
Abstract
Platelet lysates have been reported as suitable cell culture supplement for cultures of mesenchymal stromal cells (MSCs). The demand for safe and animal-free cultures of MSCs is linked to the potential application of MSCs in clinics. While the use of platelet lysates offers an alternative to animal serum in MSC cultures, obtaining supplies of fresh platelet concentrates for lysate production is challenging and raises concerns due to the already existing shortage of platelet donors. We have previously demonstrated that expired platelet concentrates may represent a good source of platelets for lysate production without competing with blood banks for platelet donors. The INTERCEPT Blood System™ treatment of platelet concentrates allows for prolonged storage up to 7 days, using highly specific technology based on amotosalen and UV-A light. The INTERCEPT system has therefore been implemented in blood processing facilities worldwide. In this study, we evaluated the suitability of INTERCEPT-treated, expired platelet concentrates, processed into platelet lysates, for the culture of MSCs compared to nontreated expired platelets. Bone marrow-derived MSCs were cultured in media supplemented with either platelet lysates from traditionally prepared expired platelet concentrates or in platelet lysates from expired and pathogen-inactivated platelet concentrates. The effects of pathogen inactivation on the ability of the platelets to support MSCs in culture were determined by evaluating MSC immunomodulation, immunophenotype, proliferation, and trilineage differentiation. Platelet lysates prepared from expired and pathogen-inactivated platelet concentrates supported MSC differentiation and immunosuppression better compared to traditionally prepared platelet lysates from expired platelet units. Pathogen inactivation of platelets with the INTERCEPT system prior to use in MSC culture had no negative effects on MSC immunophenotype or proliferation. In conclusion, the use of expired pathogen-inactivated platelet units from blood banks to prepare platelet lysates for the culture of MSCs is desirable and attainable.
Keywords
Introduction
Platelet concentrates are key blood transfusion components and must be available for use in medical treatment at any given time. The management of blood component supplies is the responsibility of blood banks worldwide with the administration of platelet inventory being especially challenging. Owing to their relatively short shelf life of maximum 5 to 7 days, depending on country-specific regulations, platelet supplies have to be renewed more rapidly than supplies of other blood components, such as red blood cell concentrates and plasma (12,46). Furthermore, the available amount of platelet concentrates should meet the patient demand while accounting for the donor availability. Owing to a combination of short shelf life and the practice of slight overproduction to meet unexpected demand, a significant proportion (10-25%) of all platelet concentrates expires before use (12,37,46).
The reason for the short shelf life of platelet concentrates is the risk of bacterial contamination (32,45). Owing to the strict screening policies for potential blood donors, the risk of transfusion-related viral infections from currently known blood-transmitted viruses is low. However, the risk of transfusion-related sepsis stemming from bacterial contamination in platelet concentrates is an ongoing problem (27,45). Since the optimal storage temperature for platelet concentrates is 22°C prior to transfusion, pathogens that have similar optimal growth temperatures, especially bacteria derived from the skin during blood collection, can multiply rapidly (21,32). Different strategies, such as screening and strict disinfection protocols, have been introduced to limit the risk of bacterial transmission via a contaminated platelet unit as much as possible. These strategies also aim at prolonging the shelf life of platelet concentrates and reducing amount of platelet products that end up expiring (30,38,45). The INTERCEPT Blood System™ (Cerus, Amersfoort, Netherlands) is one such strategy and is based on using amotosalen in combination with ultraviolet-A (UV-A) light to inactivate pathogens in platelets.
Amotosalen is a highly water-soluble synthetic psoralen that easily passes through the exterior of pathogens and cells, allowing it to interact with the helical regions of DNA and RNA. During UV-A illumination, amotosalen is photoactivated and cross-links the strands of nucleic acids, inhibiting replication and transcription in cellular entities (19). Amotosalen is not only effective for inactivation of known pathogens and cells but also for potentially emerging or currently unknown pathogens, since it interacts with nucleic acids universally (14). Structures that lack nucleic acids, such as platelets, are unaffected by amotosalen, and thus their physiology is not affected in this process (21,28). Following pathogen inactivation, the storage time for platelet units can be prolonged from 5 to 7 days according to national guidelines (14,21,26,28). This 2-day prolongation in storage time facilitates platelet inventory management and reduces expiry. Therefore, a number of blood banks have been introducing pathogen-inactivation procedures into their routine blood-processing protocols (34,40). The INTERCEPT Blood System™ for pathogen inactivation of platelet and plasma is widely used in routine, and a range of clinical studies on hemo-vigilance data demonstrate safety and efficacy in patients (19,28).
