
Abstract
The immunomodulatory properties of mesenchymal stromal cells are the subject of increasing interest and of widening clinical applications, but the reproducibility of their effects is controversial and the underlying mechanisms have not been fully clarified. We investigated the transfer of membrane vesicles, a recently recognized pathway of intercellular communication, as possible mediator of the interaction between mesenchymal stromal cells and B lymphocytes. Mesenchymal stromal cells exhibited a strong dose-dependent inhibition of B-cell proliferation and differentiation in a CpG-stimulated peripheral blood mononuclear cell coculture system. We observed that these effects could be fully reproduced by membrane vesicles isolated from mesenchymal stromal cell culture supernatants in a dose-dependent fashion. Next, we evaluated the localization of fluorescently labeled membrane vesicles within specific cell subtypes both by flow cytometry and by confocal microscopy analysis. Membrane vesicles were found to be associated with stimulated B lymphocytes, but not with other cell phenotypes (T lymphocytes, dendritic cells, natural killer cells), in peripheral blood mononuclear cell culture. These results suggest that membrane vesicles derived from mesenchymal stromal cells are the conveyors of the immunosuppressive effect on B lymphocytes. These particles should be further evaluated as immunosuppressive agents in place of the parent cells, with possible advantages in term of standardization, safety, and feasibility.
Keywords
Introduction
Mesenchymal stromal cells (MSCs) have immunomodulatory properties demonstrated in vitro, in animal studies, and in clinical applications such as the treatment of severe graft versus host disease (GVHD) (4, 16, 31). However, the underlying mechanisms have not been fully clarified (19, 21). In particular, while the suppressive effect of MSCs on activated T-cells has been extensively investigated and found to involve multiple factors (2, 17, 24), both the effect of MSCs on B-cells and the involved mechanisms are more controversial (8, 10, 22, 26, 28). Recently, it was demonstrated that several interactions between immune cells are mediated by secreted membrane vesicles (MVs) (27). Various types of secreted MVs have been described, ranging from 50 to 1,000 nm in diameter and exhibiting distinct structural and biochemical properties according to their intracellular site of origin, features probably also affecting their function(5). An increasing body of evidence indicates that they play a pivotal role in cell-to-cell communication (7); in particular, MVs can play a role in intercellular signaling by exchanging mRNA, microRNA, and proteins among cells within a defined microenvironment (29). The aim of this study was to verify the immunomodulatory properties of MVs derived by MSCs (MSC-MVs) on B-cell function. Here, we show that the immunosuppressive effect of MSCs on B-cells can be reproduced in a dose-dependent fashion by MSC-MVs secreted in the medium by cultured cells.
Materials and Methods
MSC Culture and Expansion
Commercially available bone marrow human MSCs (Lonza, Basel, Switzerland) were plated in 75-cm2 polystyrene vented tissue culture flasks (Becton Dickinson, BD, Franklin Lakes, NJ, USA) at a density of 4 × 103 cells/ cm2 in a volume of 10 ml of MesenCult basal medium (StemCell Technologies, Vancouver, BC, Canada) supplemented with 20% fetal bovine serum (FBS; HyClone Laboratories), 100 U/ml penicillin, and 100 μg/ml streptomycin (Gibco, Grand Island, NY). In order to remove endogenous MVs, FBS was centrifuged overnight at 100,000xg before use. Cultures were incubated at 37°C in a humidified atmosphere containing 5% CO2. Cells were subsequently maintained in the same medium and passaged at 80–90% confluence in a ratio 1:2 in trypsin/ EDTA solution (Invitrogen, Life Technologies, Monza, Italy), with the medium changed once a week.
Peripheral Blood Mononuclear Cell Isolation
Blood samples from healthy donors were recruited at the Blood Transfusion Center of Children's Hospital Bambino Gesù, Rome. After obtaining informed consent, peripheral blood mononuclear cells (PBMCs) were separated by Ficoll-Hypaque (Histopaque, Sigma-Aldrich Chemical C, St. Louis, MO, USA) from 50 ml of sodiumheparinized venous blood samples, washed twice in Dulbecco's phosphate-buffered saline (D-PBS, Euro-Clone, Milan, Italy), and cryopreserved in liquid nitrogen until use. The protocol, involving the use of human material, was approved by the Ethical Committee of the Children's Hospital Bambino Gesù.
