (Invitrogen). Cell nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI dihydrochloride; Molecular Probes, Leiden, the Netherlands).
In Vitro Cell Differentiation
E-iPSCs were harvested, passaged to a bacterial culture dish, and allowed to grow in suspension for 12 days in high-glucose DMEM containing 1× MEM-NEAA, 0.1% mercaptoethanol, 1% penicillin/streptomycin solution, 10% FBS, and 5% horse serum (HS; Gibco).
Reverse Transcription Polymerase Chain Reaction (RT-PCR) Analysis of Cells
Total RNA was extracted from harvested cells using the TRIzol reagent (Invitrogen) according to the manufacturer's protocol. cDNA was synthesized from extracted RNA using Moloney murine leukemia virus (M-MLV) reverse transcriptase (Invitrogen). For analysis of specific gene expression levels, the cDNA was amplified using AccuPower PCR PreMix (Bioneer Corporation, Daejeon, Republic of Korea). Primer sequences and product sizes are listed in Table 1.
List of Equine Primers Used for RT-PCR Analysis of Cells

Teratoma Formation
E-iPSCs were harvested with type IV collagenase (Gibco) in DPBS, and 5 × 106 cells were resuspended in 300 μl of Matrigel (Corning Inc., Corning, NY, USA) diluted 1:1 in PBS. The cell suspensions were injected subcutaneously into 8-week-old NOD/SCID mice (NOD. CB17-Prkdcscid/Arc; Animal Resources Centre, City of Melville, Australia). Approximately 2–3 months after the injection, masses were dissected, fixed in 10% neutral-buffered formalin (DC Chemical Co., Seoul, Republic of Korea), routinely processed, and embedded in paraffin. The sections were stained with hematoxylin (Sigma-Aldrich) and eosin (H&E; Junsei Chemicals, Tokyo, Japan).
Animals
Male Rag/mdx mice and green fluorescent protein (GFP) transgenic C57BL/6J 3-month-old mice were used for this study. Rag/mdx mice, produced by crossing mdx mice [a commonly used Duchenne muscular dystrophy (DMD) model containing a mutation leading to dystrophin deficiency] with Rag-deficient mice (immunodeficient mice that accept human grafts), were provided by Dr. Jacques P. Tremblay (CHUQ Research Center/CHUL, Quebec City, Canada). GFP transgenic mice were provided by Dr. Masaru Okabe (Osaka University, Suita, Osaka, Japan) (26). Mice were housed in a temperature (22 ± 2°C)- and relative humidity (50 ± 10%)-controlled room under 12/12-h light–dark cycles and given standard food pellets and water ad libitum.
All animal handling and experimental procedures were performed according to the National Institutes of Health (Bethesda, MD, USA) guidelines for the care and use of laboratory animals and were approved by the Kyungpook National University IACUC (Approval No. 2014-0167).
Animal Experiments Design
E-iPSC line #1-2 was used for the animal experiments. The Rag/mdx mice were divided into two groups: the notexin group (n = 7) and the notexin + E-iPSCs group (n = 7). Animals were anesthetized using zoletil (Virbac, Carros, France) and xylazine (Bayer, Leverkusen, Germany); then notexin (Lotaxan, Valence, France) with or without E-iPSCs (3 × 105 cells in saline containing notexin) was injected into the gastrocnemius muscles. A dose of 50 μl of 2 μg/ml notexin in 0.9% saline (~0.3–0.5 μg/kg) was used. Because E-iPSCs do not have fluorescence that can be utilized for cell tracking in transplanted sites and specific horse antibodies are required to detect transplanted E-iPSCs in vivo, GFP transgenic mice were used to visualize differentiated E-iPSC-derived myofibers. As with the Rag/mdx mice, GFP transgenic mice were anesthetized using zoletil and xylazine, and then notexin with (n = 2) or without E-iPSCs (n = 2) was injected into the gastrocnemius muscles. All GFP mice received cyclosporine (10 mg/kg/day; Sigma-Aldrich) subcutaneously from 1 day prior to transplantation until sacrifice. All animals were sacrificed after 15 days, and gastrocnemius muscle samples were removed for further analysis.
