
Abstract
Acute hepatic failure (AHF) is a severe liver injury leading to sustained damage and complications. Induced pluripotent stem cells (iPSCs) may be an alternative option for the treatment of AHF. In this study, we reprogrammed human dental pulp-derived fibroblasts into iPSCs, which exhibited pluripotency and the capacity to differentiate into tridermal lineages, including hepatocyte-like cells (iPSC-Heps). These iPSC-Heps resembled human embryonic stem cell-derived hepatocyte-like cells in gene signature and hepatic markers/functions. To improve iPSC-Heps engraftment, we next developed an injectable carboxymethyl-hexanoyl chitosan hydrogel (CHC) with sustained hepatocyte growth factor (HGF) release (HGF-CHC) and investigated the hepatoprotective activity of HGF-CHC-delivered iPSC-Heps in vitro and in an immunocompromised AHF mouse model induced by thioacetamide (TAA). Intrahepatic delivery of HGF-CHC-iPSC-Heps reduced the TAA-induced hepatic necrotic area and rescued liver function and recipient viability. Compared with PBS-delivered iPSC-Heps, the HGF-CHC-delivered iPSC-Heps exhibited higher antioxidant and antiapoptotic activities that reduced hepatic necrotic area. Importantly, these HGF-CHC-mediated responses could be abolished by administering anti-HGF neutralizing antibodies. In conclusion, our findings demonstrated that HGF mediated the enhancement of iPSC-Hep antioxidant/antiapoptotic capacities and hepatoprotection and that HGF-CHC is as an excellent vehicle for iPSC-Hep engraftment in iPSC-based therapy against AHF.
Keywords
Introduction
Acute hepatic failure (AHF) is a severe liver injury that will cause sustained liver damage and deterioration of liver function, leading to complications that include hepatic encephalopathy and multiorgan failure. Managing severe AHF is one of the most challenging problems in clinical medicine (47). Liver transplantation is a well-accepted treatment option for AHF at the end stage of liver disease (23). The major limitations within the field of liver transplantation include donor organ shortage, high cost, and the need for lifelong immunosuppressive medications (37). To alleviate the high demand for liver transplants, the interest in liver cell therapy has been increasing continuously in recent years (37). Among the cell types considered for cell therapy, induced pluripotent stem cells (iPSCs), which are able to differentiate into various cell types, can be generated from mouse embryonic fibroblasts or human fibroblasts via the forced expression of reprogramming factors (32,45). Thus, iPSCs have been investigated as alternative resources for restorative cell therapy of various diseases. iPSCs are capable of pluripotency and differentiation specifically into functional hepatocyte-like cells (5). An effective platform for hepatocyte generation from patient-specific iPSCs has been developed to model inherited metabolic liver disorders (41). Dental pulp tissue contains various populations of multipotent progenitors (30,36) and is a new source of pluripotent-like stem cells (1). Because of its accessibility and differentiation potential, dental pulp has gradually drawn attention in regenerative medicine. It is unclear whether the forced expression of transcription factors could reprogram dental pulp-derived cells into human iPSCs with pluripotent signatures. Moreover, it is unknown whether the resultant dental pulp-reprogrammed iPSCs could be further differentiated into iPSC-derived hepatocyte-like cells (iPSC-Heps) with potential hepatoprotective activity.
Initially identified as a potent mitogen for mature hepatocytes (35), hepatocyte growth factor (HGF) is strongly implicated in hepatic functions. HGF possesses strong antifibrotic activity and contributes to the onset or progression of liver fibrosis/cirrhosis (28,49). HGF directly promotes the differentiation of mesenchymal stem cells into hepatic lineages, and transplantation of these cells improves liver injury in rats (40). Notably, HGF is produced by stromal cells and stimulates epithelial cell proliferation, motility, morphogenesis, and angiogenesis in various organs, and all HGF-mediated effects are mediated by the tyrosine phosphorylation of its receptor, c-Met (34). HGF is a multifunctional polypeptide implicated in embryogenesis, angiogenesis, organ regeneration, and wound healing (29). The HGF/c-Met axis is also required for hepatic stem cellmediated liver regeneration in mice (13). Considering the beneficial effects of HGF in hepatocytes, it is critical to investigate whether HGF supplementation ensures the growth and engraftment of iPSC-differentiated hepatocytes or hepatocyte-like cells after transplantation.
HGF can potentially be used for the treatment of AHF (18) and CCl4-induced liver necrosis (2). Although accumulated evidence has shown the efficacy of HGF, the effects of HGF are quite variable (11,39). This variability is attributed to the rapid diffusion of HGF at the injection site, leading to insufficient HGF exposure. Notably, a hydrogel containing HGF enhances the therapeutic potential of HGF in the treatment of chronic vocal fold scarring (17). We recently developed a novel injectable carboxymethylhexanoyl chitosan (CHC) nanogel that enhances stem cell delivery and engraftment (6). Cell engraftment is a critical step in cell therapy for liver disease (12). Therefore, it is crucial to determine whether hydrogel-containing HGF improves the engraftment of iPSC-derived hepatocyte-like cells (iPSC-Heps) and leads to enhanced liver repair and the subsequent rescue of fulminant AHF. In this study, we reprogrammed human dental pulp fibroblasts into iPSCs and induced the differentiation of these iPSCs into functional iPSC-Heps. We prepared injectable CHC nanoscale hydrogel with sustained release of HGF (HGF–CHC), and we examined the growth capacity and hepatic-like functions of iPSC-Heps cultivated in HGF–CHC. We used HGF–CHC as a delivery vehicle to transplant iPSC-Heps into the liver of the thioacetamide (TAA)-induced AHF in vivo model, and we investigated the hepatoprotective efficacy of iPSC-Heps in vivo. Finally, the HGF-mediated effect was validated by the addition of anti-HGF neutralizing antibodies in this model. Our findings may elucidate the precise mechanism of iPSC-Hep-mediated hepatoprotective activity, providing an alternative therapeutic strategy for treating fulminant hepatic failure. The stable release HGF–CHC system may have therapeutic benefits in the management of AHF.
Materials and Methods
Mouse Embryonic Fibroblast Isolation, Human iPSC Generation and Culture, and Hepatic Differentiation of Human iPSCs
Mouse embryonic fibroblasts (MEFs) were prepared from the pregnant ICR mice between the 11th and 14th day postconception. The mice were purchased from National Laboratory Animal Center (Taipei, Taiwan). Briefly, the pregnant mice were sacrificed, and the uterine horns were dissected, followed by three washes with PBS (Sigma-Aldrich, St. Louis, MO, USA) in a laminar flow hood. One pair of Watchmakers' forceps was used to open each uterine wall to release the embryos. The retrieved embryos were washed three times by PBS. Subsequently, the embryos were dissected by the same tools and soft tissues (including liver, heart, and head tissues) were discarded. The clean carcasses were then transferred into a new glass Petri dish and minced using sharp Iris scissors. The tissue debris was then shifted into a 50-ml conical tube, and 10 ml of trypsin-EDTA was added (Life Technologies, Carlsbad, CA, USA). The tissue debris was then incubated for 15 min under room temperature and slowly shaken. This trypsinization was terminated by the addition of 15 ml of MEF culture medium [Dulbecco's modified Eagle's medium (DMEM; Gibco, Gaithersburg, MD, USA)] supplemented with 15% fetal bovine serum (Invitrogen/Life Technologies), nonessential amino acids (Sigma-Aldrich), and antibiotics (Invitrogen/Life Technologies). The supernatant was then transferred into a new 50-ml conical tube and centrifuged at 111.6 × g for 5 min. Finally, the pellets were resuspended with 40 ml of a fresh medium and divided evenly into T75 culture flasks. A ratio of three embryos per flask was recommended.