Coculture of PBMCs with MSCs
B-cell viability experiments were performed in a coculture system with MSCs plated in 96-multiwell culture plates (Corning-Costar, Celbio, Milan, Italy) at initial densities of 2.5×104, 1×104, or 5×103 cells/well in MesenCult basal medium supplemented with FBS (20%). For the immunomodulation experiments, MSCs were plated in 96-multiwell plates at a concentration of 5×103 cells/well and cultured in MesenCult basal medium supplemented with FBS (20%). On the following day, the medium was aspirated and replaced with PBMCs at 5×105 cells/well, corresponding to MSCs/PBMC ratios of 1:20, 1:50, and 1:100. In order to evaluate cell proliferation and differentiation, PBMCs had been prelabeled with 0.5 μM 5-chloromethylfluorescein diacetate (CMFDA, CellTracker; Molecular Probes) according to the manufacturer's guidelines and cocultured with MSCs in RPMI 1640 medium (BioWhittaker, Lona, Belgium) supplemented with 10% FBS. B-cell stimulation was achieved by incubation with 2.5 μg/ml human CpG oligodeoxynucleotides (5′-TCGTC GTTTTGTCGTTTTGTCGTT-3′) (Hycult Biotechnology). After 1 week, the cocultures of PBMCs were rescued, washed in D-PBS, and analyzed by fluorescence-activated cell sorting (FACSCanto II, BD Biosciences).
Coculture of PBMCs with MSC-MVs
MSC-MVs were isolated with a modification of the procedure of Lamparski et al. (18). Cultures of MSCs at 90% confluence were used for the isolation of MVs. The medium from culture plates with 2 × 106 seeded MSCs was collected at day 5 of culture and centrifuged at 1,000xg for 20 min to remove the debris. Ten milliliters of clarified supernatant was concentrated by centrifugation for 20 min at 2,000xg in a sterile hydrated 30-kDa MWCO Amicon Ultra Centrifugal filter (Millipore, Bedford, MA) up to a volume of 15–20 ml. The MSC-conditioned concentrated medium was diluted in 10 ml of PBS in polyallomer tubes (Beckman Coulter, Milan, Italy), then ultracentrifuged at 100,000 × g at 4°C for 1 h. At the end of the procedure, 2 ml was collected from the bottom of the tubes and concentrated by centrifuging for 20–30 min at 2,000xg in a sterile 30-kDa MWCO Amicon Ultra Centrifugal filter (Millipore) up to a volume of 15–20 μl. MVs were used either undiluted or diluted 1:3 or 1:6 in RPMI 1640 (BioWhittaker) and added to 5 × 105 CMFDA-labeled PBMCs 1 and 24 h after seeding. After 1 week in the presence of 2.5 μg/ml human CpG oligodeoxynucleotides added at the time of cell seeding, PBMCs were collected by centrifugation, washed, and analyzed by flow cytometry.
Detection of Apoptosis
For the detection of apoptosis, PBMCs cultured in medium alone, with CpG, or cocultured with MSC-MVs plus CpG were analyzed by Annexin V and 7-amino-actinomycin (7-AAD) staining. Briefly, after 4 and 7 days in culture, the cells were centrifuged at 300 × g for 5 min and incubated with anti-CD19 [1:10, phycoerythrincyanine 7 (PE-Cy7) conjugated, Becton Dickinson, BD, Franklin Lakes, NJ, USA] for 20 min at 4°C in the dark. After washing with PBS, 5 μl of Annexin V [allophycocyanin (APC)-conjugated, BD] and 5 μl of vital dye 7-AAD [peridin-chlorophyll protein complex (PerCP)-conjugated, BD] were added in a final volume of 500 μl of Annexin V Binding Buffer 1x (BB1x) according to the manufacturer's guidelines. After 15 min of incubation in the dark at room temperature, the samples were transferred on fluorescence-activated cell sorting (FACS) tubes (BD) and acquired with a FACSCanto II (BD). Flow cytometry profiles were analyzed using FACSDiva software (BD). A minimum of 20,000 events were collected per data set.
