
Abstract
Since the brain is naturally inefficient in regenerating functional tissue after injury or disease, novel restorative strategies including stem cell transplantation and tissue engineering have to be considered. We have investigated the use of such strategies in order to achieve better functional repair outcomes. One of the fundamental challenges of successful transplantation is the delivery of cells to the injured site while maintaining cell viability. Classical cell delivery methods of intravenous or intraparenchymal injections are plagued by low engraftment and poor survival of transplanted stem cells. Novel implantable devices such as 3D bioactive scaffolds can provide the physical and metabolic support required for successful progenitor cell engraftment, proliferation, and maturation. In this study, we performed in situ analysis of laminin-linked dextran and gelatin macroporous scaffolds. We revealed the protective action of gelatin–laminin (GL) scaffolds seeded with mesenchymal stem cells derived from donated human Wharton's jelly (hUCMSCs) against neuroinflammatory reactions of injured mammalian brain tissue. These bioscaffolds have been implanted into (i) intact and (ii) ischemic rat hippocampal organotypic slices and into the striatum of (iii) normal and (iv) focally injured brains of adult Wistar rats. We found that transplantation of hUCMSCs encapsulated in GL scaffolds had a significant impact on the prevention of glial scar formation (low glial acidic fibrillary protein) and in the reduction of neuroinflammation (low interleukin-6 and the microglial markers ED1 and Iba1) in the recipient tissue. Moreover, implantation of hUCMSCs encapsulated within GL scaffolds induced matrix metalloproteinase-2 and -9 proteolytic activities in the surrounding brain tissue. This facilitated scaffold biodegradation while leaving the remaining grafted hUCMSCs untouched. In conclusion, transplanting GL scaffolds preseeded with hUCMSCs into mammalian brain tissue escaped the host's immune system and protected neural tissue from neuroinflammatory injury. This manuscript is published as part of the International Association of Neurorestoratology (IANR) supplement issue of Cell Transplantation.
Keywords
Introduction
Although the last decade was markedly directed toward brain neuroprotection and restoration, no novel restorative therapy was assessed in clinical practice. One of the most promising and tested new concepts of neuroregeneration is brain stem cell-based therapy, which aims to replace cell losses in injured areas (25,31,32). The main and unresolved obstacle with cell transplantation, however, is the relatively low (5–20%) rate of graft survival reported by all research groups (15,16,21,22). Despite the various designs of transplantation modes and the different sources of stem cells used, no essential improvement in long-term survival or stable replacement and integration into mammalian brain tissue has yet been achieved. Transplanted cells have been shown to migrate into the injury site (29), but their numbers were too few to elicit their reparative ability (44,45,56). Most of the transplanted cell death occurred as a result of acute immunorejection by the host tissue shortly after grafting, which was due to innate immunological reaction, oxidative and excitotoxic stress, trophic factor withdrawal, and hypoxia (14,48,57). Moreover, grafts containing dispersed cells devoid of extracellular matrix and neighboring endogenous cells died in an apoptosis-like mechanism referred to as anoikis (18). With a view to these effects being overcome, transplanted cells were delivered to a site of injury using three-dimensional (3D) constructs. These would allow the cells to create their own extracellular integrating environment and would act also as a temporary barrier to protect them from immune system attack. In addition, artificial or natural biomaterials, such as macroporous, biodegradable polymer scaffolds, act as 3D support that provide physical cues for transplanted cells, allowing positioning and spreading during their differentiation (33).
The scaffolds already investigated can be divided into two main groups: the supportive type, with cells located mainly on the surface, and the integrative type (spongeor hydrogel-like type) with cells penetrating into the core of the scaffold (26). Supportive scaffolds revealed a marked ability to create an extracellular environment for transplanted neural stem cells (26). The integrative macroporous scaffolds provided a better physical barrier, therefore protecting grafts from immune cell afflux (1). In our study, we focused on selecting the optimal bioactive skeletons combining features of both supportive and integrative scaffolds. This was followed by studies on scaffold transplantation, together with highly hypoimmunogenic human mesenchymal stem cells (hMSCs). To generate an optimal, implantable bioscaffold, we compared such different natural materials as dextran and gelatin. Dextran, a polysaccharide derived from bacteria, consists of glucose subunits and possesses antithrombotic properties especially welcome in a stroke-injured brain (3,50). Gelatin, being a derivative of collagen, is nonimmunogenic, biodegradable, and an easy accessible protein. Due to these properties, gelatin-based scaffolds have found wide applications in various areas of tissue engineering (5,12,13,58). Moreover, the gelatin structure contains typical collagen-like chemoattractant binding domains that regulate its positive influence on cellular proliferation, adhesion, and differentiation (11,53). In vitro experiments have shown that, besides better survival, the MSCs implanted onto collagen scaffolds also displayed stronger commitment to differentiate toward osteogenic and adipogenic lineages. Neuronal differentiation of MSCs seeded on 3D collagen scaffolds has also been observed (39,43). In vivo studies in experimental models of stroke, spinal cord injury, and traumatic brain injury (TBI) have shown promising restorative effects with the use of such MSC-loaded collagen scaffold implants (52).