In addition to their hemostatic applications in clinics, platelets are now increasingly applied in the development of biomaterials and tissue engineering strategies. Applications for platelets may include orthopedics in the form of platelet-rich plasma, wound healing strategies using platelet gels, and scaffold development to support cell growth in combination with other biomaterials (3,5,9,29). Platelet lysates have also been gaining popularity as an alternative to fetal bovine serum in the culture of mesenchymal stromal cells (MSCs) (15,22,35,39).
MSCs have interesting attributes that can be applied in regenerative medicine and tissue engineering. MSCs can be isolated from various tissues, including bone marrow or the vascular fraction of adipose tissue, and can be differentiated in vitro into osteoblasts, adipocytes, and chondrocytes (trilineage differentiation potential) (23,24,41). It has also been shown that they can act during inflammation and reduce immune responses (13,25). Based on this wide range of potential applications, MSCs are not only studied in the field of musculoskeletal regeneration but also in immunology as a potential part of certain immu-notherapeutic approaches (13,17,43). Considering the potential of these cells and the desire for clinical application, current culture methods are not suitable for cells destined for human patients (6,42). Platelet lysates have been demonstrated to support MSCs and therefore suggested to be used for MSC cultures as supplement in order to avoid the risk of immunological reactions against xenogenic factors and the transmission of animal-borne pathogens linked to current culture methods (1,15,22,39).
The supply of fresh platelet concentrates produced under the strict regulations of a blood bank environment is relatively low due to an existing shortage of platelet donors (46). Nonetheless, numerous platelet concentrate units are discarded periodically due to expiry (12,46). Patient transfusion with these expired concentrates is no longer considered safe, yet growth factors and other components desirable for biomaterial development and cell growth are still amply present (2,16). The use of these expired platelet units for the manufacture of platelet lysates offers a cost-effective alternative application for a formerly discarded clinical product. Our group has previously demonstrated the effective use of platelet lysates from traditionally prepared expired platelet units for MSC culture (22).
Recently, the Blood Bank of Iceland implemented and validated the INTERCEPT Blood System™ for pathogen inactivation of platelets. In this study, we evaluated the suitability of INTERCEPT pathogen-inactivated and expired platelet concentrates, processed into platelet lysates, for the culture of MSCs and compared their bioactivity with that of nontreated expired platelet concentrates processed into platelet lysates.
Materials and Methods
Ethical Statement
The study was approved by the Landspitali University Hospital ethics committee.
Platelet Lysate Production
Four outdated units of platelet concentrates (PCs) were acquired from the blood processing department of the Blood Bank (Reykjavik, Iceland). Each platelet unit was prepared using the buffy coat method by pooling together buffy coats from eight different donors, making the total number of donors behind the four PC 32 blood donors (18 males, 14 females, age span 19-46). The buffy coats were made by separating a whole blood unit by centrifugation into red blood cell concentrate, a plasma unit, and a buffy coat unit. Two units were not pathogen inactivated, whereas the other two were pathogen inactivated using the INTERCEPT Blood System™. The expired PCs were subjected to three freeze-thaw cycles (at −80°C and 37°C, respectively) in order to sufficiently lyse the platelets. The resulting lysate was platelet depleted by centrifugation at 4,975 χ g for 20 min. The supernatant was removed and centrifuged a second time at 4,975 χ g for 20 min, while the pellet, consisting of platelet fragments, was discarded. After the second centrifugation, the supernatant was filtered through a 0.45-μm filter (Millipore, Billerica, MA, USA), and 4 IU/ml of heparin (Leo Pharma A/S, Ballerup, Denmark) was added. Lysate from the two pathogen-inactivated units were pooled together, and the lysate from the two not pathogen-inactivated PCs were pooled together. The resulting platelet lysate was aliquoted and stored at −20°C. The procurement of PCs was approved by the Icelandic ethics committee.