Histopathology
For histopathological evaluations, dissected gastrocnemius muscle samples were fixed in 10% neutral-buffered formalin, processed using standard methods, and embedded in paraffin. Paraffin-embedded sections (4 μm thick) were deparaffinized in toluene (Duksan Pure Chemicals), rehydrated in a graded alcohol series, and stained with H&E. Following H&E staining, all myofibers in the injured sections visible at original 400× magnification were measured using the Leica Application Suite program (Leica Microsystems, Wetzlar, Germany), and the number of myofibers in 10,000 μm2 was calculated. In addition, the number of centrally located nuclei, a hallmark of skeletal muscle regeneration, was counted. Five areas per section were randomly selected at the original 400× magnification, and central nucleic (CN) and peripheral nucleic (PN) myofibers were counted. The percentage of CN myofibers was calculated as follows: %CN = nCN/(nCN + nPN) × 100. Areas of calcification in the injured sections were measured using the Leica Application Suite program.
Immunohistochemistry (IHC) staining for SSEA-1 was performed to identify undifferentiated cells. Tissue sections were immunostained with the anti-mouse SSEA-1 antibody (1:200 dilution; Santa Cruz Biotechnology), and the antigen–antibody complex was visualized using the avidin–biotin–peroxidase complex (ABC; Vectastain ABC kit; Vector Laboratories, Burlingame, CA, USA).
IF Staining of Muscle Tissues
Gastrocnemius muscle tissues were collected from the GFP transgenic mice. The muscle samples were fixed in 4% PFA at RT for 15 min, in 15% sucrose for 6 h, and in 30% sucrose overnight. The fixed tissues were embedded in optimal cutting temperature (OCT) compound (Energy Beam Sciences, East Granby, CT, USA) and snap frozen in 2-methylbutane (Sigma-Aldrich, Munich, Germany) cooled by liquid nitrogen. Muscle tissues were cut into 8-μm-thick sections. The tissue sections were immunostained with the primary antibody anti-dystrophin (1:100 dilution; Abcam) as previously described (20). TRITC-conjugated anti-rabbit IgG was used as the secondary antibody, and DAPI was used as a nuclear counterstain.
RT-PCR and Quantitative RT-PCR (qRT-PCR) Analysis of Muscle Tissue
Total RNA was extracted from the frozen muscle tissue samples using the TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA) following the manufacturer's protocol. cDNA was synthesized using M-MLV reverse transcriptase (Thermo Fisher Scientific). The expression levels of specific genes were analyzed by RT-PCR using the cDNA with AccuPower PCR PreMix. Primer sequences and product sizes are listed in Table 2. The optical density (OD) of the PCR product bands was quantified with ImageJ, and the relative expression of the target genes was normalized to that of glyceraldehyde 3-phosphate dehydrogenase (GAPDH; relative expression of mRNA = OD of the target gene/OD of GAPDH).
List of Primers Used for RT-PCR Analysis of Muscle Tissues

Rotor-Gene Q (Qiagen, Hilden, Germany) was used for qRT-PCR. The expression of the following genes was analyzed: Pax7, myogenin, Wnt3a, and β-catenin. The expression of the target genes was normalized to GAPDH. The ΔΔ threshold cycle (ΔΔCt) method was used to analyze relative expression. The primers for Pax7 (QT00147728), myogenin (QT00112378), Wnt3a (QT00250439), β-catenin (QT00160958), and GAPDH (QT01658692) were obtained from Qiagen.
Statistical Analysis
Data were expressed as mean ± SEM and were analyzed for statistical significance using the unpaired Student's t-test. The statistical significance value was set at p < 0.05. Statistical analysis of the data was performed using the SPSS Statistics 21 software (IBM, Armonk, NY, USA).
Results
Characterization of Equine ADSCs
For characterization of the E-ADSCs using FACS analysis, the cells showed a positive reaction for stromal cell-related makers, which are CD29, CD44, and CD90, whereas the cells were negative for CD31 (endothelial cell-related marker) and CD34 and CD45 (hematopoietic cell-related marker) (Fig. 1A).