For our experiments, we chose MSCs derived from human umbilical cord Wharton jelly (hUCMSCs). These stem cells are easily accessible from the umbilical cord matrix, possess multilineage differentiation potential, and express characteristic MSC surface proteins markers [e.g., cluster of differentiation 90 (CD90), CD73, CD44]. Furthermore, these cells have strong immunomodulating properties exerted in vitro on cell populations of both adaptive and innate immunity (17). Since the bioscaffolds containing cells are proposed to create a neurogenic niche, the scaffolds were coated with laminin (LM) (400–900 kDa), the main component of extracellular matrix in brain parenchyma. These glycoproteins contain various functional domains; among them are domains that are responsible for receptor-mediated cell attachment (6). LMs interact with cells via receptors belonging to the integrin family, which are responsible for initiating there specific signals. These highly complex downstream signaling pathways that originate at the focal adhesion sites (24) regulate complex cellular behavior such as survival, proliferation, migration, differentiation (41), neurite outgrowth, and cell adhesion (2,46). Accordingly, our studies, like others, aimed to functionalize scaffolds by using LM peptide fragments, which would be able to reproduce partial effects of the native extracellular matrix molecule.
Materials and Methods
Production of Dextran–Laminin (DL) and Gelatin–Laminin (GL) Cryogel Scaffolds
DL Scaffolds
The method of preparation of the dextran macromer (dex-MA), used in the production of dextran cryogels, has been described elsewhere (5). Dextran cryogels were prepared as follows. A degassed solution of 4 ml 5% (w/v) dex-MA (Pharmacia, Stockholm, Sweden) was cooled on ice for 15 min. Initiator system (5 μl tetramethylethylenediamine and 4 mg ammonium persulfate; Sigma-Aldrich, Stockholm, Sweden) was added, and then the solution was poured into 2-ml plastic syringes and stored overnight at −20°C. Frozen samples were thawed at room temperature and washed with deionized water, followed by 0.05 M NaOH (Sigma-Aldrich) and then a final deionized water wash. Cryogels obtained were incubated in 0.1 mg/ml LM solution (see below) before a final wash with deionized water.
LG Scaffolds
A solution of 0.01% (w/v) LM from Engelbreth–Holm–Swarm murine sarcoma basement membrane (Sigma-Aldrich) and 4.4% (w/v) cold water fish skin gelatin (Sigma-Aldrich) was incubated on ice for 15 min. Glutaraldehyde (Sigma-Aldrich) was added to the solution to final concentrations of 0.1% or 0.5% (v/v) for low and high degrees of cross-linking, respectively. This solution was stirred for 10 s; 0.5 ml of the solution was transferred quickly into each 20 × 7 mm i.d. glass tube and then closed at the bottom with a silicon cap. Solutions in tubes were frozen within 20–30 min in a cooling chamber (Arctest, Espoo, Finland) at −12°C. Samples were kept frozen at this temperature for 8 h before thawing at room temperature. Caps were removed, and the cryogels were washed by passing 10 ml of deionized water through each tube. Two milliliters of freshly prepared NaBH4 (Sigma-Aldrich) solution (0.1 M in sodium carbonate buffer, pH 9.2) was passed through each cryogel followed by extensive washing with deionized water. Cryogels were cut with a sharp blade into small cubes (2 × 2 × 2 mm) and stored in 20% ethanol at 4°C.
Scanning Electron Microscopy of Cryogel Scaffolds
Cryogel scaffolds were fixed in 2.5% glutaraldehyde in 0.1 M phosphate buffer saline (PBS; Lonza, Basel, Switzerland) at pH.7.2 overnight and then postfixed in 1% osmium tetroxide (Sigma-Aldrich) for 1 h. Samples were dehydrated in a series of ethanol washes (30%, 50%, 75%, and 99.5%) before being critical point dried. The dried samples were coated with gold/palladium (Sigma-Aldrich, Stockholm, Sweden) (40:60) and examined using a JEOL JSM-5600LV scanning electron microscope (Jeol, Sollentuna, Sweden).