Detection of Immunoglobulin Production
PBMCs were cocultured in 96-multiwell plates with or without incubation with MSC-MVs in RPMI 1640 (BioWhittaker) supplemented with 10% FBS (HyClone). After 1 week, the plates were centrifuged at 300 × g for 5 min, and the supernatants were collected and tested by enzyme-linked immunosorbent assay (ELISA) in order to assess immunoglobulin production. The 96-multiwell ELISA plates (Corning) were coated with purified goat anti-human IgA or IgG or IgM diluted in PBS and incubated overnight at 4°C (IgA, 10 μg/ml; IgG, 15 μg/ml; IgM, 2.5 μg/ml; Jackson ImmunoResearch Laboratories, West Grove, PA, USA). After three washes in PBS/Tween (0.1%), the supernatants from cell cultures were added to the plates (50 μl/well) and incubated at 37°C for 1 h in a humidified atmosphere. After washing, the plates were incubated for 1 h at 37°C with 50 μl of peroxidase-conjugated goat anti-human IgA (1:1,000), IgG (1:2,000), or IgM (1:1,000) diluted in PBS (Jackson ImmunoResearch Laboratories). o-Phenylenediamine tablets (Sigma-Aldrich), diluted in PBS according to the manufacturer's guidelines, were used as chromogenic substrate to develop the assay. The reaction was stopped after 30 min by adding 50 μl/well of SDS (10%). At the end of the procedure, the plates were read with a microplate spectrophotometer (Benchmark Plus) at 460 OD.
Flow Cytometry
At the end of the experiments, PBMCs were harvested from culture plates, centrifuged at 300xg for 5 min, and resuspended in PBS/FBS (2%). Single-cell suspensions were incubated in the dark for 20 min at 4°C with directly conjugated monoclonal antibodies (mAbs) directed against the following human surface molecules: CD19 (1:7, Cy5-conjugated), CD27 (1:7, PE-conjugated), CD38 (1:30, PE-Cy7-conjugated), IgM (1:100, Cy5-conjugated), CD86 (1:5, APC-conjugated), CD3 [1:7, fluorescein isothiocyanate-conjugated (FITC)], CD56 (1:7, FITC-conjugated), CD19 (1:20, APC-Cy7-conjugated). All antibodies were purchased from BD. After labeling, cells were washed twice in PBS/FBS (2%), and data were acquired with a FACSCanto II (BD). Flow cytometry profiles were analyzed using FACSDiva software (BD). A minimum of 20,000 events were collected per data set.
For selected experiments, the MVs derived from supernatant of 2×106 seeded MSCs grown at confluence were isolated with the procedure described above and added with 5 μl of Annexin V (APC-conjugated; BD) in conjunction with 5 μl of vital dye 7-AAD (PerCP-conjugated; BD) in a final volume of 500 μl of Annexin V Binding Buffer 1x (BB1x) according to the manufacturer's guidelines. After 15 min of incubation in the dark at room temperature, the MV samples were transferred to Troucount tubes (BD) containing calibration beads in order to gate the MVs by morphological parameters (forward scatter FSC-H; side scatter, SSC-H).
MSC-MV Cytokine Content
Pelletted MSC-MVs were mixed with Triton X-100 (1% final concentration), and the lysate was assayed with the FluoCytomix Multiple Analyte Detection kit (eBioscience, San Diego, CA, USA) according to the manufacturer's instructions. Assayed human cytokines included interferon (IFN)-γ, interleukin (IL)-1β, IL-2, IL-4, IL-5, IL-6, IL-8, IL-10, IL-12 p70, tumor necrosis factor (TNF)-α, and TNF-β. MSC-MVs were also maintained in culture medium at 37°C, and cytokine release was measured in medium after 2, 4, and 7 days.
Imaging Analysis of MSC-MV Association with PBMCs
MVs were isolated from the medium of 2×106 MSCs as described above and labeled with 2.5×10−6 M PKH26 (25) (Sigma, St. Louis, MO, USA) according to the manufacturer's instructions. The labeled MVs were gently added to the PBMC suspension (5×105 cells/well) in RPMI plus 10% FBS. After 1 h of incubation at 37°C in a humidified atmosphere with 5% CO2, the cell population was washed in PBS and split in two aliquots for flow cytometry and for confocal microscopy analysis.
Immunofluorescence Analysis
PBMCs incubated with PKH26-labeled MVs were rinsed in PBS, fixed in 4% formaldehyde, blocked with PBS/FBS (5%) for 30 min, and single-labeled with the following monoclonal antibodies: anti-CD86 (1:10), anti-CD3 (1:10), and anti-CD19 (1:10), all conjugated to allophycocyanin (BD), as well as anti-CD56 antibody (1:30, FITC-conjugated; BD).