As for the human iPSC generation and culture, this research followed the tenets of the Declaration of Helsinki, and all samples were obtained after patients had given informed consent. Human iPSCs were reprogrammed from dental pulp stromal cells isolated from a 54-year-old donor during dental extraction. The study was approved by the Institutional Ethics Committee/Institutional Review Board of Taipei Veterans General Hospital. iPSCs were reprogrammed via the transduction of pMXs vectors (Addgene, Cambridge, MA, USA; https://www.addgene.org/vector-database/3674/) encoding the transcription factors, including octamer-binding transcription factor 4 (Oct4), sex-determining region Y-box 2 (Sox2), Kruppel-like factor 4 (Klf4), and c-Myc. Plat-A cells (Cell Biolabs, Inc., San Diego, CA, USA), which were used for plasmid transfection, were incubated overnight at a density of 2.5 × 106 cells per 100-mm dish (Corning Inc., Corning, NY, USA). The next day, 10 μg of each pMXs plasmid was transfected into the Plat-A cells with 10 ml of fresh DMEM using TransIT®-LT1 (Mirus, Madison, WI, USA). Forty-eight hours after the transfection, virus-containing medium was collected for target cell infection. In preparation for viral infection, 5 × 104 target cells were seeded per well into six-well plates (Corning Inc.) 1 day prior to transduction. Supernatants containing equal amounts of each of the four retroviruses were filtered through 0.45-μm filters (Millipore, Bedford, MA, USA) and supplemented with 10 μg/ml polybrene (Sigma-Aldrich), and the medium in each of the six-well plates was replaced with the virus-containing medium. The six-well plates were centrifuged at 550 × g for 1 h, and then the medium was replaced. On day 7 postinfection, target cells were passaged onto mitotically inactivated mouse embryonic fibroblast (MEF originally from mouse embryos) feeder layers and cultured using human embryonic stem cell (ESC) (19) medium (DMEM/F12; Gibco) supplemented with 20% KnockOut serum replacer (KSR; Invitrogen/Life Technologies), 0.1 mM nonessential amino acids (Invitrogen/Life Technologies), 1 mM l-glutamine (Invitrogen/Life Technologies), 0.1 mM β-mercaptoethanol (Sigma-Aldrich), 10 ng/ml recombinant human basic fibroblast growth factor (Invitrogen/Life Technologies), and antibiotics (Gibco). The drugs SB431542 (2 μM; Stemgent, Cambridge, MA, USA), PD0325901 (0.5 μM; Stemgent), and thiazovivin (0.5 μM; Santa Cruz Biotechnology, Santa Cruz, CA, USA) were added to the culture medium to aid colony formation (26). The drug-containing medium was replaced daily until iPSC colonies were detected. Undifferentiated iPSCs were maintained on mitotically inactivated MEFs (50,000 cells/cm2) in the human ESC medium. To prevent MEF contamination, human iPSCs or human ESCs (TW1 line; Biomedical Technology and Device Research Laboratories, ITRI, Hsinchu, Taiwan) were transferred to a feeder-free/serum-free culture system in CSTI-8 medium (Cell Science and Technology Institute Inc., Tokyo, Japan) without KSR supplementation. Briefly, the 100-mm Petri dish was coated with fibronectin (Sigma-Aldrich). Human iPSCs were dissociated with 2 U/ml dispase (Invitrogen/Life Technologies) and were added to a precoated dish with CSTI-8 medium (Cell Science & Technology Institute Inc.) and without KSR supplementation. In some experiments, the iPSCs were transfected with pCX-eGFP to constitutively express green fluorescence. The morphology of human iPSCs cultured with the use of the serum-free/feeder-free system was comparable to iPSCs cultured using the conventional MEF feeder system.
The hepatic differentiation protocol of human iPSCs was conducted as described previously with minor modifications (43). This protocol was composed of four stages: endoderm induction, hepatic specification, hepatoblast expansion, and hepatic maturation. For endoderm induction, human iPSCs were incubated with RPMI-1640 media (Invitrogen/Life Technologies), supplemented with 0.5 mg/ml albumin fraction V (Sigma-Aldrich) and 100 ng/ml activin A (PeproTech, Rocky Hill, NJ, USA). In the following 2 days, the culture was further treated with 0.1% and 1% insulin–transferrin–selenium (Invitrogen/Life Technologies), respectively. Subsequently, the differentiated human iPSCs were shifted to hepatocyte culture medium (HCM) (Cambrex, Baltimore, MD, USA) containing 30 ng/ml fibroblast growth factor 4 and 20 ng/ml bone morphogenetic protein 2 (PeproTech) for 4 days. At the next step, the differentiated cells were first incubated in HCM containing 20 ng/ml HGF and 20 ng/ml keratinocyte growth factor, followed by incubation in HCM containing 10 ng/ml oncostatin-M (R&D, Minneapolis, MN, USA) plus 0.1 μM dexamethasone (Sigma-Aldrich) for 5 days, and finally, in DMEM containing N2, B27, 1 mM/l glutamax, 1% nonessential amino acids, and 0.1 mM β-mercaptoethanol (Invitrogen/Life Technologies) for another 3 days.
Preparation of CHC Thermogelling Solutions
CHC was purchased from Advance Delivery Technology Inc. (Hsinchu, Taiwan). Glycerol and human HGF were purchased from Sigma-Aldrich. β-Glycerol phosphate disodium salt hydrate (β-GP) was obtained from Merck. Amphiphatic CHC hydrogels were synthesized and characterized as described previously (27). Briefly, CHC polymer powder was dissolved in distilled water and freeze dried to form sponge-like powder for a subsequent dissolution in culture medium. Then, 0.6 g of CHC sponge-like powder was added in 20 ml medium under stirring in an ice bath, until the powder was totally dissolved in the medium to form a viscous solution. Subsequently, 50 μg of β-GP with or without HGF was dissolved in 1 ml of the CHC/medium solution under stirring on ice to prepare CHC hydrogels and HGF–CHC hydrogels (final HGF concentration: 50 ng/ml), respectively. The final solution with thermally induced sol–gel transition was observed when the temperature increased from 4°C and formed a solid gel at 37°C.
The incorporation of HGF into the CHC hydrogels generated the HGF–CHC complex, which can sustain the release of HGF into the culture medium. The in vitro HGF release test was carried out via a modified dissolution test. Briefly, a known amount of HGF–CHC hydrogel was placed in a dialysis membrane, followed by adding 2 ml of the culture medium. The membrane was then placed in a water bath of the same culture medium and maintained at 37 ± 0.5°C. The samples were magnetically stirred at 100 rpm. At predetermined time intervals, an aliquot of sample was taken from the water bath, and the amount of HGF inside was measured by a Human HGF Quantikine ELISA Kit (Life Technologies, Bethesda, MD, USA). The same amount of the fresh culture medium was added back into the water bath after sample collection at each time point to maintain the release condition of the whole system.
Microarray Analysis and Bioinformatics
Total RNA was extracted from cells using Trizol reagent (Life Technologies) and the Qiagen RNAeasy (Qiagen, Valencia, CA, USA) column for purification. Total RNA was reverse-transcribed with Superscript II RNase H-reverse transcriptase (Gibco BRL) to generate Cy3- and Cy5-labeled (Amersham Biosciences Co., Piscataway, NJ, USA) cDNA probes for the control and treated samples, respectively. The labeled probes were hybridized to a cDNA microarray containing 10,000 gene clone-immobilized cDNA fragments. Fluorescence intensities of Cy3 and Cy5 targets were measured and scanned separately using a GenePix 4000B Array Scanner (Axon Instruments, Burlingame, CA, USA). Data analysis was performed using GenePix Pro 3.0.5.56 (Axon Instruments) and GeneSpring GX 7.3.1 software (Agilent, Palo Alto, CA, USA). The average linkage distance was used to assess the similarity between two groups of gene expression profiles as described below. The difference in distance between two groups of sample expression profiles to a third was assessed by comparing the corresponding average linkage distances [the mean of all pairwise distances (linkages) between members of the two groups concerned]. The error of such a comparison was estimated by combining the standard errors (the standard deviation of pairwise linkages divided by the square root of the number of linkages) of the average linkage distances involved. Classical multidimensional scaling was performed using the standard function of the R program to provide a visual impression of how the various sample groups are related.