Isolation and Culture of hUCMSCs
Samples of human umbilical cord tissue were obtained from the Departement de Gynecologie Obstetrique at the Centre Hospitalier St Joseph–St Luc and The Hopital Privé Natecia, Lyon, France. Cord units from both sexes were collected following the receipt of written consent from the parents. The sections of umbilical cord tissue were between 5 and 20 cm long, and the cells were not pooled so that each unit of cells was from one single donor. Tissues were placed in a sterile 50-ml tube (Corning B.V. Life Sciences, Amsterdam, The Netherlands) filled with 20 ml of sterile PBS with antibiotics (penicillin, streptomycin, and amphotericin B) (Lonza). Tissues were stored at room temperature and processed at most 48 h after the collection time. Umbilical cord sections were washed in PBS with antibiotics, and the Wharton's jelly tissue was explanted from the cord. Pieces of Wharton's jelly were placed in a six-well plate dish (Nunc Thermo Scientific, Langenselbold, Germany) in mesenchymal stem cell growth medium (MSCGM™) BulletKit® medium (Lonza), which contains a mixture of MSC basal medium supplemented with MSC growth supplement, l-glutamine, and GA-1000 antibiotic-containing serum (Lonza) or ABCellBio expansion medium (serum free) supplemented with glutamax and antibiotics (ABCellBio, Paris, France). Cultures were kept in the incubator at 37°C, 5% CO2, and 95% humidity. Cells migrating from Wharton jelly tissues were cultured in vitro until they reached 80% confluence. After expansion, hUCMSCs were cryopreserved in Cryo3 medium (Stem Alpha, St Genis L'Argentière, France) supplemented with 10% dimethyl sulfoxide (Sigma) and stored at −80°C. All experiments were performed using hUCMSCs at passage 4–5. After thawing, cells were cultured for 3–5 days to about 70–80% confluence before harvesting.
Characterization of hUCMSC Analyses
hUCMSCs cultured in 24-well plates (Nunc Thermo Scientific) were characterized by flow cytometry and immunocytochemical analyses. For flow cytometry analysis, hUCMSCs were detached from the surface by enzymatic reaction with trypsin (PAA: The cell culture company, Les Mureaux, France), resuspended in PBS, and centrifuged. For single antibody staining, 0.5 × 106 cells were used. hUCMSCs were incubated with antibodies (Table 1) for 30 min at room temperature. After incubation, cells were washed with PBS to remove the antibodies before being resuspended in 500 μl of 5% fetal calf serum (Lonza)/PBS and analyzed with a Becton Dickinson fluorescence-activated cell sorter (FACS, Canto; Becton Dickinson, Le Pont de Claix Cedex, France). Flow cytometry data analysis was performed using FACS Diva software (Becton Dickinson).
Table of Primary Antibodies Used for Immunocytochemistry and FACS Staining

First letter in the isotype description indicates host animal (M, mouse; R, rabbit; G, goat), second letter indicates polyclonal (P) or monoclonal (M) antibody, abbreviation at the end indicates immunoglobulin isotype of an antibody including isotype subtype. CD44, cluster of differentiation 44; ED1, antibody clone for CD68; GFAP, glial fibrillary acidic protein; Iba1, ionized calcium-binding adapter molecule 1; NeuN, neuronal nuclei; NF200, heavy neurofilament 200 kDa; NuMA, nuclear mitotic apparatus protein; MAP2, microtubule-associated protein 2; O4, oligodendrocyte marker O4; TUJ1, antibody clone for neuronal β III tubulin.
For immunocytochemical studies, hUCMSC cultures were washed with PBS and fixed with Accustain (Sigma-Aldrich) for 15 min at room temperature. For intracellular protein staining, cell membranes were permeabilized using a 1% Triton X-100 in PBS solution (Sigma-Aldrich) for 1 h at room temperature. Primary antibodies were diluted in PBS before incubation with cells (Table 1). Subsequently, the secondary antibodies were used against primary antibodies hosts' isotype: Alexa Fluor 488 or Alexa Fluor 594 against primary antibody at dilution 1:1,000 (Invitrogen, Warsaw, Poland). Epifluorescence images were captured by inverted microscope Eclipse Ti-S (Nikon, Warsaw, Poland) equipped with cooled digital camera DS-Ri1 (Nikon). These images were analyzed with NIS-Elements software (Nikon).
Seeding of hUCMSCs on 3D Cryogel Scaffolds
Imaging of hUCMSCs on Scaffolds
For characterization of hUCMSCs on GL scaffolds, cells were collected and seeded via pipeting 5 × 106 cells/ml into scaffolds. Seeded scaffolds were cultured for 4 days under static culture conditions [ABCell Bio expansion (serum-free) medium; 37°C, 21% O2, 95% humidity] prior to being tested for cell viability with Invitrogen Live/Dead assay containing calcein AM (viable cells) and propidium iodide (dead cells). Scaffolds were cryosectioned using a cryostat (Thermo Scientific, Langenselbold, Germany) into 60-μm slices and then analyzed using epifluorescence microscopy.