Cell Culture
Bone marrow-derived MSCs from two donors (males, ages 24 and 26, passage 1) were obtained from Lonza (Walkersville, MD, USA) and cultured in DMEM/F-12 + Glutamax medium (Gibco, Grand Island, NY, USA), 0.1% penicillin/streptomycin (Gibco), supplemented with either 10% lysate from expired pathogen-inactivated platelet concentrate (hPLpi) or 10% lysate from expired traditionally produced platelet concentrate (hPLex). Cells were maintained in culture for three passages before harvesting and induction of in vitro differentiation.
MSCs for osteogenic differentiation were seeded at a density of 3,000 cells/cm2 in hMSC Differentiation BulletKit® Osteogenic media (Lonza). Chondrogenic differentiation was performed in pellet cultures. MSCs were seeded in microtubes (Sarstedt, Nümbrech, Germany) with 250,000 cells per tube and centrifuged at 150 χ g for 5 min in hMSC Differentiation BulletKit® Chondrogenic media (Lonza). The lids of the microtubes were punctured with a sterile needle (Misawa, Tokyo, Japan) to facilitate gas exchange. For adipocytic differentiation, MSCs were seeded at a density of 10,000 cells/cm2 with StemPro® Adipogenesis Differentiation medium (Gibco).
Immunophenotyping and Proliferation
After three passages of culture in either 10% hPLpi or 10% hPLex, cells were harvested and stained with 10 ng/ml of cluster of differentiation 45 (CD45), CD105, CD73, CD90, and human leukocyte antigen (HLA)-DR antibodies (Becton Dickinson, Franklin Lakes, NJ, USA). MSC immunophenotype was evaluated by flow cytometry using a FACSCalibur instrument (Becton Dickinson).
For cell proliferation studies, 25,000 MSCs were seeded in a microplate and proliferation evaluated with an XTT assay (ATCC, Manassas, VA, USA) over a period of 7 days.
Mixed Lymphocyte Reaction Assay
The immunomodulatory potential of MSCs was evaluated by mixed lymphocyte reaction (MLR) assay in a direct coculture setup. Mononuclear cells (MNCs) were isolated from three discarded buffy coat units (males, ages 23, 33, and 35), obtained at the Blood Bank, using histopaque-1077 (Sigma-Aldrich, St. Louis, MO, USA) and following the manufacturer's instructions. MNC count was determined using a hematology analyzer (Cell-Dyn Ruby, Abbott, Wiesbaden, Germany) and a coculture of MSCs and MNCs initiated with a ratio of 1:10. MLR in the cocultures was stimulated with 2 μg/ml of phytohemaglutinin (PHA; Sigma-Aldrich). Stimulated MNCs in the absence of MSCs were used as controls. Briefly, MSCs were seeded and allowed to adhere for 24 h, after which media was changed and nonadhering cells removed. Then MNCs were added and coculture initiated. After 72 h of coculture, the MNCs were carefully removed from the coculture as not to disturb the adhering layer of MSCs. The MNC cellular proliferation was evaluated via XTT assay (ATCC) according to the manufacturer's instructions.
Evaluation of Glycosaminoglycan Content
After 14 and 28 days of chondrogenic differentiation, glycosaminoglycan (GAG) content was evaluated as a marker of extracellular matrix (ECM) production. Chondrocytic pellets were washed with phosphate-buffered saline (PBS; Gibco) and then digested for 3 h at 65°C in papain extraction reagent containing 0.1 M sodium acetate (Sigma-Aldrich), 0.01 M Na2EDTA (Carl Roth GmbH + Co. KG, Karlsruhe, Germany), 0.005 M cysteine HCL (Sigma-Aldrich), and 8 μl of crystallized papain (Sigma-Aldrich) per ml solution. GAG was then quantified with Blyscan assay (Biocolor, Carrickfergus, UK) following the manufacturer's instructions.