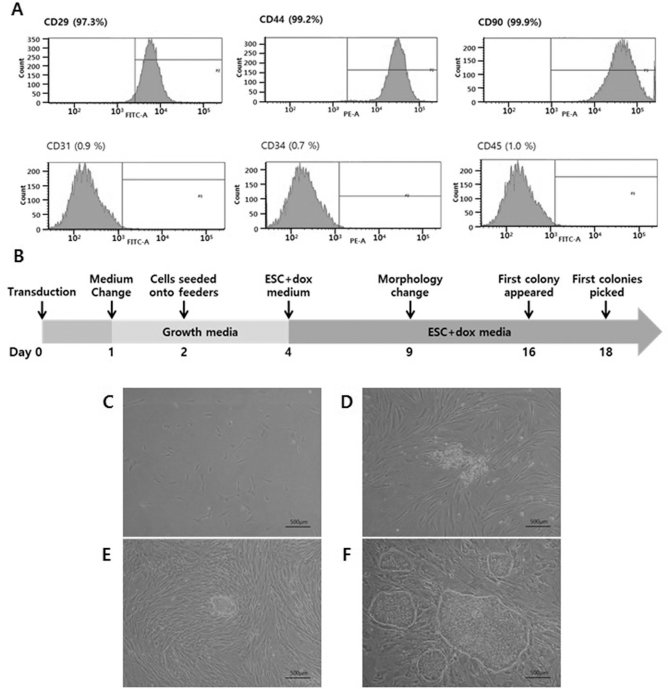
Characterization and reprogramming of equine ADSCs (E-ADSCs). (A) The cells showed a positive reaction for stromal cell-related markers, which are CD29, CD44, and CD90, whereas the cells were negative for CD31, CD34, and CD45. (B) Schematic diagram illustrating the process of generation of E-iPSCs from equine ADSCs. (C) Photomicrograph of ADSCs. (D) Morphology changes in E-ADSCs transduced with lentiviral vector in ESC media + Dox after 9 days. (E) Initial colonies that appeared after 16 days. (F) Morphology of established iPCSs. Scale bars: 500 μm.
Generation of E-iPSCs
Initially, we reprogrammed ADSCs from an adult horse; a schematic diagram depicting the process of E-iPSC generation is shown in Figure 1B. E-ADSCs plated in a 100-mm culture dish at a density of 1 × 105 cells were transduced with a TetO inducible OSKM polycistronic lentiviral vector (4) (Fig. 1C). Transduced cells were cultured in growth media for cell stabilization and then in ESC media with Dox to induce the expression of the Oct4, Sox2, Klf4, and c-Myc genes. Nine days after transduction, the cells exhibited morphology changes (Fig. 1D). Clearly defined colonies appeared on days 15–16 after transduction (Fig. 1E). Colonies that grew to an adequate size were picked on days 18–30 after transduction and individually expanded by passaging. Established E-iPSC colonies displayed distinctly flat, tight, and well-defined structures, which somewhat resembled primed pluripotent stem cells (Fig. 1F). Experiments were performed twice independently, and a total of six (3 + 3) E-iPSC lines were successfully established, maintained in culture, and characterized in detail. The reprogramming efficiency was estimated at 0.003%.
In Vitro E-iPSCs Characteristics
The E-iPSCs showed high AP activity, which is considered to be a marker of pluripotency in ESCs (25) (Fig. 2A). In addition, immunofluorescence and RT-PCR analysis were used to determine the expression of endogenous pluripotency markers in the E-iPSC lines. E-iPSCs exhibited high expression levels of key pluripotent markers including Oct4, Sox2, Nanog, Lin28, and Rex, whereas the parental E-ADSCs showed low expression of these markers (Fig. 2B). The E-iPSCs were also positive for expression of the Oct4, Sox2, SSEA-1, and Nanog proteins as determined by immunofluorescence staining (Fig. 2C and D). Taken together, the pluripotent marker expression analysis results demonstrate the successful reprogramming of equine cells into iPSCs.

In vitro characterization of E-iPSCs. (A) E-iPSC colonies (E-iPSC line #1-2) were positive for alkaline phosphatase staining (scale bar: 500 μm). (B) Gene expression levels of key pluripotency markers determined using RT-PCR with equine-specific primers. (C) Oct4 and Sox2; (D) SSEA-1 and Nanog detected by immunofluorescence staining in E-iPSC line #1-2 (scale bars: 200 μm).
In Vitro and In Vivo E-iPSC Differentiation Potential
To induce differentiation in vitro, E-iPSC line #1-2 was placed in a low-adherence bacterial petri dish in the absence of LIF. The cells formed embryoid bodies (EBs) after 6, 9, and 12 days in culture (Fig. 3A); the EBs grew in size and developed fluid-filled cysts in a time-dependent manner. Total RNA was extracted from equine EBs on days 6, 9, and 12, and RT-PCR was performed to analyze gene expression (Fig. 3B). The expression of TBX-4 (paraxial mesodermal marker) and Pax6 (ectodermal marker) increased in the EBs on day 6 and then decreased on days 9 and 12. The expression of bone morphogenetic protein 4 (BMP4; mesodermal marker) and α-fetoprotein (AFP; endodermal marker) exhibited a time-dependent increase.