Implanted Scaffolds
hUCMSCs were collected from culture flasks by trypsinization before being counted and concentrated according to the desired seeding cell number into 500 μl of Lonza MSCGM™ BulletKit® medium (density 1 × 106 cells/ml) with 10 mM of 5-chloromethyl-fluorescein-diacetate cell tracker (CMFDA; Invitrogen) added. Cells were incubated at 37°C for 30 min and then washed twice with PBS. A small volume of cell suspension (1 ml) was centrifuged for 3 min at 120 × g with these scaffolds. During centrifugation, hUCMSCs penetrated into these 3D scaffolds through large pores (<100 μm) and resided in the medium (60–100 μm) and small (20–60 μm) pore regions, as previously described by us (28). These 3D cellular devices were transplanted immediately after centrifuging.
Organotypic Hippocampal Slice Cultures
Organotypic hippocampal slice cultures (OHCs) were prepared according to Stoppini (49). Slices were prepared from 7- to 9-day-old Wistar rats of both sexes (breeding house of the Medical Research Centre, Warsaw, Poland). Pups were sacrificed by decapitation; the hippocampi were dissected, and slices of 350 μm thickness were prepared with a McIlwain tissue chopper (The Mickle Laboratory Engineering Co., Guildford, UK). Slices were then transferred onto 0.4-μm porous Millicell membrane inserts (Millipore, Mosheim, France) and placed in a six-well plate. Cultures were maintained in 1 ml of serum-containing medium [50% Dulbecco's modified Eagle's medium (DMEM)-F12 (Gibco, Warsaw, Poland), 25% Hanks' balanced salt solution (HBSS) (Gibco), 17 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (Sigma-Aldrich, Poznan, Poland), 5 mM glucose (Sigma-Aldrich, Poznan, Poland), 1 mM l-glutamine (Biochrom, Berlin, Germany), 25% horse serum (Sigma-Aldrich, Poznan, Poland), and 0.5% gentamicin (Gibco)] at 37°C for 2–3 days. Thereafter, cultures were transferred to serum-free medium [50% DMEM-F12, 25% HBSS, 17 mM HEPES, 5 mM glucose, 1 mM l-glutamine, 25% Neurobasal-A (Gibco), 0.5% B27 (Gibco), and 0.5% gentamicin] and kept at 37°C in 5% CO2 with a medium change every second day. Scaffold transplantations were performed after 10 days. All experiments were approved by the local government commission for animal health. All possible efforts were made to minimize animal suffering and the numbers of animals used.
Model of OGD Injury to OHCs
Oxygen glucose deprivation (OGD) was performed on 10-day-old OHCs. These slices were transferred to six-well plates with 1 ml of solution containing 10 mM mannitol (Sigma-Aldrich, Poznan, Poland) saturated with 95% N2/5% CO2 and placed in an anaerobic chamber equilibrated to 37°C. Then these cultures were exposed to 40 min of 95% N2/5% CO2 gas flow. Control cultures were maintained for the same time under a normoxic atmosphere in glucose-containing solution. After OGD injury, these OHCs were returned to their normal culture conditions.
Ouabain Model of Brain Injury
Male Wistar rats of weight 250 g were used. Animals were kept in standard conditions with 12:12 photoperiod and unlimited access to water and food. All procedures were approved by the First Warsaw Local Ethics Committee and were consistent with EU guidelines. Rats were anesthetized with ketamin (Biowet, Gorzow Wielkopolski, Poland) (10%; 90 mg/kg body weight given intraperitoneally) and xylazine (Biowet) (2%; 10 mg/kg body weight given intraperitoneally), situated in a stereotactic apparatus (Stoelting, Bydgoszcz, Poland), and in the right hemisphere cranium a burr hole was drilled. Using accurate coordinates (A 0.0, L 3.0, D 5.0 mm) and a 10-μl Hamilton syringe (Hamilton, Reno, N V, USA) with 15-mm-long needle (gauge 33) (Hamilton), 1 μl of 5 mmol ouabain (Sigma) was injected into the brain (0.1 μl/min) using a microinfusion pump (Stoelting). The needle was left in situ for 5 min to avoid leakage of ouabain. After that time, the needle was removed, and the skin was closed with sutures.
Transplantation of Scaffold Supported hUCMSCs Into OHCs
The GL scaffolds (empty or filled with hUCMSCs) were cut into 0.4 μm x 0.4 μm x 1 μm samples, using a McIlwain tissue chopper, before being transplanted by micropipette, under microscope control, into the CA1 region of intact OHC slices or those 10 days after OGD. Seven days after transplantation, the hippocampal slices were fixed in 4% paraformaldehyde (PFA) (MP Biomedicals, Illkrich, France) for 45 min. After washing in PBS, free-floating slices were moved to PBS in 24-well culture plates.