Real-Time Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
Real-time RT-PCR was performed as previously described (4). For real-time RT-PCR analysis, the total RNA of cells was extracted by using the RNAeasy kit (Qiagen). Briefly, the total RNA (1 mg) of each sample was reversely transcribed using 0.5 μg of oligo dT and 200 U Superscript II RT (Invitrogen). The amplification was carried out in a total volume of 20 μl of reaction volume containing 0.5 mM of each primer, 4 mM MgCl2, 2 ml LightCycler FastStart DNA Master SYBR green I (Roche Diagnostics, Pleasanton, CA, USA), and 2 ml of 1:10 diluted cDNA. The quantification of the unknown samples was performed by LightCycler Relative Quantification Software, version 3.3 (Roche Diagnostics). In each experiment, the GAPDH housekeeping gene was amplified as a reference standard. PCR reactions were prepared in duplicate and heated to 95°C for 10 min followed by 40 cycles of denaturation at 95°C for 10 s, annealing at 55°C for 5 s, and extension at 72°C for 20 s. All PCR reactions were performed in duplicate. Standard curves (cycle threshold values vs. template concentration) were prepared for each target gene and for the endogenous reference (GAPDH) in each sample. The primers and cycling conditions for RT-PCR are shown in Table 1.
The Sequences for the Primers of RT-PCR

Detection of CYP450 Activity
For analysis of CYP450 activity, CYP1A2 and CYP3A4 enzyme activities were assayed directly in iPSC-Heps, ESC-Heps, and iPSCs as previously described (42). Briefly, those cells assigned for CYP450 activity measurement were separated into three groups: control untreated cells (Group I), omeprazole-treated cells for CYP1A2 measurement (Group II), and rifampicin-treated cells for CYP3A4 measurement (Group III). Group I: Untreated cells were cultured in IMDM (Invitrogen/Life Technologies, Carlsbad, CA, USA) alone. Group II: Cells were cultured in IMDM supplemented with 100 μM omeprazole for inducing CYP450 isotypes 1A2. Group III: Cells were cultured in IMDM supplemented with 25 μM rifampicin for inducing CYP450 isotypes 3A4. All conditions were incubated for 3 days, and the culture media were changed daily. The metabolism of these cells were measured using the P450-Glo™ Assays for 1A2 and 3A4 following the instruction of the manufacturer for the CYP 450 induction test (V8421 and V9001; Promega, Madison, WI, USA). After 72 h of each treatment, cells were washed twice and then switched to IMDM containing a luminogenic CYP substrate 3 μM luciferin-IPA (Group III) or to Krebs–Henseleit Buffer (Sigma-Aldrich) containing 6 μM luciferin-1A2 (Group II), for 30–60 min. Subsequently, 25 μl of the incubation medium was shifted to a 96-well white luminometer plate (Nunc, Roskilde, Denmark), and 25 μl of luciferin detection reagent was added to each well. After a further incubation at room temperature for 20 min to stabilize the luminescent signal, the luminescence was recorded using a luminometer. The net signals were calculated by subtracting background luminescence values (no-cell control) from test compound-treated and untreated (vehicle control) values. The percent changes were calculated by dividing net treated values by the net untreated value and multiplying by 100.
Determination of Intracellular Production of Reactive Oxygen Species (ROS), Malondialdehyde (MDA), and Nitrate/Nitrite
The detection for intracellular ROS production was conducted using the probe 2′,7′-dichlorofluorescin diacetate (DCFH-DA; Molecular Probes, Eugene, OR, USA) as previously described by Lee et al. (20). After accumulation within the cells, DCFH-DA will be hydrolyzed by cytoplasmic esterases and form 2′,7′-dichlorofluorescin, which subsequently will react with hydrogen peroxide to produce 2′,7′-dichlorofluorescein. After incubating with 5 μM DCFH-DA in culture medium for 30 min at 37°C, the cells were washed and resuspended in 0.5 ml PBS and assigned for flow cytometric analysis.
MDA, a biomarker for assessing lipid peroxidation in the kidney tissue, was assayed using a commercial kit for thiobarbituric acid reactive substances (Cayman Chemical, Ann Arbor, MI, USA). Optical density at 530 nm (OD530) was determined using an ELISA reader (Bio-Rad Laboratories, Hercules, CA, USA). Nitrite and nitrate are the primary oxidation products of NO subsequent to reaction with oxygen, and therefore the nitrite/nitrate concentration was used as an indicator of NO synthesis. Nitrite/nitrate levels in kidney tissues were measured after enzymatic conversion of nitrate to nitrite using nitrate reductase (Cayman Chemical). Subsequently, the total nitrite was assayed by adding 100 μl Griess reagent (0.05% naphthalethylenediamine dihydrochloride and 0.5% sulfanilamide in 2.5% phosphoric acid; Sigma-Aldrich) to each sample. OD550 was measured, and the total nitrite/nitrate concentration of each sample was calculated by comparison against the OD550 of a standard solution of sodium nitrate prepared in saline.
Determination of LDL Uptake and Glycogen Synthesis
The ability of cells for LDL uptake was evaluated by the uptake capability of 1,10-dioctadecyl-1-,3,3,30,30-tetramethyl-indo-carbocyanine perchlorate conjugated to acetylated-LDL (DiI-Ac-LDL; AbD Serotec, Oxford, UK). Cells were incubated with 20 μg/ml DiI-Ac-LDL at 37°C for 24 h, and the incorporation of DiI-Ac-LDL into cells was visualized by fluorescence microscopy.
As for the determination of glycogen synthesis, cells were initially fixed in 4% paraformaldehyde (Sigma-Aldrich) and then permeabilized by 0.1% Triton X-100 (Sigma-Aldrich) for 10 min. Subsequently, the samples were oxidized with 1% periodic acid (Sigma-Aldrich) for 5 min, rinsed three times in deionized (d)H2O, followed by the treatment of Schiff's reagent (Sigma-Aldrich) for 15 min and further washed using (d)H2O for 5–10 min. Samples were then counterstained with Mayer's hematoxylin (Sigma-Aldrich) for 1 min, rinsed in (d)H2O, and then visualized by a light microscope.
Animal Model of Liver Injury, Liver Functional Test, and Histological Quantification of Liver Injury
The induction of liver injury in the mouse model was conducted as previously described (24). All mice were caged at 24°C with a 12-h light–dark cycle and were allowed free access to water and food. This study was approved by Taipei Veterans General Hospital Animal Committee and followed the principles of Laboratory Animal Care. Male BALB/c nude mice (National Laboratory Animal Center, Taipei, Taiwan), 8 weeks old and weighing 25–30 g, were used for the induction of fulminant hepatic failure, which was achieved by intraperitoneal injection of TAA (200 mg/kg body weight, dissolved in 0.9% saline; Sigma-Aldrich). Various cell doses of iPSC-Heps using either PBS or HGF–CHC as delivery vehicle were intrahepatically transplanted into the mice with liver injury 4 h after the TAA administration, and 24, 48, or 72 h after TAA administration, hepatic damage was validated by histological examination. Meanwhile, blood samples were collected to determine hepatic biochemical parameters. Recipient survival was continuously monitored for 2 weeks after TAA administration.