Alkaline Phosphatase Activity Assay
The quality of osteogenic differentiation was evaluated after 7, 14, and 21 days by alkaline phosphatase (ALP) assay (Sigma-Aldrich) via the conversion of p-nitrophenolphosphate to p-nitrophenyl. Cells were washed with PBS and then lysed with 0.02% Triton X (Sigma-Aldrich). Half of the cell lysate was incubated in a solution of p-nitrophenol phosphate at 37°C for 30 min until a yellow color change was observed. The color change was evaluated in a spectrophotometer (Thermo Scientific, Vantaa, Finland) at an optical density of 400 nm and the ALP activity determined using the Beer-Lambert law. The other half of the cell lysate was evaluated for protein content using a BCA assay (Thermo Scientific, Rockford, IL, USA) for normalization of ALP activity.
Gene Expression Analysis
Osteogenic and adipogenic samples for gene expression were homogenized by thorough vortexing in RLT buffer (Qiagen, Hamburg, Germany) containing 1% β-mercaptoethanol (Sigma-Aldrich). Chondrocytic pellets were homogenized in RLT buffer containing 1% β-mercaptoethanol using the gentleMACS™ Dissociator (Miltenyi Biotec, Bergisch Gladbach, Germany). RNA was then isolated using an RNeasy mini kit (Qiagen) following the manufacturer's instructions. cDNA was prepared using GeneAmp® RNA PCR (Applied Biosystems, Foster City, CA, USA), according to the manufacturer's instructions. Real-time qPCR was performed on all samples with runt-related transcription factor 2 (RUNX2, Hs00231692_m1), secreted phosphoprotein 1 (SPP1, Hs00167093_m1), adiponectin (ADIPOQ, Hs00605917_m1), and sex-determining region Y box 9 (SOX9, Hs01001343_g1) as genes of interest. The two most stable reference genes were carefully chosen after screening of several reference genes. Thymine adenine thymine adenine (TATA) box-binding protein (TBP, Hs00427620_m1) and YWHAZ (Hs03044281_g1) were found to be most suitable and both used for normalization of expression in all samples. TaqMan® Gene Expression Assays were used in all experiments (Applied Biosystems).
Statistical Analysis
GraphPad® version 5 (GraphPad Software, La Jolla, CA, USA) and Microsoft Office Excel 2007 (Microsoft Inc., Seattle, WA, USA) were used for all data analysis except gene expression data. Two-way ANOVA was used to analyze statistical significance followed by Bonferroni correction. Relative gene expression was evaluated using REST-384© version 2 followed with a pairwise fixed reallocation randomization test to determine statistical significance (33). Two reference genes, TBP and YWHAZ, were used for normalization in each sample and relative values presented. Two independent donors of MSCs were used in all experiments. Data are presented as mean ± standard error. A value of p <0.05 was considered statistically significant.
Results
Immunomodulatory Properties Are Increased by hPLpi
Surface antigen expression was evaluated by flow cytometry after three passages of culture in either hPLpi or hPLex. Expression of the hematopoietic markers CD45 and HLA-DR was not observed. Both cultures expressed the nonhematopoietic markers CD73, CD90, and CD105. No differences in expression were observed between the different types of platelet lysates (Table 1).
Surface Antigen Expression of MSCs Cultured With 10% hPLex or 10% hPLpi

MSCs, mesenchymal stromal cells; hPLex, human platelet lysate from expired platelet concentrates; hPLpi, human platelet lysate from pathogen-inactivated platelet concentrates; gMFI, geometric mean fluorescence intensity; CD105, cluster of differentiation 105; HLA, human leukocy-ate antigen; NS, not significant.
Cell proliferation was comparable between cells in hPLex and hPLpi, with no significant difference observed (Fig. 1A).