Transplantation of hUCMSCs Into the Rat Brain
Adult male Wistar rats were used as the recipient. Stereotaxic surgery was performed under anesthesia with ketamin (10%; 90 mg/kg body weight given intraperitoneally) and xylazine (2%; 10 mg/kg body weight given intraperitoneally). hUCMSCs (2 × 104 in 2 μl saline) were injected with a needle (15 mm long) attached to a 10-μl Hamilton syringe into the corpus callosum, using a microinfusion pump (0.5 μl/min). The coordinates used were A 0.0, L 4.0, V 3.0. The bregma was adjusted to the same horizontal plane, and the ventral coordinates were calculated from the dura. After needle removal, the skin was repositioned and resealed.
Transplantation of Scaffold-Supported hUCMSCs Into Ouabain-Injured Rat Brain
At 3 days after ouabain brain injury, rats were anesthetized as described above and received transplantation of scaffolds. The GL scaffolds (empty or filled with hUCMSCs) were placed into the cortex of rat brain above the ouabain lesion site with ophthalmic tweezers (F.S.T., Heidelberg, Germany). Following the transplantation procedure, the scalp skin was repositioned, and the skin flap was closed with a suture (Silkam Braun, Tuttingen, Germany).
Brain Tissue Preparation and Fixation
At 7 days after the transplantation procedure, the rats were deeply anesthetized with pentobarbital (65 mg/kg intraperitoneally) (Sigma-Aldrich, Poznan, Poland). The brains were removed, immediately frozen with dry ice, and stored at −70°C. Before sectioning, brains were left at −20°C overnight. Coronal tissue sections (20 μm thick) were cut in a cryostat, mounted on superfrost microscope slides (Marienfield, Lauda, Germany), and then stored at −70°C until immunohistochemistry was performed.
Immunohistochemistry and Confocal Microscopy Analysis of Brain Tissue
For immunofluorescence analysis, the sections were air dried at room temperature (RT) for 30 min and fixed with 4% PFA in PBS (pH 7.4) for 15 min. Prior to incubation with primary Ab, nonspecific binding was blocked with normal goat serum (Life Technologies, Warsaw, Poland) or bovine serum albumin (Sigma-Aldrich, Poznan, Poland) (1:10, diluted with 0.1% Triton X-100) for 1 h. Then the primary antibodies were applied, and brain slices were incubated overnight at 4°C. For characterization of hUCMSCs, CD73 (Santa Cruz, Heidelberg, Germany), CD90 (Santa Cruz), fibronectin (DakoCytomation, Glostrup, Denmark), and vimentin (Dako, Warsaw, Poland) were used. To determine the neural fate of implanted hUCMSCs, specific antibodies were used: anti-heavy neurofilament protein (NF200; Sigma, 1:400), anti-β tubulin-III (clone TUJ1; Covance, Warsaw, Poland, 1:750), anti-microtubule-associated protein 2 (MAP-2; Sigma-Aldrich, Poznan, Poland, 1:1,000), nestin (R&D, Warsaw, Poland), and neuronal nuclei marker (NeuN, Chemicon Millipore, Warsaw, Poland) as neuronal markers; anti-glial fibrillary acidic protein (GFAP) (Cappel, MP Biomedicals, Warsaw, Poland, 1:100) and anti-S100β (Abcam, Cambridge, UK, 1:1,000) as astrocyte markers; and anti-A2B5 (ganglioside GQ1c; Chemicon, Millipore, 1:100) or anti-oligodendrocyte marker 4 (O4; Sigma-Aldrich, Poznan, Poland, 1:100) as oligodendrocyte markers. Characterization of the host immune response made use of the following antibodies: anti-CD68 (clone ED1) (Serotec, Biokom, Warsaw, Poland, 1:100) and goat polyclonal anti-ionized calcium-binding adapter molecule 1 (Iba1; Abcam, 1:500) for macrophages/microglia. Additionally, with a view to the presence of transplanted hUCMSCs in the rat brain tissue being identified, use was made of primary antibodies: anti-nuclear mitotic apparatus protein (NuMA; Calbiochem, Millipore, 1:50) against human nuclear antigens and anti-Ki67 (Leica, Novocastra, Zalesie Gorne, Poland, 1:100) as a marker of proliferating cells. Following rinsing in PBS, rat brain sections were exposed to goat anti-mouse IgG1 Alexa Fluor 546, goat anti-mouse IgG2a Alexa Fluor 546, goat anti-mouse IgG2b Alexa Fluor 546 (Invitrogen, 1:500), goat anti-mouse IgG3 Texas Red conjugated (Southern Biotechnology, Birmingham, AL, USA, 1:500), or goat anti-rabbit IgG (H+L) Alexa Fluor 546 conjugate (Invitrogen, 1:500) secondary antibody, for 60 min at RT in the dark. Cell nuclei were stained with 5 μM Hoechst 33258 (Sigma-Aldrich, Poznan, Poland). The adjacent sections were used as negative controls. All procedures for negative controls were processed in the same manner, except that primary antibodies were omitted. A confocal laser scanning microscope (Zeiss LSM 510; Poznań, Poland) was used to obtain detailed images of the positively stained cells. A helium–neon laser (543 nm) was used for excitation of Alexa Fluor 546. An argon laser (488 nm) was used for the excitation of CMFDA, and a diode of 405 nm was used to excite Hoechst. Following acquisition, images were processed using the ZEN 2008 software package (Zeiss).