Biochemical parameters were measured using standard clinical methods. After anesthesia by ketamine (10 mg/100 g body weight; Sigma-Aldrich), intracardiac aspiration of blood was performed. A 0.8- to 0.9-ml blood sample from the heart was collected into a pyrogen-free syringe (Terumo, Tokyo, Japan) containing ~75 units of heparin sodium (Sigma-Aldrich), then placed in an ice bath and transported immediately to the laboratory. Serum ammonia and biochemistry tests, including alanine aminotransferase (ALT), aspartate aminotransferase (AST), and total bilirubin were measured by Vitro DT chemistry system (Johnson & Johnson, New Brunswick, NJ, USA).
To examine the histological quantification of liver injury, the harvested livers were fixed in 4% paraformaldehyde (Sigma-Aldrich), dehydrated using graded ethanol, and then embedded in paraffin. The paraffin blocks were section and stained with hematoxylin and eosin (H&E; Sigma-Aldrich) using standard histological techniques. TAA injection resulted in liver parenchyma damage mainly around the central veins. The necrotic area in injured liver was distinguishable from normal liver tissue in terms of the brighter color and the presence of inflammatory cell infiltrates. The liver sections were examined, and photos were taken under a microscope at 10× magnification. The necrotic areas of the injured liver were determined by measuring five independent fields per liver using a computerized morphometry system. The percentage of relative necrotic area (%) was calculated by dividing the necrotic area by total observed area.
Ex Vivo Green Fluorescent Protein (GFP) Imaging
Ex vivo GFP imaging was performed as described previously (7). The animal experiment was performed following the “Principles of laboratory animal care” of Taipei Veterans General Hospital and National Yang-Ming University. A 470-nm excitation filter was used with a lamp supply of optical lighting of 150 W (Southern California Services, Los Alamitos, CA, USA) as an excited light source (470 nm) to project on the foci of GFP-positive cells within ex vivo transplanted tissues or living mice. The GFP images and photography were obtained from the recordings of a digital camera (Olympus, Tokyo, Japan) through the optical configuration of a dissected microscope (SZ60; Olympus) with a 515-nm viewing (emission) filter.
Immunofluorescence Staining
An avidin–biotin complex-based method was used for immunohistochemical staining of differentiated iPSCs. Following washes with 3% hydrogen peroxide, sodium azide and antigenicities were retrieved using a microwave. Each slide was then treated with antibodies for human Oct4 (0.4 μg/ml) (Santa Cruz Biotechnology), human Nanog (0.4 μg/ml) (Santa Cruz Biotechnology), human SSEA4 (0.4 mg/ml) (Santa Cruz Biotechnology), DAPI (0.2 μg/ml) (Santa Cruz Biotechnology), human HNF-3β (0.4 μg/ml) (Chemicon International, Temecula, CA, USA), and human AFP (0.4 μg/ml) (Santa Cruz Biotechnology), and human albumin (Santa Cruz Biotechnology). Immunoreactive signals were detected with a mixture of biotinylated rabbit anti-mouse IgG and Fluoresave (0.4 μg/ml) (Calbiochem, La Jolla, CA, USA) and a confocal microscope (FV300; Olympus).
Western Blot Assay
The extraction of proteins from cells and Western blot analysis were performed as described. Samples (15 μl) were boiled at 95°C for 5 min and separated by 10% SDS-PAGE. The proteins were wet transferred to Hybond-ECL nitrocellulose paper (Amersham, Arlington Heights, IL, USA). The following primary antibodies were used: mouse anti-Bax, mouse anti-Bcl-2, mouse antibody against active caspase 3 (anti-CPP32 antibody), human Bcl-2, Bax, and caspase 3, and human anti-α-tubulin (all antibodies were at 0.2 μg/ml and from Santa Cruz Biotechnology). Immunoreactive protein bands were detected by the ECL detection system (Amersham Biosciences Co., Piscataway, NJ, USA).
Statistical Analysis
The results are expressed as mean± SD. Statistical analyses were performed using the t-test for comparing two groups, and one-way or two-way ANOVA, followed by Bonferroni's test, was used to detect differences among three or more groups. Results were considered statistically significant at p < 0.05.
Results
Preparation of HGF-Releasing CHC Hydrogel
Thermosensitive injectable hydrogels have drawn much attention in drug release, cell encapsulation, and tissue engineering (3,10,14,21). These hydrogels, when combined with drugs or proteins, provide sustained systemic or local therapeutic effects and act as in vivo scaffolds for cell delivery (10,15,25,44). The implant procedure for injectable hydrogels requires minimal invasiveness, which has a great advantage over conventional implantation surgery (9,10,50). Other advantages include easy handling, reduced pain, less complications, reduced surgery time and healing period, minimal scarring, and the capacity to conform to irregular defects (50). CHC hydrogels are a promising system for sustained drug delivery (27), and recently we developed a novel injectable CHC hydrogel that enhances stem cell delivery and engraftment into an injured cornea (6). Here we investigated the application of CHC hydrogel containing exogenous HGF to the treatment of fulminant AHF.
We first prepared a CHC nanoscale hydrogel containing HGF by incorporating HGF peptides into CHC hydrogels (Fig. 1A). The kinetics of HGF release from HGF–CHCs with different HGF concentrations were calculated from the cumulative percentages of released HGF at different time points over 2 days. The HGF–CHC complex steadily secreted HGF into the culture media in a dose-dependent manner and sustained for at least 10 days (Fig. 1B). The HGF concentration of 50 ng/ml was selected as the optimal concentration.

Characterization of HGF-CHC. (A) Design of the nanocapsule structures formed by the amphiphatic CHC hydrogel. (B) Left: dose-dependent HGF release. Effect of HGF-CHC delivery on hepatic necropsy and necrotic area in a TAA (200 mg/kg)-injured liver. Right: representative H&E images comparing the effect of CHC alone and CHC containing 50 ng/ml HGF. (C) Comparison of the efficacy of HGF using either CHC or PBS as delivery vehicles on the hepatic necrotic area in a TAA-injured liver. (D) Dose-dependent survival of recipients of HGF delivered by CHC. (B) *p<0.05 versus 12.5 ng/ml HGF. (C) *p<0.05 versus 0 ng/ml HGF with corresponding treatment (PBS or CHC). (D) *p < 0.05 versus 0 ng/ml HGF.
We then tested the delivery efficiency of HGF–CHC in TAA-induced AHF in BALB/c nude mice. The TAA-injured liver was discolored to dark red with obvious enlargement and swelling. Intrahepatic injection of HGF–CHC led to a moderate dose-dependent reduction of the hepatic necrotic area (Fig. 1C) and decreased the hepatic biochemical parameters AST and ALT (Table 2). A significant improvement in survival was observed among recipients receiving 50 ng/ml HGF (Fig. 1D). Collectively, these data demonstrated the mild hepatoprotective capacity of sustained HGF release from the HGF–CHC complex. Considering the utility of CHC hydrogel as delivery vehicle for stem cells (6), the HGF–CHC with the highest levels of HGF release was selected for stem cell delivery in subsequent experiments.