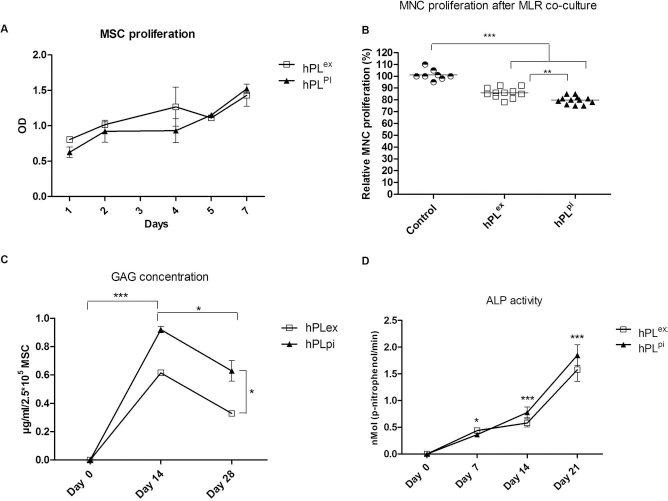
MSCs demonstrated good proliferation, immunosuppression, and expressed lineage-specific biomarkers after culture in both hPLex and hPLpi. (A) Proliferation of MSCs was comparable in both hPLex and hPLpi supplemented media. (B) Both MSCs from hPLex and hPLpi significantly reduced MNC proliferation by 14.09% ± 1.31% and 20.25% ± 0.98%, respectively. hPLpi MSCs exerted significantly more suppression than hPLex MSCs (n = 12). (C) GAG concentration increased from day 0 to day 14 and then decreased toward day 28 in all cultures. GAG concentration was consistently higher in hPLpi chondrocytic cultures than in hPLex cultures (n = 6). (D) ALP activity increased in a stepwise manner with a significant increase from day 7 onward for hPLpi and day 14 onward for hPLex (n = 6). *p < 0.05, **p < 0.01, ***p < 0.001.
To evaluate whether MSCs maintained their abilities to suppress MNC proliferation in vitro, MSCs were cocultured with PHA-stimulated MNCs. All MSC cultures significantly suppressed MNC proliferation in cocultures compared with MNC proliferation in cultures without MSCs (14.09% ± 1.31% suppression with hPLex and 20.25% ± 0.98% suppression with hPLpi, p < 0.001, n = 12). Furthermore, MSCs from hPLpi were found to be 6.16% ± 1.61% more suppressive than MSCs from hPLex (p < 0.01, n = 12) (Fig. 1B).
Lineage-Specific Biomarkers Are More Strongly Expressed in hPLpi-MSCs During Differentiation
The amount of GAGs per 250,000 MSCs in a chondrocytic pellet was chosen as an indicator for chondrogenesis and was measured after 14 and 28 days of differentiation. The amount of GAG was significantly higher at day 14 compared to day 28 (0.62 ± 0.02 μg/ml/2.5 χ 105 MSCs for hPLex and 0.92 ± 0.02 μg/ml/2.5 χ 105 MSCs for hPLpi at day 14 vs. 0.33 ± 0.01 μg/ml/2.5 χ 105 MSCs for hPLex and 0.63 ± 0.07 μg/ml/2.5 χ 105 MSCs for hPLpi, p < 0.05, n = 6). GAG concentration was significantly higher in chondrocytic pellets from hPLpi MSCs than hPLex at both time points, with a mean difference of 0.3 ± 0.04 μg/ml/2.5 χ 105 MSCs (p < 0.05, n = 6) (Fig. 1C).
The activity of ALP was evaluated as a marker of osteoblast formation, which is indicative of the progress of osteogenic differentiation. ALP activity in osteogenic cultures that had been expanded in hPLex increased throughout the differentiation period, with statistically significant increases between day 0 and day 7 [0.0001 ± 0.00 p-nitrophenol/min (pnp/min) at day 0 vs. 0.44 ± 0.051 pnp/min at day 7, p < 0.05, n=6) and between days 14 and 21 (0.58 ± 0.077 pnp/min at day 14 vs. 1.58 ± 0.219 pnp/min at day 21, p < 0.001, n = 6) (Fig. 1D). However, no significant increase was observed in hPLex cultures between days 7 and 14. Osteogenic cultures that had been expanded in hPLpi showed a significant increase in ALP activity between all time points (0.0001 ± 0.00 pnp/min at day 0, 0.36 ± 0.037 pnp/min at day 7, 0.778 ± 0.099 pnp/min at day 14 and 1.85 ± 0.194 pnp/min at day 21, p < 0.001, n=6) (Fig. 1D). The choice of platelet lysate did not significantly affect the ALP activity observed at each individual time point.