In vitro and in vivo differentiation potential of E-iPSCs. (A) E-iPSCs were cultured in bacterial culture dishes to induce embryoid body (EB) formation. Microscopic morphology of equine EBs at 6, 9, and 12 days (scale bars: 200 μm). (B) Expression of mesodermal (BMP4 and TBX-4), endodermal (AFP), ectodermal (Pax6), and pluripotency (Oct4) markers in equine EBs at 6, 9, and 12 days determined by RT-PCR. (C) Tumors included differentiated derivatives of the three germ layers, including keratinized epithelium (ectoderm), cartilage (mesoderm), and gut-like epithelium (endoderm) (H&E, original magnification: 200×, scale bars: 100 μm).
E-iPSCs were injected subcutaneously into NOD/SCID immunocompromised mice. Following 2–3 months of cell implantation, teratomas containing structures derived from the three germ layers, including keratinized epithelium (ectoderm), cartilage (mesoderm), and gut-like epithelium (endoderm), were successfully developed (Fig. 3C). This result indicates that the generated E-iPSCs possess pluripotent capacity.
Histopathological Evaluation of Injured Muscles In Vivo
Muscle-injured Rag/mdx mice were used to determine whether E-iPSCs possess myogenic differentiation potential in vivo. A myotoxic venom (notexin) with or without E-iPSCs was injected into the gastrocnemius muscles of Rag/mdx mice, and muscle samples were histopathologically examined 15 days after injury.
The H&E staining demonstrated that the muscles from the E-iPSC-injected mouse group (notexin + E-iPSCs group) had enhanced regeneration compared to those from the notexin group. However, all mice from both groups exhibited incomplete muscle regeneration. Fewer dystrophic calcification areas were observed in the notexin + E-iPSCs compared to the notexin group, suggesting that the muscle fibers remained intact (Fig. 4A and B). In addition, the percentage of CN myofibers and the number of myofibers per unit area (10,000 μm2) were measured and calculated based on the H&E staining results. The notexin + E-iPSCs group had a significantly increased percentage of CN myofibers, a hallmark of skeletal muscle regeneration, compared to the notexin group (Fig. 4C and D), and an increased number of myofibers per unit area was detected in the E-iPSC-transplanted group (Fig. 4E). Moreover, 54.14% of the muscle samples (4/7 E-iPSC-transplanted muscles) from the notexin + E-iPSCs group contained undifferentiated cells (Fig. 4F); IHC staining demonstrated that these cells expressed SSEA-1 (Fig. 4G). Although undifferentiated cells were present in E-iPSC-injected muscles, the muscle differentiation capacity of the E-iPSCs was confirmed using muscles from GFP transgenic mice. E-iPSCs were injected into injured muscles of GFP transgenic mice to visualize the differentiated E-iPSC-derived myofibers. Several non-GFP-expressing E-iPSC-derived myofibers expressed dystrophin, suggesting that E-iPSCs possess the capacity for muscular differentiation (Fig. 4H).

Histopathological evaluation of injured muscles. (A, B) H&E staining of gastrocnemius muscles. Fewer calcification areas were observed in the E-iPSC-transplanted group than in the control group (original magnification: 50×, scale bars: 500 μm). (C, D) Gastrocnemius muscles from the E-iPSC-transplanted group had an increased percentage of central nuclei myofibers compared to the notexin group (original magnification: 400×, scale bars: 50 μm). (E) The number of myofibers per 10,000 μm2 was elevated in gastrocnemius muscles from the notexin + E-iPSCs group. (F) Undifferentiated cells were observed in the muscle tissue of four mice (original magnification: 100×, scale bars: 200 μm). (G) These cells expressed SSEA-1 as detected by IHC staining (original magnification: 400×, scale bars: 50 μm). (H) A number of GFP-negative myofibers expressed dystrophin (white arrows) (original magnification: 400×, scale bars: 50 μm). Values represent the mean ± SEM (*p < 0.05).