Dose Effect of CHC-Mediated HGF Administration on Hepatic Biochemical Parameters in TAA-Treated Recipients

Results were expressed as mean ± SD from 10 recipients.
p < 0.05 versus HGF 0 ng/ml.
iPSC Generation From Human Dental Pulp
Dental pulp is a source of multipotent progenitors and pluripotent stem cells (1,30,36). Owing to its accessibility and differentiation potential, dental pulp has gradually drawn attention in regenerative medicine. We isolated dental pulpderived stromal cells (DP-SCs) from dental pulp tissue of donors. We reprogrammed these DP-SCs into iPSCs via transfection with retroviral vectors encoding Oct4/Sox2/ Klf4/c-Myc and investigated the pluripotency and stemness properties of these reprogrammed cells. Seven days after transfection, the cells were passaged onto mitotically inactivated MEFs and cultured with ESC media containing SB431542, PD0325901, and thiazovivin. During the reprogramming process, these cells progressively formed colonies with increasing size. After these colonies were cultivated on the MEF feeder cells in KSR-based media, they stained positive for alkaline phosphate (ALP), and they exhibited a morphology that was indistinguishable from human ESCs (Fig. 2A). These DP-iPSCs were subsequently transferred to feeder-free culture in HESF V2 media without KSR supplementation, as described previously (6). In the serum-free and feeder-free system, the DP-iPSCs remained stable for at least 30 passages. RT-PCR showed that the passage 30 DP-iPSCs expressed various stemness genes, including Oct4, Sox2, Nanog, REX, and DPPA2, identical to the genes detected in human ESCs (Fig. 2B). Immunofluorescence also confirmed the stemness signature and revealed the strong expression of Oct4, Nanog, SSEA-3, SSEA-4, Tra1–60, and Tra1–81 in the passage 30 DP-iPSCs (Fig. 2C). Based on their functions in the Gene Ontology database, the differentially expressed genes in human ESCs and a subset of the 10th, 20th, and 30th passage DP-iPSCs had similar expression patterns in these cells (data not shown). To test pluripotency in vivo, DP-iPSCs were implanted into the subcutaneous space of immunocompromised mice. After 6 weeks, ex vivo biopsies and histological analysis indicated the formation of structures from all three dermal lineages, including ectodermal (nestin: marker for neural progenitor), mesodermal (SMA: marker for smooth muscle), and endodermal (AFP: marker for hepatocyte) differentiated cells, respectively (Fig. 2D). These data demonstrated that human DP-SCs were reprogrammed into iPSCs, which possessed ESC-like characteristics and were greatly expanded in vitro and could be capable of multilineage differentiation.
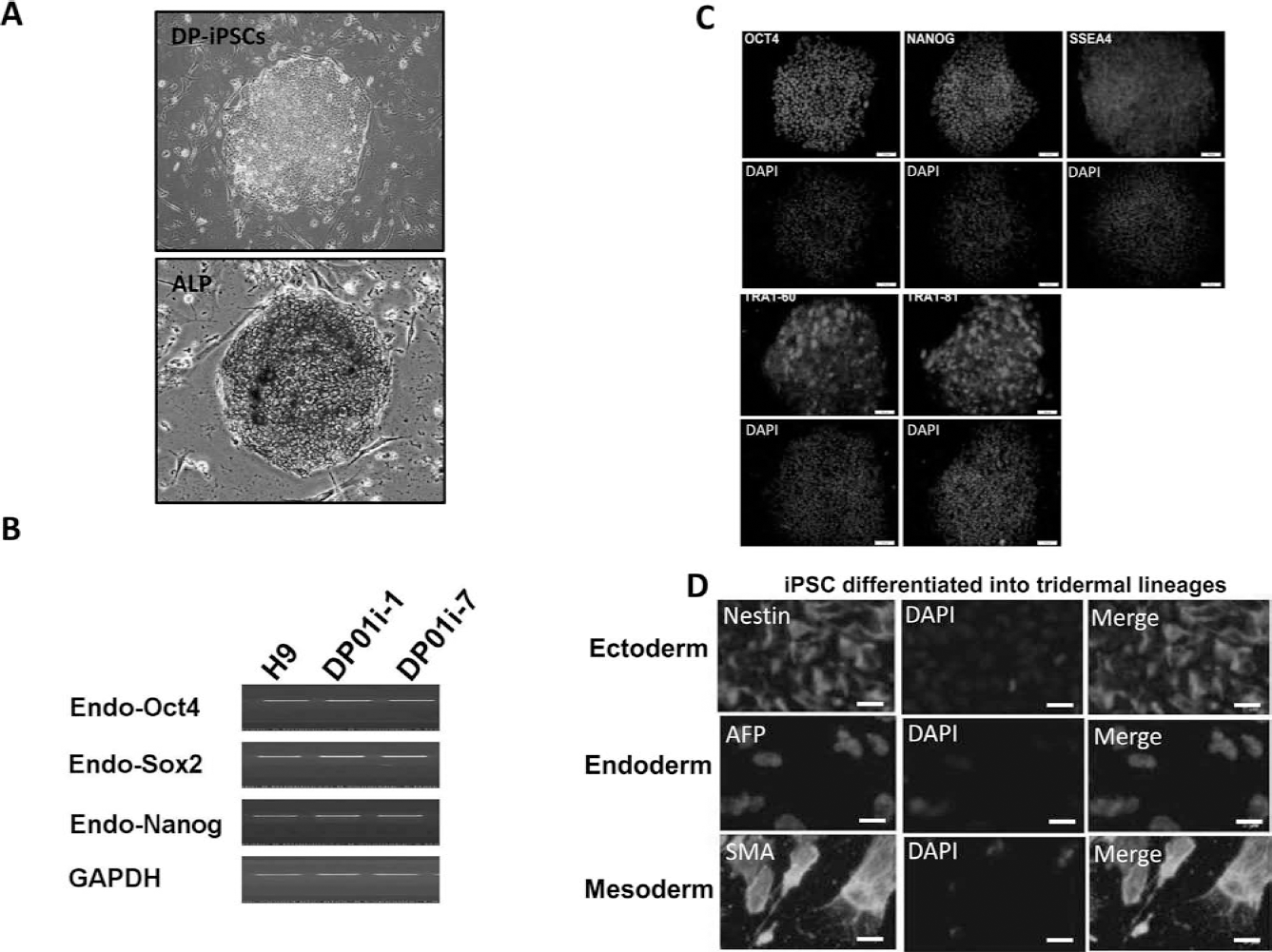
The reprogramming of DP-SCs into iPSCs. Under a serum-free and MEF feeder-free culture system, these MP-iPSCs were stably cultured through at least 30 passages (upper) and retained high ALP activity (lower) (A). Passage number does not affect the capacity of iPSCs to form colonies. (B) RT-PCR results indicating an ESC-like gene expression pattern in DP-iPSC clones (i.e., DP01i-1, DP01i-4, DP01i-7, DP01i-10) and the human ESC line H9. The expression of ESC-like genes, including endogenous stemness genes Oct4, Sox2, Klf4, and c-Myc, and Nanog, REX, DPPA2, DPPA4, ESG1, GDF3 were measured. (C) Immunofluorescence assays showed that iPSC colonies expressed Oct4, Nanog, SSEA3, SSEA4, Tra-1–60, and Tra-1–81. (D) DP-iPSCs, 2 × 105, were implanted into the subcutaneous space of immunodeficient mice (n = 6). After 6 weeks, ex vivo biopsies and histological analysis revealed three dermal lineage formations (striated muscle-like tissue, columnar epithelium-like tissues, keratinocyte-like formation, and neuroepithelium-like tissue) in the subcutaneous grafts. DAPI: nuclear staining. (A) Scale bar: 200 μm; (C and D) scale bar: 100 μm.