RNA Isolation, Reverse Transcription, and qRT-PCR Method
Total RNA isolated after removal of the scaffolds from the OHCs was extracted using TRIzol reagent (Invitrogen). Contaminating genomic DNA was eliminated by DNase (Qiagen, Hamburg, Germany) digestion according to the manufacturer's instructions. The extracted RNA was quantified by reading absorbance at 260 nm, and the purity was evaluated from 260/280 absorbance ratio. One microgram of RNA sample was used for cDNA preparation, using High Capacity RNA-to-cDNA kit (Applied Biosystems, Life Technologies, Warsaw, Poland). Real-time qPCR analyses were performed in an ABI Prism 7500 Sequence Detection System using 50 ng of cDNA, designed specific primers, and SYBR Green PCR Master Mix (Applied Biosystems). Reaction parameters were as follows: 2 min at 50°C, 10 min at 95°C, 40 cycles of 15 s at 95°C, and 1 min at 60°C. The dissociation curve was plotted to determine the specificity of amplification. The fluorescent signals from specific transcripts were normalized against that of the β-actin gene, and threshold cycle values (ΔCt) were quantified as fold changes by the 2-ΔΔCt method (33). The primers used were F: TCCGCAAGAGACTTCCAGCCA and R: AGCCTCCGACTTGTGAAGTGGT for interleukin-6 (IL-6) transcript detection and F: TTCTACAATGAGCT GCGTGTG and R: GGGGTGTTGAAGGTCTCAAA for β-actin. All procedures applied in performing and reporting qRT-PCR were provided according to the MIQE guidelines. Statistical analysis of the raw data was provided by one-way ANOVA followed by Bonferroni's multiple comparison test. The values were considered as significant when p < 0.05. Data were presented as mean±SEM.
In Situ Zymography
In order to localize activity of matrix metalloproteinase-2 and -9 (MMP-2 and MMP-9), we conducted in situ zymography in the brain sections. Frozen nonfixed coronal brain sections (20 μm thick) were thawed and incubated for 3 h at 37°C in a dark chamber in 100 ml of reaction buffer containing 100 mg/ml of FITC-labeled DQ-gelatin (Molecular Probes, Eugene, OR, USA), which was quenched intramolecularly. Gelatin-FITC cleavage by tissue gelatinases releases peptides whose fluorescence is representative of net proteolytic activity. Sections were washed with PBS, and samples were fixed in cold 4% PFA for 30 min. Samples were then mounted in Mowiol (Sigma-Aldrich, Poznan, Poland) and analyzed using fluorescence microscopy.
Results
Characterization of hUCMSCs Prior to Scaffold Encapsulation
hUCMSCs started to migrate out of Wharton jelly pieces at 3–14 days in vitro, depending on the particular cord unit, and then proliferated extensively as plastic adherent entities until 8–10 cell passages (Fig. 1A). FACS analysis of these cells revealed the positive expression of CD44, CD73, and CD90; the main mesenchymal markers; and lack of CD45 and CD133 hematopoetic lineage markers (Fig. 1B–F). We found also the expression of some markers characteristic for other tissue-specific lineage, that is, nestin and β tubulin-III (Fig. 1J, K), although hMSCs maintained in the expansion medium did not reveal the markers of more advanced neuronal differentiation like NF200 or MAP-2 (Fig. 1L, M).