MSCs Expanded in hPLpi Maintain Their Differentiation Potential
In osteoblasts, no time-dependent change in RUNX2 expression was observed, and no significant statistical difference between hPLex and hPLpi expanded cultures was noted (Fig. 2A). The late osteogenic marker gene SPP1 increased significantly over time (0.97 ± 0.32-fold for hPLex and 3.19 ± 0.34-fold for hPLpi, p < 0.001) (Fig. 2B). At day 7, a 0.47 ± 0.30-fold difference in SPP1 expression was observed between the cultures, with hPLpi osteoblasts showing significantly lower expression than hPLex osteoblasts (p < 0.001). By day 21, no significant difference was observed between hPLex and hPLpi osteoblasts (Fig. 2B).

Lineage-specific gene expression in differentiating cultures. (A) RUNX2 did not increase from day 7 to day 21, and no significant difference in expression between hPLex and hPLpi osteoblasts was found. (B) SPP1 expression increased significantly from day 7 to day 21 in both hPLex and hPLpi osteoblasts. At day 7, hPLex osteoblasts expressed SPP1 significantly higher than hPLpi. (C) SOX9 expression increased significantly from day 14 to day 28 in both hPLex and hPLpi chondrocytes. (D) No statistically significant difference was found in ADIPOQ expression between hPLex and hPLpi. All expression values have been normalized to two reference genes, TBP and YWHAZ. *p < 0.05, **p < 0.01, ***p < 0.001.
The analysis of chondrocytic pellets after 14 and 28 days of differentiation revealed a significant increase in SOX9 expression over time (6.83 ± 0.22-fold for hPLex and 6.12 ± 0.2-fold for hPLpi, p < 0.001). No significant difference was found in SOX9 expression in chondrocytes based on treatment (Fig. 2C).
Gene expression in adipocytes was evaluated at a single time point. After 14 days, ADIPOQ (adiponectin) had a 1.79 ± 0.96-fold higher expression in hPLpi adipocytes than in hPLex adipocytes. However, the difference observed was not statistically significant (Fig. 2D).
Trilineage-specific transcription factors were evaluated in all samples, independent of the differentiation stimulus, that is, RUNX2 (osteogenesis), SOX9 (chondrogenesis), and ADIPOQ (adipogenesis). After 7 days of osteogenic differentiation, osteoblasts expressed the osteogenic marker gene RUNX2 significantly more strongly than SOX9 or ADIPOQ (0.22 ± 0.00 and 0.27 ± 0.15-fold SOX9 expression in hPLex and hPLpi, respectively, and 0.04 ± 0.01 and 0.02 ± 0.09 fold ADIPOQ expression relative to RUNX2 in hPLex and hPLpi, respectively, p < 0.001) (Fig. 3A). No significant difference in the expression of RUNX2 and SOX9 was observed at day 21 in osteoblasts, whereas the expression of ADIPOQ was significantly lower than the other two marker genes (0.12 ± 0.04 and 0.15 ± 0.00-fold ADIPOQ expression in hPLex and hPLpi, respectively, relative to RUNX2, p < 0.001) (Fig. 3B).
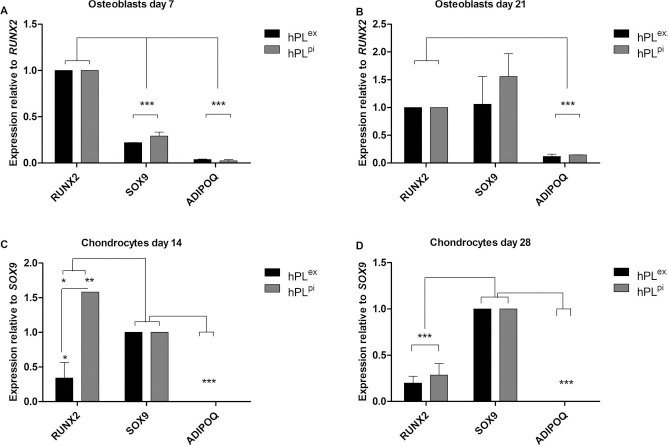
Comparison of expression of lineage-specific genes in osteoblasts and chondrocytes. (A) Osteoblasts had significantly stronger RUNX2 expression than SOX9 and ADIPOQ expression at day 7 of differentiation. (B) At day 21, RUNX2 and SOX9 expression in osteoblasts was comparable, while ADIPOQ expression was still significantly lower. (C) In chondrocytes at day 14 RUNX2 expression in hPLex and hPLpi was found to be significantly different. RUNX2 expression in hPLex was significantly lower compared to SOX9 expression while the RUNX2 expression in hPLpi was significantly stronger compared to SOX9. (D) SOX9 expression was significantly stronger than RUNX2 and ADIPOQ expression at day 28 in chondrocytes. All expression values have been normalized to two reference genes, TBP and YWHAZ. *p < 0.05, **p < 0.01, ***p < 0.001.