Differentiation of DP-iPSCs Into Functional iPSC-Heps
To further evaluate the potential of hepatic-specific differentiation in DP-iPSCs, we employed the hepatic differentiation protocol (refer to the Hepatic Differentiation of Human iPSCs section in Materials and Methods) and shifted DP-iPSC-derived embryoid bodies into differentiation media. These cells underwent morphological changes and gradually exhibited more elongated and cuboidal shapes and eventually differentiated into hepatocyte-like cells (DP-iPSC-Heps) 28 days postinduction. Using microarray analysis, we compared the gene expression profiles in normal liver, ESC-Heps, DP-iPSC-Heps, DP-iPSCs, H9 ESC lines, and fetal liver (Fig. 3A). The gene expression profile of DP-iPSC-Heps was similar to the profiles of normal liver and ESC-Heps (Fig. 3A). Multidimensional scaling analysis also indicated that the gene expression pattern of DP-iPSC-Heps was closer to the patterns of normal liver, fetal liver, and ESC-Heps than to those of DP-iPSCs and ESCs (Fig. 3B). Similar to mature ESC-Heps, iPSC-Heps acquired regular functions for glycogen storage (Fig. 3C, top) and LDL uptake (Fig. 3C, bottom) during differentiation. Immunofluorescence staining indicated that several hepatic-specific markers, including α-fetoprotein (AFP), cytochrome c p450 enzyme CYP2E1, and nuclear protein hepatocyte nuclear factor 3β (HNF-3β), and human bile canaliculi marker 9B2 (Fig. 3D) were recruited to DP-iPSC-Heps after hepatic differentiation; the magnitude of expression was similar to the expression in ESC-Heps (Fig. 3D). To evaluate whether the cytochrome c p450 enzyme was active, we measured the enzyme activity of CYP1A2 and CYP3A4 in DP-iPSC-Heps receiving the administration of omeprazole 100 μM or rifampicin 25 μM, respectively. The endpoint catalytic activity after incubating these cells with substrates was assessed using a luminometer. Similar to the elevation of enzyme activity in ESC-Heps stimulated by omeprazole or rifampicin, the enzyme activities of CYP1A2 and CYP3A4 were elevated by seven- to 10-fold in treated DP-iPSC-Heps, comparing with those untreated DP-iPSC-Heps (Fig. 3E). Taken together, we demonstrated that these DP-iPSC-Heps, derived from human dental pulp, exhibited mature hepatocyte functions, such as glycogen synthesis, and expressed cytochrome P450 enzyme, similar to human ESC-Heps.

In vitro differentiation of MP-iPSCs into iPSC-Heps. (A) Gene expression microarray analysis in normal liver, ESC-Heps, DP-iPSC-Heps, iPSC, H9 ESC lines, and fetal liver. (B) Multidimensional scaling analysis further showed that the expression pattern of DP-iPSC-Heps was closer to the patterns of normal liver, fetal liver, and ESC-Heps than to DP-iPSCs and ESCs. (C) Comparison of mature hepatocyte functions, including glycogen synthesis and LDL uptake, between DP-iPSC-Heps and ESC-Heps at day 0 or day 28 postdifferentiation. (D) Quantification of the immunofluorescence results of hepatocyte-specific proteins in DP-iPSC-Heps and ESC-Heps at day 0 or day 28 postdifferentiation. (E) CYP1A2 and CYP3A4 activities were induced after the addition of corresponding enzyme inducers for 72 h. These enzyme activities were then analyzed using P450Glo™ assay kit with different luciferin substrates. After incubation with luciferin substrates, luciferase activities were measured. (C and D) *p < 0.05 versus D0 in corresponding cells. (E) *p < 0.05 versus corresponding untreated cells.
Biology of DP-iPSC-Heps Cultivated in CHC–HGF
To evaluate the utility of HGF–CHC as a vehicle for iPSC-Hep delivery, we assessed the fate of iPSC-Heps cultivated in HGF–CHC (Fig. 4A). Cultivation of iPSC-Heps within HGF–CHC did not affect the kinetics of HGF release (Fig. 4B). Therefore, we selected HGF–CHC with an HGF concentration of 50 ng/ml as the optimal condition for DP-iPSC-Heps cultivation. To trace the growth of the DP-iPSC-Heps, some iPSCs were initially transfected with pCX-eGFP to constitutively express green fluorescence. During cultivation within the HGF–CHC, the GFP signal from GFP-labeled iPSC-Heps gradually increased, indicating DP-iPSC-Hep growth (Fig. 4C, left). An MTT assay also confirmed the proliferation of DP-iPSC-Heps (Fig. 4C, right). Quantitative RT-PCR revealed that several hepatic-specific genes, including HNF-3β, AFP, ALB, TAT, TTR, AAT, and HNF-4α, were stably expressed and retained in the HGF–CHC for at least 7 days (Fig. 4D). Collectively, HGF–CHC allowed a sustained HGF release that maintained the biology and regular growth of DP-iPSC-Heps in the hydrogel.

Cultivation of DP-iPSC-Heps in HGF-CHC in vitro. (A) Scheme for DP-iPSC-Hep cell encapsulation in HGF-CHC and morphology of HGF–CHC-cultivated iPSC-Heps. (B) The kinetics of HGF release showed that embedded DP-iPSC-Heps did not affect the dose-dependent release of HGF. (C) Detection of increasing GFP signal revealed the regular growth of DP-iPSC-Heps at days 0, 3, 5, and 7. MTT assay showing the viability of DP-iPSC-Heps in the presence of HGF–CHC. (D) Quantitative RT-PCR results indicating various hepatic-specific marker genes expressed in the presence of HGF–CHC. (A) Scale bar: 100 μm. (B) *p < 0.05 versus 12.5 ng/ml HGF. (C) *p < 0.05 versus day 0.
Improved Hepatoprotection by HGF–CHC-Mediated Delivery
To examine the bioavailability of HGF–CHC, we compared the efficacy of DP-iPSC-Hep transplantation using either PBS or HGF–CHC as a delivery vehicle in immunocompromised mice with TAA (200 mg/kg)-induced AHF. Briefly, GFP-labeled DP-iPSC-Heps were trypsinized and then premixed with cold HGF–CHC hydrogel or PBS (Fig. 5A). Four hours after TAA administration, the HGF–CHC/DR-iPSC-Hep mixture was injected into the liver, and the warm ambient temperature in the injected tissue facilitated a sol-to-gel transition within approximately 10 min, as described previously (6). Twenty-four hours after TAA administration, the treatment efficacy of DP-iPSC-Heps using PBS or HGF–CHC on TAA-injured liver was determined. Gross necropsy findings and histological examination confirmed the induction of TAA-induced hyperacute severe liver injury (Fig. 5B, upper left). PBS-delivered DP-iPSC-Heps at 106 cells/kg body weight moderately attenuated TAA-induced liver injury and decreased the necrotic area (Fig. 5B, upper right). Delivery of HGF–CHC alone mildly alleviated liver injury (Fig. 5B, lower left). Of the four treatment groups, HGF–CHC-delivered DP-iPSC-Heps exhibited the most prominent efficacy, significantly ameliorating liver injury and reducing the necrotic area (Fig. 5B, lower right). Transplantation of DP-iPSC-Heps using HGF–CHC as a delivery vehicle produced a cell dose-dependent reduction of the TAA-injured hepatic necrotic area (Fig. 5C) and improved TAA-induced lethality (Fig. 5D). Remarkably, compared with DP-iPSC-Heps delivered by PBS only, HGF–CHC-delivered DP-iPSCs were more effective for reducing the necrotic area and promoting recipient survival at any given cell dose (Fig. 5C, D). Taken together, these findings suggested that HGF–CHC as a delivery vehicle enhanced the hepatoprotective activity of iPSC-Heps during TAA-induced hepatic failure.