Gene Expression Levels in Injured Muscles
RT-PCR analysis was performed to evaluate the effect of muscle injury on gene expression levels. The E-iPSC-transplanted group (notexin + E-iPSC group) showed increased expression of Pax7, a specific marker for muscle satellite cells, compared with the notexin group (Fig. 5A and B). Transplantation of E-iPSCs also affected the expression of myogenic differentiation markers (MyoD and myogenin); MyoD and myogenin expression levels were higher in the injured muscles of the E-iPSC-transplanted group compared to those in the group not transplanted with cells (Fig. 5A, C, and D). Furthermore, the qRT-PCR results also showed increased expression of Pax7 and myogenin in the notexin + E-iPSCs group (Fig. 5E and F).
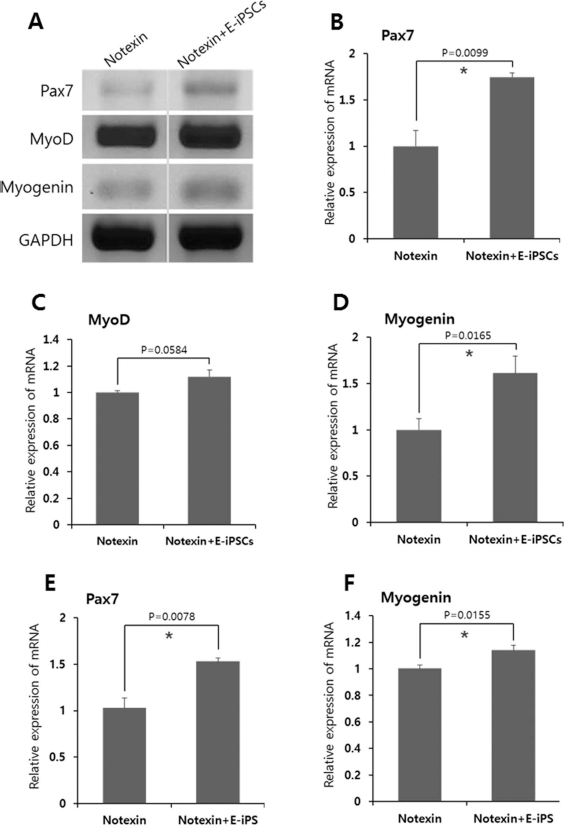
RT-PCR and qRT-PCR analysis of myogenic markers in injured muscle tissues of Rag/mdx mice 15 days after notexin injection. (A–D) RT-PCR analysis showed that the notexin + E-iPSCs group had elevated expression of myogenic markers (Pax7, MyoD, and myogenin) compared to the notexin group. (E, F) The qRT-PCR results also showed increased Pax7 and myogenin expression in the notexin + E-iPSCs group. Values represent the mean ± SEM (*p < 0.05).
Wnt signaling plays an essential role during embryonic muscle development and in the maintenance of skeletal muscle homeostasis in adults. In the skeletal muscles, canonical Wnt signaling regulates the differentiation of muscle progenitor cells (satellite cells), and noncanonical signals mediate the self-renewal of satellite cells and the growth of myofibers (39). The expression levels of canonical Wnt signaling-associated factors including Wnt3a, glycogen synthase kinase 3b (GSK3b), β-catenin, lymphoid enhancer binding factor 1 (Lef1), and transcription factor 7 (Tcf7) levels were significantly increased in the notexin + E-iPSC group compared to those in the notexin group (Fig. 6A–F). qRT-PCR analysis demonstrated that Wnt3a expression was significantly increased in the notexin + E-iPSC group compared with that in the notexin group (Fig. 6G) and that the expression of β-catenin was higher in the E-iPSC-transplanted group (Fig. 6H), although these results were not significant.