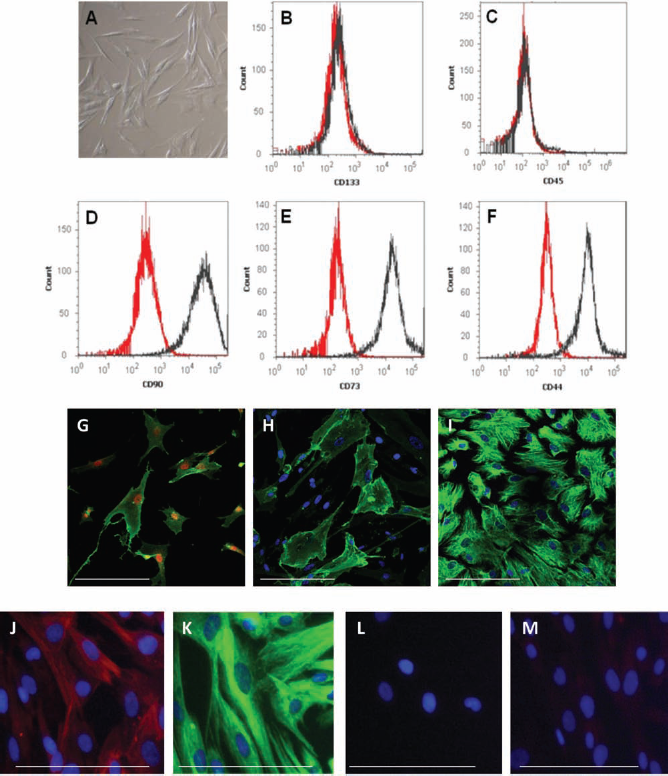
hUCMSC flow cytometry analysis of mesenchymal, hematopoietic, and neuroectodermal marker expression. Naive human umbilical cord-derived mesenchymal stem cells (hUCMSCs) cultured in serum-free medium revealed a fibroblast-like morphology (A). Representative fluorescence-activated cell sorting (FACS) analysis shows hUCMSCs negative for hematopoietic lineage markers: cluster of differentiation 45 (CD45) and CD133 (B and C, respectively) and positive for mesenchymal cell markers: CD90, CD73, and CD44 (D, E, and F, respectively). FACS data are presented as overlay of positive labeling (red) and negative labeling (isotype control) (black). Additionally, using immunocytochemical analysis, the positive expression of mesenchymal [CD73 (G green), CD90 (H green), vimentin (I green)] and neuroepithelial [nestin (J red) and β tubulin-III (K green)] markers were verified. More advanced neuronal markers [i.e., MAP2 (L, red) and NeuN (M, red)] were not detected; cell nuclei costained with Hoechst (blue; H–L) or NuMa (red; G). Scale bar: 50 μm.
Cryogel Scaffold Structure
All the cryogel scaffolds obtained had elastic spongy properties and highly porous structures with a pore size distribution in the range of 20–160 μm, as was demonstrated by scanning electron microscopy. The pore size has been optimized by us previously for human stem cell adhesion and neural differentiation (34). However, mean pore size was larger in gelatin cryogels than in dextran cryogels (Fig. 2A, B). Gelatin-based cryogels cross-linked with different concentrations of glutaraldehyde have similar microstructure (porosity, mean pore size, and wall thickness) but differ significantly in mechanical properties and in the rate of in vitro degradation (8). Densely cross-linked GL cryogels (prepared with 0.5% glutaraldehyde) were more stable mechanically and are characterized by an 80-fold slower rate of degradation in vitro than weakly cross-linked GL cryogels (prepared with 0.1% glutaraldehyde), which are completely solubilized in 0.025% trypsin/EDTA within 5–10 min (27).
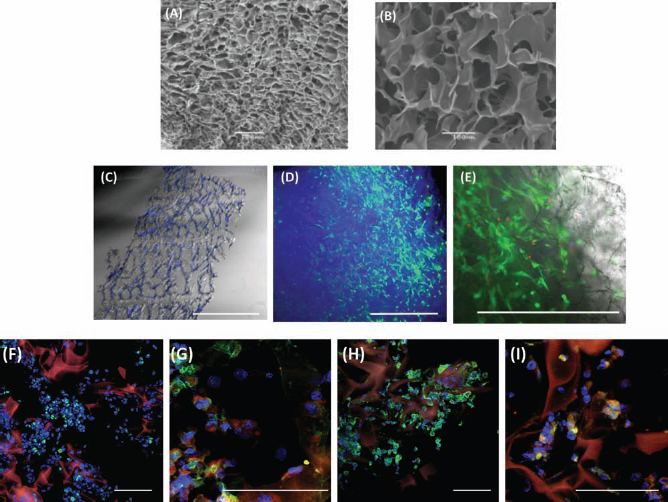
3D Protista gelatin–laminin cryogel scaffolds used for culturing of cryopreserved hUCMSCs. Scanning electron micrographs of cryogels composed with dextran–laminin (DL) (A) and densely cross-linked gelatin–laminin (GL) (B). Micrograph of the 60-μm cryosection of scaffold after 4 days with 5 × 106 cells/ml in static culture with ABCell Bio expansion (serum-free) medium (cells stained with Hoechst) (C). Cells showed approximately 75% cell viability in the scaffold when cultured with ABCell Bio expansion (serum-free) medium (D) 4x and (E) 10x. Living cells were stained with calcein AM (green, E), dead cells were stained with ethidium (red, E), and total cell nuclei were stained with Hoechst (blue). According to the images of sections (F–I), hUCMSCs appeared to occupy all available spaces after they had penetrated into sponge porosity. Cell aggregates inside the scaffolds were stained positively with Ki67 (F), nestin (green), and GFAP (red) (G). During in vitro culture in the presence of morphogens, hUCMSCs present inside the scaffolds started to express markers of immature neuroblasts NF200 (green), mature neurons MAP2 (red) (H), and astrocytes S100-β (green) (I). Scale bar: 100 μm.