In chondrocytes at day 14, RUNX2 and SOX9 were found to have different expressions in hPLex and hPLpi (Fig. 3C). In hPLex, the RUNX2 expression was 0.34 ± 0.23 times lower than SOX9 expression (p < 0.05). However, RUNX2 expression was 1.58 ± 0.00 times stronger than SOX9 in hPLpi chondrocytes (p < 0.01). The difference in RUNX2 expression in hPLex and hPLpi chondrocytes was 1.24 ± 0.22-fold (p <0.05). This difference was only observed at 14 days. At day 28, RUNX2 had significantly lower expression relative to SOX9 in both hPLex and hPLpi (0.2 ± 0.07 and 0.29 ± 0.13-fold expression relative to SOX9 for hPLex and hPLpi, respectively). The expression of ADIPOQ in chondrocytes was close to the detection limit in chondrocytes at both time points (Fig. 3D).
The expression of RUNX2 and SOX9 in adipocytes was significantly lower than ADIPOQ expression (0.02 ± 0.01 and 0.01 ± 0.002-fold RUNX2 expression and 0.02 ± 0.01 and 0.003 ± 0.00-fold SOX9 expression relative to ADIPOQ in adipocytes for hPLex and hPLpi, respectively, p < 0.001) (Fig. 4).
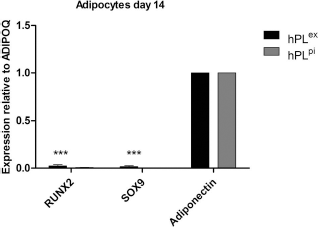
Comparison of expression of lineage-specific genes in adipocytes. ADIPOQ expression was significantly stronger than RUNX2 and SOX9 expression in adipocytes after 14 days of differentiation. All expression values have been normalized to two reference genes, TBP and YWHAZ. *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
In this study, we evaluated the suitability of expired PCs processed into platelet lysates for the culture of MSCs after pathogen inactivation with the INTERCEPT Blood System™ compared to lysates from nontreated platelets. Bone marrow-derived MSCs were cultured in media supplemented either with traditionally expired platelet lysates or with platelet lysates produced from expired and pathogen-inactivated PCs. The effects of pathogen inactivation on immunomodulation, immunophenotype, proliferation, and trilineage differentiation of MSCs were determined. Platelet lysates prepared from expired and pathogen-inactivated PCs performed comparably to traditionally prepared platelet lysates from expired platelet units. Pathogen inactivation with INTERCEPT had no negative effects on immunophenotype, immunomodulation, trilineage differentiation, or proliferation.
Pathogen inactivation of platelets did not have a statistically significant effect on MSC immunophenotype. Both hPLex and hPLpi MSCs significantly decreased PHA-stimulated MNC proliferation during a MLR, demonstrating direct MSC participation in immunosuppression. However, hPLpi MSCs demonstrated significantly stronger immunosuppression than hPLex MSCs. During pathogen inactivation, white blood cells (WBCs) are inactivated as well as pathogens. It has been demonstrated previously that WBCs in pathogen-inactivated PCs secrete inflammatory cytokines to a significantly lower extent than WBCs in untreated PCs and are also less effective in provoking immune responses (7,44). It has also been shown that WBCs in photochemically pathogen-reduced PCs fail to cause alloimmunization following allogenic transfusion in mice (20). The immunosuppressive abilities of MSCs are currently of major interest and are being studied intensively as potential candidates for future immunotherapies (36,43). When formulating a new supplement for MSC cultures, it is therefore important that the cells maintain this important characteristic. Lower WBC contamination and their limited bioactivity following pathogen inactivation may lead to lower concentrations of inflammatory cytokines in platelet lysates produced from pathogen-inactivated PCs. Such lysates are therefore less likely to cause immune responses and do not interfere with the immunosuppression exerted by the MSCs.