All antibodies were diluted in PBS/bovine serum albumin (BSA) (1%) and incubated for 1 h. Negative controls were performed using PBMC samples without incubation with MVs, as well as omitting the primary antibody in the staining procedure.
Nuclei were counterstained with 1 μg/ml Hoechst 33342 (Invitrogen, Molecular Probes, Eugene, OR, USA). After washing in PBS, the samples were mounted with ProLong antifade reagent (Invitrogen).
Confocal Laser Microscopy and Image Processing Analysis
Confocal imaging was performed on an Olympus Fluoview FV1000 confocal microscope equipped with FV10-ASW version 2.0 software, Multi Ar (458–488 and 515 nm), 2x He/Ne (543 and 633 nm), and 405-nm diode lasers, using a 60x (1.35 NA oil) objective. Optical single sections were acquired with a scanning mode format of 1,024×1,024 pixels, with a 207 nm/pixel size, sampling speed of 40 μs/pixel, and 12 bits/pixel images. Fluorochrome unmixing was performed by acquisition of automated sequential collection of multichannel images in order to reduce spectral crosstalk between channels. The pinhole aperture was 1 Airy unit.
Z-reconstruction of serial single optical sections was performed with a scanning mode of 1,024× 1,024 pixels with an electronic zoom at 2, corresponding to 103 nm/ pixel, sampling speed of 20 μs/pixel, and Z stack of 0.40 μs/ slice. Images were processed using Photoshop software version 9.0 (Adobe Systems Inc., San Jose, CA, USA).
Statistical Analysis
Each experiment was run in duplicate, and the mean of the two results was calculated. Data were analyzed in terms of medians and minimum and maximum values. A nonparametric analysis of variance (Kruskal–Wallis test) followed by multiple comparisons by mean ranks was performed to compare quantitative data among groups. Linear regression analysis was performed to evaluate dose–response relationships. A value of p < 0.05 was considered statistically significant.
Results
Coculture of PBMCs with MSCs
We first investigated the effects of MSCs on B-cell viability upon stimulation with CpG. We cocultured CMFDA-labeled PBMCs (C) isolated from n = 8 healthy donors with MSCs. After 7 days of incubation, we analyzed the CD19-positive cells by cytometric analysis. A significant inhibition of B-cell viability was observed at the MSC/C ratio of 1:20 (Fig. 1A, D). We next investigated whether coculture with MSCs affects B-cell proliferation and differentiation. After 7 days in the previously described experimental conditions, double-stained CD19/CMFDA and CD19/CD27 cells were gated in flow cytometry analysis. Maximum inhibition of B-cell proliferation (Fig. 1B, D) and differentiation (Fig. 1C, D) was observed at 1:20 MSCs/C ratio.

Coculture of CpG-stimulated PBMCs with MSCs. (A–C) Representative density plots. Cytometric analysis was performed by morphological parameter (forward scatter, FSC-H) of chloromethylfluorescein diacetate (CMFDA)-labeled peripheral blood mononuclear cells (PBMCs) (C) or PBMCs after coculture with mesenchymal stromal cells (MSCs) (MSCs/C) at ratios of 1:20, 1:50, and 1:100, respectively. (A) Positive CD19 cells were gated in R1 from PBMCs cultured with or (C) without MSCs. (B) In the top left quadrant (Q1), the percentage of CD19/CMFDA-positive cells is shown for PBMCs cultured with or (C) without MSCs. (C) Double-positive CD19/CD27 Plasma cells (PCs) are shown for PBMCs cultured with or (C) without MSCs. (D) Inhibition of viability, proliferation, and differentiation of CpG-stimulated B-cells in PBMCs cultured with or (C) without MSCs. Graphs show individual data and medians of three independent experiments.
Effect of MSC-MVs on B-Cell Proliferation and Differentiation
MSC-MVs were added to PBMCs at 1 and 24 h after seeding. Statistically significant inhibition of B-cell proliferation was observed in the presence of MSC-MVs (Fig. 2A, top; B). The differentiation of plasma cells (PCs) from B lymphocytes was also evaluated by double staining with CD19 and CD27. A strong inhibition of PCs was observed in the presence of MSC-MVs (Fig. 2A, bottom; C). MSC-MVs also inhibited the production of IgM, IgG, and IgA by PBMCs (Figure 3).