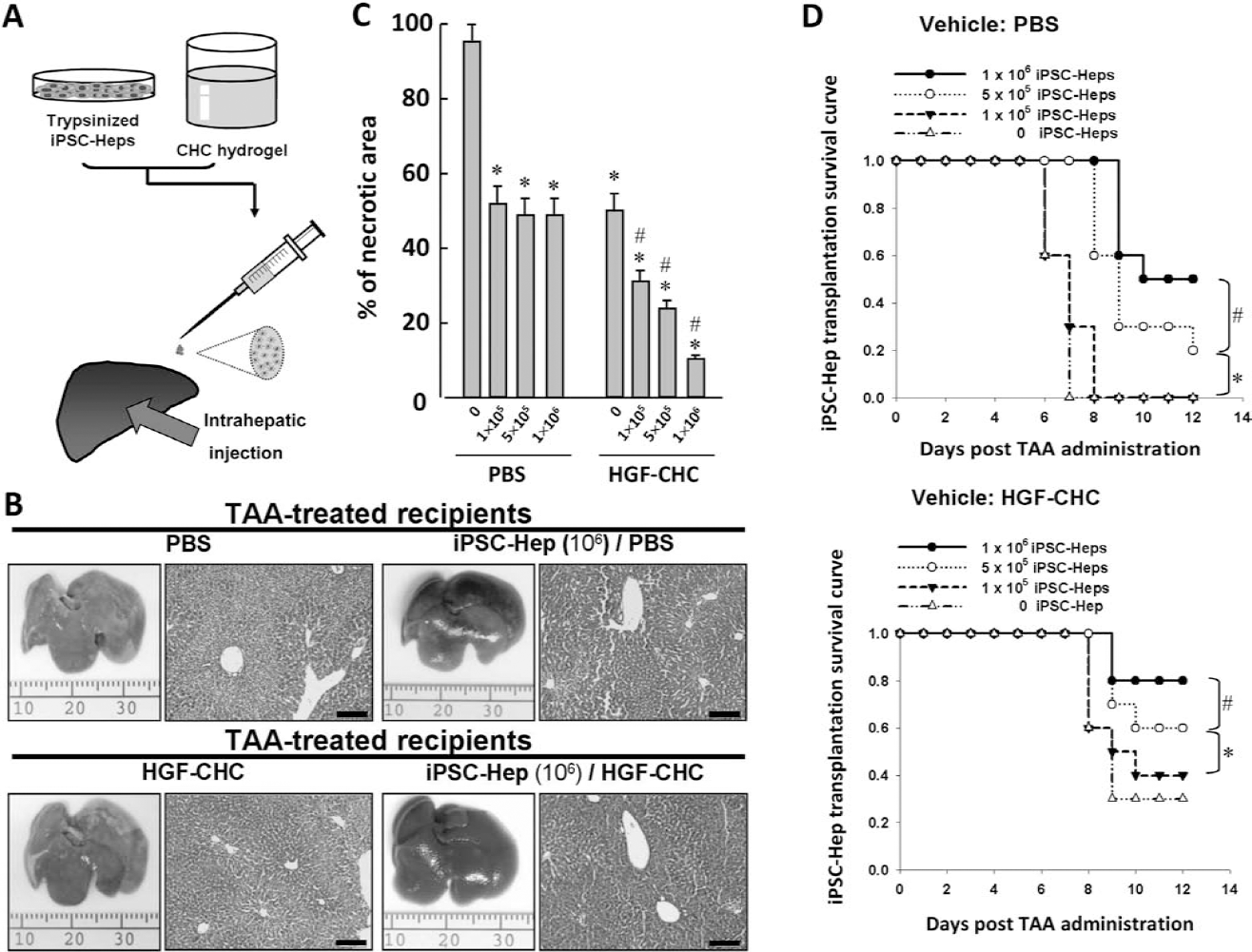
HGF–CHC-delivered DP-iPSC-Heps showed higher hepatoprotection than PBS-delivered cells. (A) Procedure for HGF–CHC-based intrahepatic delivery of DP-iPSC-Heps. (B) The reduction of hepatic necrotic area in recipients of DP-iPSC-Heps using PBS or HGF–CHC as delivery vehicles. (C) Dose-dependent reduction of the TAA-injured hepatic necrotic area in transplantation of DP-iPSC-Heps using HGF–CHC as a delivery vehicle. (D) The survival of recipients of DP-iPSC-Heps using PBS or HGF–CHC as delivery vehicles. (B) Scale bar: 200 mm. (C) *p < 0.05 versus 0 iPSC-Heps using PBS as delivery vehicles. #p < 0.05 versus iPSC-Heps at the same cell dose delivered with PBS. (D, upper and lower), *p < 0.05 versus 0 iPSC-Heps using PBS as delivery vehicles. #p < 0.05 versus 5 × 105 iPSC-Heps.
Involvement of Antioxidant/Antiapoptotic Effects in Hepatoprotection
We previously reported that MEF-reprogrammed iPSCs and iPSC-Heps possessed antioxidant enzymes that reduced oxidative stress in vivo (24). In addition, iPSCs had an antiapoptotic effect in an acute kidney injury rat model (22). Using quantitative RT-PCR, we detected the expression of various antioxidant enzymes, including MnSOD, Cu/ZnSOD, Catalase, GPX, and PRX in human DP-iPSC-Heps. The DP-iPSC-Heps expression of antioxidant genes was significantly higher than in human DP-SCs, but was moderately lower than in DP-iPSCs (Fig. 6A). Considering the multifaceted actions of HGF in hepatocytes and hepatic progenitors (2,13,18,28,29,34,35,40,49), we presumed that HGF is involved in one or more mechanisms that enhance the iPSC-Hep hepatoprotective effect. We investigated the involvement of HGF in the hepatoprotective effect of DP-iPSC-Heps. To elucidate the contribution of HGF, anti-HGF neutralizing antibody (nAb) was injected via the tail vein once per day 24 h before TAA administration and 24 and 48 h post-TAA administration (i.e., once per day for 3 days). Gross necropsy findings indicated that HGF–CHC alone moderately ameliorated TAA-induced liver injury (Fig. 6B, upper left), and the combination of HGF–CHC plus DP-iPSC-Heps further improved this injury (Fig. 6B, lower left). Although some areas with focal liver injury were observed in the liver from TAA-treated recipients of HGF–CHC-delivered iPSC-Heps, most liver tissue exhibited a regular color with a smooth surface (Fig. 6B, lower left). Remarkably, after exogenous administration of HGF nAb, the liver lost its smooth surface and exhibited a pale color, and several fibrosis-like changes were observed in the TAA-injured liver (Fig. 6B, upper right and lower right). As shown by H&E staining in TAA-treated recipients of HGF–CHC or HGF–CHC-delivered DP-iPSC-Heps, administration of anti-HGF nAbs confirmed the contribution of HGF, regardless of the presence of DP-iPSC-Heps (Fig. 6B). MDA is a product of lipid peroxidation. Both MDA and nitrate/nitrite indicate oxidative damage (8). Consistent with previous findings, these iPSC-Heps significantly suppressed the production of MDA, nitrate/nitrite, and ROS in the livers of TAA-treated recipients (Fig. 6C). Notably, HGF–CHC alone reduced these oxidative substances and exhibited a synergistic antioxidant effect in the presence of iPSC-Heps (Fig. 6C), whereas this synergism and the antioxidant effect of iPSC-Heps were completely blocked by HGF neutralization (Fig. 6C). In addition, marked increases in the Bax/Bcl-2 ratio and in caspase 3 (CPP32) expression were detected in the TAA-treated livers receiving PBS only (Fig. 6D). Transplantation of iPSC-Heps led to mild reductions in both the Bax/Bcl-2 ratio and caspase 3 expression in the TAA-treated liver. The addition of HGF–CHC further suppressed the expression of these apoptosis-related proteins, which was fully restored by the administration of anti-HGF nAbs (Fig. 6D). Taken together, our findings demonstrated that the hepatoprotective mechanism of iPSC-Heps delivered with HGF-enriched hydrogel includes the enhanced removal of oxidative substances and antiapoptotic activities.