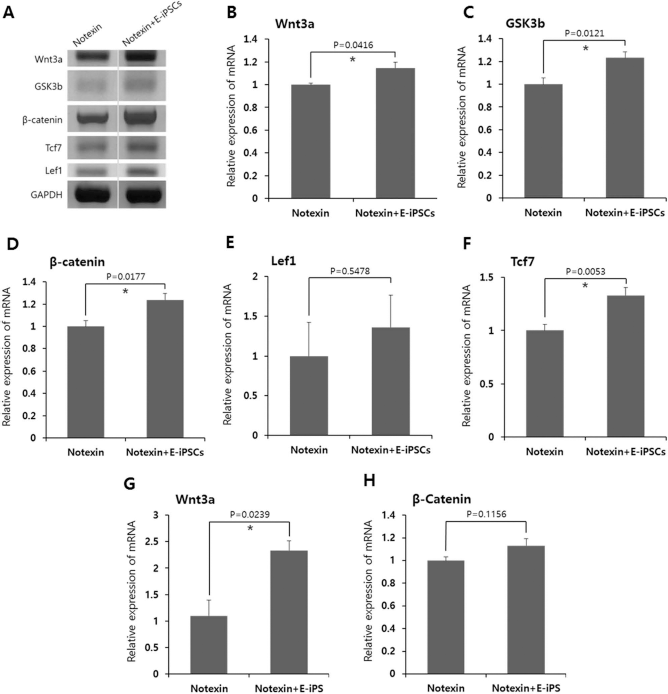
RT-PCR and qRT-PCR analysis of Wnt signaling-associated factors in injured muscle tissues of Rag/mdx mice 15 days after notexin injection. (A–F) The relative mRNA expression of Wnt signaling-associated factors (Wnt3a, GSK3b, β-catenin, and Tcf7) was significantly upregulated in the E-iPSC-transplanted group compared to the control group. (G, H) qRT-PCR analysis showed that Wnt3a and β-catenin expression levels were higher in the E-iPSC-transplanted group. Values represent the mean ± SEM (*p < 0.05).
Discussion
In this study, we successfully established an E-iPSC line derived from adult horse ADSCs. Horse-derived iPSCs have been previously described in several studies (2,17,24,33); these studies reported that iPSCs could be generated from equine fibroblasts and keratinocytes. However, unlike these previous studies, we used ADSCs, a type of adult stem cells. ADSCs have several advantages as a reprogramming source, such as abundance in the body, ease of collection, and the ability to maintain stable growth and proliferation (15,38). In addition, we used a TetO inducible lentiviral vector for the induction of OSKM expression. TetO systems exhibit high maximal expression levels comparable to the maximal levels obtained with strong constitutive mammalian promoters such as cytomegalovirus (CMV). In contrast to other inducible mammalian expression systems, gene regulation using the TetO system is highly specific; therefore, the interpretation of the results is not complicated by pleiotropic effects or nonspecific induction (42). We generated E-iPSC lines from E-ADSCs using the TetO induction system, which expressed pluripotent markers and showed differentiation potential into three germ layers (Figs. 1–3). Equine ESC isolation has been previously reported by several researchers (14,19,32). The characteristics of the E-iPSCs established in the present study are similar to those of the equine ESCs isolated in those previous studies. In particular, the morphology of the E-iPSC colonies and EBs was identical to that of equine ESC colonies and EBs. The E-iPSC colonies were flat and formed by monolayered and tightly packed compact cells; they also exhibited a large nucleus-to-cytoplasm ratio. In addition, when the E-iPSCs were cultured in suspension, the cell aggregates developed into cystic EBs composed of two layers of cells with heterogeneous cellular particles within the cavity. Although not conclusive for iPSC validation, EB formation suggests that the cells have the potential to differentiate into multiple tissues.
The characteristics of equine pluripotent cells are both similar and different to those of human and mouse ESCs. Murine pluripotent cells, the representative naive stem cells, typically grow in multilayer compact piled-up colonial groups and form dome-shaped colonies. In addition, murine pluripotent cells initiate spontaneous differentiation at the periphery of the colony. In contrast, the morphology of human-derived pluripotent cell colonies, the representative primed stem cells, is generally flatter in appearance, with fewer layers, and spontaneous differentiation tents to initiate at the center of the colony (12). As mentioned above, equine ESCs/iPSCs form flat monolayered tightly packed colonies with well-defined edges. In addition, spontaneous differentiation tends to commence at the center of the colonies when cultured without feeder cells. Equine pluripotent cells are more similar to human ESCs in terms of morphological and differentiation characteristics. Specific culture conditions are recommended for the propagation and maintenance of pluripotent cells; the basic requirements for cell culture suggest that LIF is critical for maintaining the undifferentiated state and self-renewal of murine ESCs (40). Conversely, LIF is not required for human ESCs; basic fibroblast growth factor (bFGF) plays a role similar to that of LIF in humans (18,41). In addition, equine cells are similar to mouse and human cells in terms of gene and protein expression. Mouse ESCs express SSEA-1 but not SSEA-4, while human ESCs exhibit equal expression of these stage-specific embryonic antigens (12). Both equine pluripotent cells, including the E-iPSCs established in this study, express SSEA-1 (2,17,19,24).