Effect of MSC-MVs on CpG-stimulated PBMCs. (A) Representative density plots of cytometric analysis of CMFDA-labeled PBMCs cultured in medium alone, with CpG, or with MSC-membrane vesicles (MVs) and CpG. Lymphocytes were gated in Q1 by morphological parameters (FSC-H; SSC-H) as living cells (left). They were subsequently analyzed either for the proliferation rates by CMFDA/ CD19 staining (top right) or for the differentiation stage of plasma cells (PCs) by selecting CD19/CD27-positive events (bottom right). (B) Inhibition of proliferation of CMFDA/CD19-positive lymphocytes from a culture of PBMCs cultured in medium alone, with CpG, or with MSC-MVs and CpG. (C) Inhibition of differentiation of CD19/CD27-positive plasma cells (PCs) from a culture of PBMCs cultured in medium alone, with CpG, or with MSC-MVs and CpG. Graphs show individual data and medians of four independent experiments.

Inhibition of immunoglobulin production in CpG-stimulated PBMCs after incubation with MSC-MVs. ELISA assay of supernatants from a culture of PBMCs cultured in medium alone, with CpG, or with MSC-MVs and CpG. Graphs show individual data and medians of IgM, IgG, or IgA concentrations (102 ng/ml) in four independent experiments.
Apoptosis
The percentage of Annexin V/7-AAD-positive B lymphocytes (approximately 2% of the total B lymphocytes) was not statistically different among cells cultured with medium alone, with CpG alone, or with CpG and MSC-MVs both after 4 days and after 7 days in culture, indicating that the suppressive effect of MSC-MVs was not associated with B-cell death. For the sake of simplicity, only data at day 7 are shown (Figure 4).

Detection of apoptosis. Annexin V/7-amino-actinomycin (7-AAD) analysis of proliferating CD19-positive cells maintained for 7 days in medium alone, with CpG, or treated with MSC-MVs plus CpG. Graphs show individual data and medians of three independent experiments.
Characterization of MSC-MVs
A representative density plot of the MSC-MVs population isolated as described above is shown in Figure 5. MSC-MVs are 7-AAD-negative and express Annexin V.
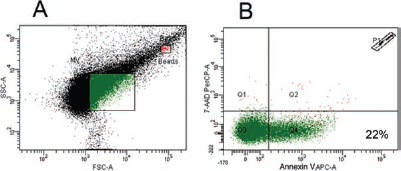
Flow cytometry analysis of MSC-MVs. Representative scatter plot obtained by flow cytometry of MSC-MVs. (A) The MSC-MVs were gated using size calibration beads to define the proper gate of events within 0.5- to 1-μm dimensions. (B) MSC-MVs derived from the gate shown in (A) were identified in quadrant Q4 by Trucount beads (P1), Annexin V positivity, and 7-AAD negativity. The percentage of Annexin V-positive/7-AAD-negative MSC-MVs is indicated in Q4.
Dose-Dependent Effect of MSC-MVs on B Lymphocytes
Serial dilutions of the MSC-MV suspension, obtained as described above, were added to 5×105 PBMC cultures. A linear correlation was found between MSC-MV concentration and the inhibiting effect on B lymphocytes, with respect to both cell proliferation and differentiation (Fig. 6A). A dose-dependent inhibitory effect of MSC-MVs was also observed on IgM, IgG, and IgA production (Fig. 6B).

Dose-dependent inhibitory effect of MSC-MVs on B lymphocytes. (A) Inhibition of proliferation (left) and differentiation (right) of CpG-stimulated PBMCs after culture with undiluted MSC-MVs and at 1:3–1:6 dilutions. (B) Immunoglobulin ELISA assay of supernatants from a culture of PBMCs alone (not shown) or incubated with MSC-MVs at 1:3 or 1:6 dilutions. A significant dose-dependent inhibition of IgM, IgG, and IgA production was observed. The results were obtained from four independent experiments.
Cytokine Content of MSC-MVs
Both IL-6 and IL-8 were detectable in MSC-MV lysate and were released in culture medium. Both lysate and culture medium tested negative for IFN-γ, IL-1β, IL-2, IL-4, IL-5, IL-10, IL-12 p70, TNF-α, and TNF-β.
Analysis of MSC-MV Association with PBMCs
Flow cytometry analysis of PBMCs preincubated with PKH26-labeled MSC-MVs revealed that these particles were associated with a subset of CD86/CD19-positive cells corresponding to B lymphocytes. No association of labeled MSC-MVs was found either with CD3-positive cells (corresponding to T lymphocytes) or with CD56-positive cells (corresponding to natural killer cells) (Fig. 7).