HGF promoted antioxidant and antiapoptotic activities in iPSC-Heps. (A) The expression levels of antioxidant enzyme genes in DP-SCs, DP-iPSCs, and DP-iPSC-Heps, measured via quantitative RT-PCR. (B) H&E staining showing the necrotic areas of the liver sections from TAA-treated recipients of HGF–CHC or HGF–CHC-delivered DP-iPSC-Heps. Administration of anti-HGF nAbs indicated the contribution of HGF effect, regardless of the presence of DP-iPSC-Heps. (C) Changes in MDA content (left), nitrite/nitrate content (middle), and ROS levels (right) in TAA-treated recipients of DP-iPSC-Heps delivered by PBS alone or HGF–CHC. (D) Western blot indicated the protein expression levels of Bax, Bcl-2, and active caspase 3 (CPP32) in the livers from TAA-treated recipients of DP-iPSC-Heps delivered by PBS alone or HGF–CHC. (B) Scale bar: 200 mm. *p < 0.05 versus HGF–CHC without HGF nAb in corresponding groups. #p < 0.05 versus the same treatment without iPSC-Hep delivery. (C) *p < 0.05 versus untreated group without iPSC-Hep delivery. #p < 0.05 versus the same treatment without HGF nAb. (D) *p < 0.05 versus PBS, #p < 0.05 versus HGF–CHC/iPSC-Hep without HGF nAb.
Discussion
Somatic cells such as skin fibroblasts (45) or human keratocytes (6) have been shown to have the capacity to be reprogrammed into pluripotent stem cells. The wisdom teeth were usually removed from patients whose jaws are not large enough to accommodate wisdom teeth (16). Accumulated studies have demonstrated that the adult third molar (wisdom tooth) can be a rich source of dental pulp stem cells (38). Since dental pulp cells are easily obtained from extracted teeth and can be expanded in vitro under simple conditions, the potential of dental pulp cells has been widely demonstrated as a source of iPSCs for use in regenerative medicine (38,46). In this study, we reprogrammed DP-SCs into iPSCs, which differentiated into tridermal lineages, including the hepatocyte-specific lineage iPSC-Heps. These pluripotent properties and stemness signature of DP-iPSCs were consistent with other studies reported previously (38,46).
Liver failure can result from acute insults or chronic persistent damages. The ultimate therapy for liver failure is orthotopic liver transplantation (23). Owing to the shortage of donor livers, developing a cell therapy stratagem to reduce damage, prevent progression, and restore liver function is clinically relevant (37). Patient-derived iPSCs could be used to produce autologous hepatocytes from donors themselves. However, whether iPSCs reprogrammed from dental pulpderived somatic cells could serve as hepatic cell resources for liver repair remains uncertain. Our findings have demonstrated that these DP-iPSC-Heps exhibited mature hepatocyte functions, such as glycogen synthesis, and expressed cytochrome P450 enzyme, similar to human ESC-Heps. Although cell therapy with iPSC-Heps is one approach for AHF treatment, the extremely low retention and survival of implanted cells is a major hurdle. Therefore, the use of absorbable biomaterials coupled with certain hepatotropic and hepatoprotective factors, such as HGF, could promote the survival of cells implanted at the injured sites. We developed an injectable amphiphatic CHC hydrogel with sustained HGF release (HGF–CHC) and used it as the delivery vehicle for DP-iPSC-Heps. HGF–CHC delivered via the intrahepatic route potentially improved liver function and rescued transplant recipients from TAA-induced lethality. This mechanism increased iPSC-Hep viability, antioxidant activity, and antiapoptotic effects. Finally, anti-HGF nAbs blocked the protective effects of HGF–CHC alone or with iPSC-Heps and impaired the antioxidant and antiapoptotic properties of iPSC-Heps in TAA-treated recipients. These findings demonstrated that the functional iPSC-Heps delivered via HGF–CHC have potential benefits for the management of AHF.
HGF was initially identified as a potent hepatotropic factor and hepatocyte mitogen (35). After binding to its specific receptor c-Met, HGF evokes a variety of biological functions, including mitogenesis, morphogenesis, antiapoptosis, and angiogenesis (Fig. 7). In addition, endogenous HGF is required for the repair of injured livers, kidneys, lungs, hearts, etc. (34). HGF is critical for the regression of fibrosis in numerous organs (31,33,48). In the present study, the effects of HGF implanted via the CHC hydrogel in the TAA-injured liver were diverse. Previously, we showed that murine iPSC-Heps possess moderate anti-oxidant activity and suppress oxidative substances and ROS in vivo (24). Here we detected antioxidant enzymes expressed in human DP-iPSC-Heps. In the presence of HGF–CHC, iPSC-Heps reduced oxidative substances and the expression of apoptosis-related proteins and eventually facilitated liver recovery. HGF neutralization studies demonstrated the essential role of HGF in the hepatoprotective actions of iPSC-Heps. Although HGF is a potent mitogen involved in hepatocyte proliferation, tissue repair during hepatic failure (18), and CCl4-induced liver fibrosis (2), HGF neutralization also blocked its hepatotropic effect. Moreover, HGF directly promotes mesenchymal stem cells to differentiate into the hepatocyte lineage, and transplantation of mesenchymal stem cells cultured with HGF effectively treats liver injury in rats (40). In addition, HGF is used to supply iPSC-derived hepatic cell cultures to maintain cell viability and functions. We demonstrated that HGF in a hepatic differentiation cocktail could facilitate iPSC differentiation toward hepatocyte-like lineages with mature liver functions. In addition, HGF-releasing CHC hydrogel promoted iPSC-Hep growth and maintenance while in the gel, indicating the CHC hydrogel could be used as delivery vehicle in vivo to improve the antioxidant capacities of transplanted iPSC-Heps in damaged liver tissues. Taken together, our findings demonstrate and emphasize the crucial role of HGF in iPSC-Hep-based therapy for the treatment of AHF.

Scheme for the multifaceted functions of HGF that enhances the DP-iPSC-Hep hepatoprotective effect. HGF is able to evoke a variety of biological functions, including mitogenesis, morphogenesis, antiapoptosis, and angiogenesis. Using a vehicle that can steadily release HGF effectively enhances the DP-iPSC-Hep hepatoprotective effect in AHF. This HGF-mediated effect may involve enhancement of hepatocyte engraftment and liver regeneration, rescuing severe liver damage and recovery of liver function.
Conclusions
We demonstrated the generation of iPSCs from human dental pulp-derived fibroblasts. The reprogrammed iPSCs exhibited pluripotency and were differentiated into tridermal lineages and functional hepatocyte-like cells (iPSC-Heps) with mature hepatocyte functions. Our in vivo studies further revealed that the antioxidant capacities of DP-derived iPSC-Heps was improved by the presence of HGF–CHC, such that HGF retained its cytoprotective effect. Therefore, the HGF–CHC delivery system coupled with iPSC-Heps is a potential bioactive bio-material strategy for hepatic or other tissue repair and restoration.
Footnotes
Acknowledgments
This study was assisted in part by the Division of Experimental Surgery of the Department of Surgery Taipei Veterans General Hospital. We thank Dr. Cheng-Chung Liao (Proteomic Research Center, National Yang-Ming University) for his outstanding proteomic analysis, Dr. Pei-Chan Hsieh for her valuable assistance in experimental designs and technology, Dr. Hsin-Yang Li and Li-Ing Sung (Institute of Biotechnology, National Taiwan University) for the critical review of this manuscript. This study was funded by NSC-(100–2120-M-002–011, 100–2325-B-010 -010, 100–2321-B-010–020), the Joint Projects of VGHUST & UTVGH (VN100–03 & VN101–06), Taipei Veterans General Hospital (Stem Cell Project E99–101), Yen-Tjing-Ling Medical Foundation (CI-99/100), National Health Research Institutes (NHRI-EX102–10258SI), the Department of Health Cancer Center Research of Excellence (DOH101-TD-C-111–007), the Genomic Center Project of National Yang-Ming University (Ministry of Education, Aim for the Top University Plan), and the grant 103F003C08 and CY10410 from the Cheng Hsin General Hospital. The authors declare no conflicts of interest.