Disorders affecting skeletal muscle tissue and peripheral nerves are common in horses (3,11,21). In addition to myopathic disorders that can adversely affect athletic performance, severe degenerative myopathy can be fatal in horses (11). Regenerative medicine and stem cell therapies constitute promising approaches for the treatment of musculoskeletal injuries and diseases in horses. MSCs, ESCs, and iPSCs have been widely studied in horses as a source for stem cell therapy. E-iPSCs in particular have attracted attention as a promising tool for regenerative therapies in the field of veterinary medicine.
To determine whether E-iPSCs have the potential for myogenic differentiation, we performed in vivo animal experiments. E-iPSC transplantation into injured muscles of Rag/mdx mice reduced the area of calcification and increased the percentage of CN myofibers, a hallmark of skeletal muscle regeneration. Dystrophic calcification occurs in areas of necrosis; dead and dying cells are no longer able to regulate the influx of calcium into the cytosol resulting in an accumulation of calcium in the mitochondria. An increased number of myofibers per unit area was detected in the E-iPSC-transplanted group. In addition, transplanted E-iPSCs also affected the expression of myogenic markers; Pax7, MyoD, and myogenin expression levels were higher in the E-iPSC-injected group. Pax7 is a specific marker for muscle satellite cells (muscle progenitor cells). Increased Pax7 expression following E-iPSC transplantation may signify the presence of more Pax7-positive cells. In other words, E-iPSC transplantation may replenish the muscle progenitor cell pool in injured muscles. The histopathological and gene expression results showed that the injection of E-iPSCs promoted muscle regeneration in injured skeletal muscles of Rag/mdx mice. Moreover, transplanted E-iPSC-derived myofibers were observed with dystrophin immunofluorescence staining, although several muscles in the E-iPSC-injected group had undifferentiated cells. This result suggests that E-iPSCs possess muscle differentiation capacity in vivo.
Interestingly, Wnt signaling-associated factors including Wnt3a, GSK3b, β-catenin, Lef1, and Tcf7 were significantly upregulated in the notexin + E-iPSCs group compared to those in the notexin group. Wnt signaling plays an essential role in the maintenance of skeletal muscle homeostasis (39) and is involved in the regulation of muscular differentiation and satellite cell self-renewal during the regeneration of adult skeletal muscles following injury. In the early stages of muscle regeneration after injury, Wnt5a, Wnt5b, and Wnt7a become upregulated, whereas Wnt4 is downregulated. In later stages, Wnt7b and Wnt3a are additionally expressed (1,30). Addition of Wnt7a to regenerating muscles expands the satellite cell pool and induces hypertrophy through the Akt/mTOR signaling cascade, whereas application of Wnt3a to injured muscles induces the differentiation of satellite cells leading to depletion of the progenitor cell pool and muscle hyperplasia (increased levels of new myofibers) (39). RT-PCR analysis of mTOR, Raptor, and forkhead box protein O1 (FoxO) expression levels revealed no differences between the groups (data not shown). However, the number of myofibers per unit area was increased in the E-iPSC-injected group. These results suggest that transplantation of E-iPSCs increases the expression of Wnt3a and subsequently induces hyperplasia of the skeletal muscles. Wnt3a is involved in the stem cell differentiation signal complex; Wnt3a not only maintains the pluripotency of stem cells (37) but also promotes differentiation of stem cells into muscle (16). Wnt3a might affect both the maintenance of nondifferentiation conditions and myogenesis of E-iPSCs in muscles.
Conclusions
This study shows that E-iPSCs can be successfully generated from equine ADSCs and that E-iPSCs transplanted into injured muscles have the capacity to induce muscle regeneration. We believe that these findings will pave the way for a better understanding of E-iPSC technology and the development of muscle regeneration therapies in veterinary medicine; however, challenges such as safety assurance will need to be addressed in future studies.
Footnotes
Acknowledgments
This research was supported by the Bio-industry Technology Development Program, Korea Institute of Planning and Evaluation for Technology in Food, Agriculture, Forestry and Fisheries (312062-5). The authors declare no conflicts of interest.