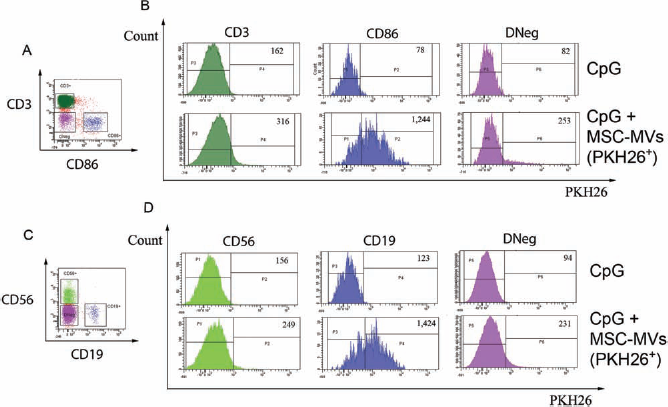
Fluorescence-activated cell sorting (FACS) analysis of the association between PKH26-labeled MSC-MVs and selected cell phenotypes in CpG-stimulated PBMCs. (A) Density plots of CD3+, CD86+, and double-negative lymphocytes (DNeg), selected in three different gates. (B) Histograms of fluorescence intensity (IF) of CD3-FITC-, CD86-APC-positive gated lymphocytes analyzed before (top) or after incubation with PKH26-labeled MSC-MVs (bottom). (C) Density plots of CD56+, CD19+, and double-negative lymphocytes (DNeg), selected in three different gates. (D) Histograms of IF of CD56-FITC-, CD19-APC-Cy7-positive gated lymphocytes analyzed before (top) or after incubation with PKH26-labeled MSC-MVs (bottom). After incubation with PKH26-labeled MSC-MVs, the levels of IF (plotted values in the top right quadrant) increased in CD86+ cells (IF = 1,244 vs. basal level IF = 78) and CD19+ cells (IF = 1,424 vs. basal level IF = 123).
In order to further evaluate the association of MSC-MVs with specific cell phenotypes, we performed an immunofluorescence analysis of PBMC samples preincubated or not with labeled MSC-MVs by confocal laser scanning microscopy (Fig. 8). A panel of fluorescently labeled antibodies directed to CD3, CD19, CD56, and CD86 molecules was used, and the colocalization analysis revealed a consistent association of MSC-MVs with CD19- and CD86-positive cells, whereas no association was observed with CD3- and CD56-positive cells (Fig. 9). The analysis of the localization of red fluorescently labeled MSC-MVs within cells was based on the Z-reconstruction of serial single optical sections of CD19- and CD86-positive cells (Fig. 9). The MSC-MV distribution in the lateral and axial dimensions, visualized by xz- and yz-axis projections, confirmed their intracellular localization in the cytoplasm of CD19- and CD86-positive cells (Fig. 9).

Confocal microscopy of the association between PKH26-labeled MSC-MVs and selected cell phenotypes in CpG-stimulated PBMCs. PBMCs immunolabeled with antibodies against CD3, CD19, and CD86 conjugated to allophycocyanin (pseudocolored in green) and FITC-conjugated CD56 (green), with (right) and without (left) incubation with PKH26-labeled MVs (red). Nuclei were counterstained with Hoechst. MSC-MVs were only associated with CD19-positive (arrowheads) and CD86-positive (arrows) cells. Scale bar: 10 μm.

Confocal microscopy analysis of microphotographs of Z-reconstruction. The analysis of microphotographs of Z-reconstructions (a, d) performed by confocal laser scanning microscopy of CpG-stimulated PBMCs incubated with PKH26-positive MVs and stained with anti-CD19 (a–c) and anti-CD86 (d–f) antibodies. xz- and yz-axis projections (b–c, e–f) obtained from multiple consecutive optical sections, showing the intracellular localization of red fluorescently labeled MVs in CD19-positive (c, in green) and CD86-positive (f, in green) cells. Nuclei were stained with Hoechst. Scale bars: 5 μm (a, d) and 2 μm (b–c, e–f).