It was observed that MSC differentiation in the presence of an osteogenic stimulus induced the expected increase in ALP activity and osteogenic marker gene expression. ALP is a commonly used biomarker for bone turnover, and increased activity is consistent with higher osteoblast activity (18). ALP activity was observed to increase earlier in hPLpi osteoblasts (from day 7) than in hPLex (a similar increase was not observed until day 14), indicating superior formation of osteoblasts in hPLpi osteoblasts. Concerning gene expression of osteogenic marker genes, the early marker RUNX2 was elevated after 7 days, and its high expression compared to SOX9 (chondrogenesis) and ADIPOQ (adipogenesis) points to true commitment toward the osteoprogenitor lineage.
The late osteogenic marker SPP1 increased around day 21. SPP1 is a direct downstream target of RUNX2, the master transcription factor of osteogenic differentiation, and plays an important role in the mineralization process (8,11). The increase in SPP1 is in concert with the increase in ALP activity demonstrated, since both indicate 1) that mineralization is in process and 2) successful osteogenic differentiation with a sufficient quality of osteoblasts.
During chondrogenic differentiation, the amount of GAGs initially increased, but then decreased toward the end of the differentiation period, closely following the natural course of cartilage development. Chondrogenesis is marked by initial cellular proliferation and production of ECM proteins like GAG, leading to hypertrophy and ECM degradation during later stages, prior to endo-chondral ossification (47). While both hPLex and hPLpi chondrocytes exhibited this pattern, hPLpi chondrocytic cultures were found to produce significantly more ECM as measured by GAG concentration. The ECM components of cartilage are essential to support the functionality of the tissue (31). Since GAGs represent the second largest group of ECM macromolecules in cartilage, high levels are indicative of good quality differentiation (10). This was further supported by the high expression of SOX9 in both hPLpi and hPLex chondrocytes during late differentiation. SOX9 is the master transcription factor for chondrogenesis, and high expression points to commitment directed toward the chondroprogenitor cell lineage (4). Adipogenesis was confirmed by the strong expression of ADIPOQ and the absence of both RUNX2 and SOX9 expression. No spontaneous adipogenesis due to lack of stimulation was observed in cultures induced toward osteogenesis or chondrogenesis, as evidenced by the lack of ADIPOQ gene expression in these cultures. Generally, no effect on trilineage differentiation could be attributed to the pathogen inactivation of platelet concentrates processed into platelet lysates.
Being able to obtain cell culture supplements from clinically approved PCs that are no longer useful for transfusion is an important step toward animal-free cell culture that is both safe and beneficial. Improvement of current culture strategies is a priority, especially when intended for human treatment. The use of platelet lysates from safe sources should be strongly considered.
Here, we demonstrated that pathogen inactivation of platelets using the INTERCEPT Blood System™ prior to lysate production does not negatively interfere with the physiology of MSCs in vitro. The expired inactivated platelet concentrates are able to support MSCs in culture, allowing them to maintain their differentiation and immunomodulatory abilities to an even better extent than when cultured in lysates from nonpathogen-inactivated PCs.
Blood banks are a source of clinical-grade PCs, but are forced to discard considerable quantities of them due to platelet expiry. We conclude that the use of expired INTERCEPT™ pathogen-inactivated platelet for platelet lysate production is a practical possibility for MSC culture and should be considered as an acceptable alternative for the expansion of MSCs according to good manufacturing practice.
Footnotes
Acknowledgment
The authors want to thank the Icelandic Student Innovation Fund and Landspitali University Hospital Research Fund for partially funding the project. We would also like to acknowledge Ragna Landro and Bjorn Hardarsson for technical support and Kristine Wichuk for editing and reviewing of the manuscript. The authors declare no conflict of interest.