Discussion
The immunomodulatory effect of MSCs on B-cells is still controversial, and the mechanisms involved are unclear (8, 10, 22, 26, 28, 30). Because of the documented species differences (23), we will discuss previously published work with human cells only. The role of soluble factors is generally recognized, and it was found to be more relevant in mediating the effect on B than on T lymphocytes (1). It has been proposed that the conflicting results between different laboratories could derive from differences in the methods used to isolate and characterize MSCs (21). For our studies, we relied on a commercial MSC preparation, which in our hands demonstrated good batch-to-batch reproducibility in the present experimental setup (data not shown). Also, we studied the effect of MSCs in culture with PBMCs rather than with purified B-cells, since the former is a more physiological system, including all immune-competent cells.
We observed that MSCs exert a strong inhibitory effect on B-cell proliferation and differentiation in PBMCs, in agreement with previous reports (8, 10, 26) but not with others (22, 28). Both effects could be fully reproduced with MSC-MVs. MSC-MVs affected also B-cell lineage function, as shown by the strong inhibition on immunoglobulin secretion. Also, a linear relationship was observed between MSC-MV concentration and inhibition of B-cell proliferation and differentiation.
It has been reported that the suppressive effect of MSCs on B-cells was still present when separating PBMCs and MSCs by a permeable membrane (1, 10). However, such inhibition was absent (10) or only partial (1) when using MSC supernatant. It was thus hypothesized that the inhibitory effect of MSCs required cell-to-cell contact or the release of paracrine signals from B-cells in order to be fully expressed (10). However, the present results show that the MSC effect on both B-cell proliferation and differentiation can be fully reproduced by MSC-MVs in a dose-dependent fashion. We thus hypothesize that the concentration of MSC-MVs in the microenvironment surrounding B-cells could determine the degree of inhibition and could play a role as a regulatory factor in vivo, since MSCs migrate to inflammation sites (11). Experimental differences affecting MSC-MV availability to B-cells can probably explain previous conflicting results.
In order to evaluate whether other cell types present in PBMCs were involved in the observed phenomenon, we labeled MSC-MVs with a fluorescent dye and found them to be associated only with stimulated B-cells, both by FACS analysis and confocal microscopy.
These data suggest that the suppressive effect of MSCs on B-cells is largely mediated by MV release. The role of MVs as conveyors of immune response has been demonstrated in several pathways, involving all immune cell phenotypes (27). MVs derived by MSCs have been previously described as possible mediators of the antiapoptotic (13) and proregenerative effects (3, 15) associated with MSC administration. However, their role in mediating MSC-induced B-cell suppression was not described so far. We found that MSC-MVs contain IL-6 and IL-8. IL-6 production by MSCs has been described previously (22). Clearly, the identification of these cytokines in MSC-MVs does not allow to draw any immediate hypothesis on the mechanisms underlying their immunosuppressive effect. Even if in our experimental setup these particles were found to be preferentially associated with stimulated B-cells, the observed effect could result from an interplay with other cell types present in PBMCs. In order to characterize the MSC-MVs preparations, we used Annexin V binding as a morphological/functional marker. It is known (14) that due to phospholipid asymmetry, some MVs express phosphatidylserine (PS) on the outer surface and thus bind Annexin V. In our experience, approximately 22% of MSC-MVs bound Annexin V, in agreement with the findings of Connor et al. (9). The biological significance, if any, of these different MV populations is presently unknown.
MSCs were reported to have both inhibitory (8, 10, 26) and stimulatory (22, 28) effects on B-cell proliferation, differentiation, and antibody production. Under some conditions, MSCs can function as antigen-presenting cells, thus enhancing the immune response (6). In one study involving interaction with B-cells, MSCs were found to exhibit opposite effects (i.e., either inhibitory or stimulatory), depending on the magnitude of the stimulus used to trigger the B-cells or on the different cell donors (22). Such unpredictable outcomes raise concern on the possibility to control the effect of administered MSCs in complex autoimmune disorders. MSC-MVs could possibly represent a safer and more reproducible therapeutic tool than MSCs. The clinical use of MVs was found to be both feasible and safe in phase I trials involving MVs derived from dendritic cells for immunotherapy of advanced cancer (12, 20). Clearly, additional studies are needed to explore the role of MSC-MVs as modulators of immune response before they can be proposed for clinical use in place of the parent cells.
Footnotes
Acknowledgments
This work was supported by the Italian Ministry of Health. We thank Paolo Parini for help with the statistical analysis and Rita Carsetti and Ezio Giorda for the FACS analysis. The authors declare no conflict of interest.