3D Cryogel Scaffolds Support Neural Induction of hUCMSCs
These hUCMSCs were cultured on GL cryogels for 2 weeks to reveal if hUCMSCs could survive and proliferate within such skeletons. As a result, serial observations under an inverted fluorescence microscope have been made. The hUCMSCs seeded into the GL scaffolds showed approximately 70–80% cell viability. According to the images of the sections, hUCMSCs appeared to occupy all available spaces evenly, after they had penetrated into the sponge pores. Immediately after transplantation, the cells were suspended evenly in the internal spaces of the scaffold and were stained positively with proliferation marker Ki67, nestin, and GFAP. After 1 week of culture, some cells appeared to form multicellular 3D aggregates (niches) of spherical shape. These aggregates seemed to be anchored in the center of the scaffold. They expressed typical markers of immature neuroblasts (e.g., NF200). After 2 weeks, we observed markers characteristic for mature neurons and glial cells (MAP2 and S100β, respectively) (Fig. 2).
GL Scaffolds Provoke Lower Inflammatory Response of Brain Tissue Than Transplanted Ex Vivo DL Scaffolds
To investigate the brain tissue response for a xenograft, we used a model of rat OHCs. This model is presumably free of systemic immunological influence and permits the study of short-term interaction of transplanted material with host tissue, without affecting this process by the presence of circulating blood cells. Seven days after GL or DL scaffold transplantation, a local immune response was detected in the brain tissue. Immunofluorescence analysis revealed macrophage (ED1+) and astrocyte (GFAP+) activation around and inside the transplanted 3D scaffolds. The reaction was significantly stronger after DL than after GL scaffold transplantation (Fig. 3A–D). Accordingly, in the host tissue around the DL graft, many cells were necrotic or apoptotic (results not shown), whereas around the GL scaffold hippocampal tissue seemed to be intact. Only a few inflammatory cells were detected around the graft.

Ex vivo tissue response following scaffold transplantation into rat organotypic slice culture (OHC). After 7 days of empty scaffold coculture with rat tissue, OHC slices were immunostained with CD68 (clone ED1) or glial fibrillary acidic protein (GFAP) antibodies and analyzed using a confocal laser scanning microscope. Immunofluorescence analyses revealed that a DL scaffold (A, B), compared with a GL scaffold (C, D), evoked a significant increase in macrophage and astrocyte numbers [expressing ED1 (A, C) or GFAP (B, D); green] occurring in the host tissue close to the scaffolds (area defined by dotted lines). In the next step, the tissue response following transplantation of gelatin–laminin scaffolds encapsulated with hUCMSCs into an OGD OHC was examined. Analyses of immunostaining after 7 days revealed a significant reduction in ED1 (E, H; red), GFAP (F, I; green), and ionized calcium-binding adapter molecule 1 (Iba1) (G, J; green)-positive cells within the hUCMSC population present in 3D implants (H–J) in comparison with untreated OGD-injured tissue (scaffold autofluorescence in orange) (E–G). Cell nuclei were stained with Hoechst (blue). Scale bar: 200 μm.
Anti-inflammatory Effect of Scaffold-Supported hUCMSCs Following Their Transplantation Into an OHC Model of the Injured Brain
hUCMSC bioscaffolds revealed relatively low immunogenic properties and additionally suppressed neuroinflammatory response in injured brain tissue. Astrogliosis (cells expressing GFAP), usually observed in OGD-injured hippocampal tissue (Fig. 3F), was significantly less intense when the tissue was exposed to hUCMSCs encapsulated in these scaffolds (Fig. 3I). OGD-induced microglia/macrophage activation as revealed by ED1, Iba1 expression (Fig. 3E–G) was diminished by transplanted scaffold-based hUCMSCs (Fig. 3H–J). A significant reduction in GFAP-, ED1-, and Iba1-positive cells was observed not only around transplanted 3D grafts but also in the more distant hippocampal CA1 region, which is most sensitive to OGD. To investigate further the anti-inflammatory effect of hUCMSC transplantation, the expression of IL-6 was examined at mRNA level in OHC slices. qRT-PCR revealed that administration of hUCMSCs encapsulated in GL scaffolds significantly decreased IL-6 mRNA level in brain tissue samples since the level of IL-6 was doubled after DL scaffold compared with GL scaffold transplantation (12.79 and 6.41, respectively) (Fig. 4). In the light of the results obtained, GL scaffolds were chosen for the further in vitro and in vivo experiments.

RT-PCR analysis of IL-6 mRNA level in oxygen–glucose-deprived organotypic slices following empty or hUCMSC-seeded scaffold transplantation. Data are presented as mean ± SEM and considered as significant when p < 0.05. IL-6, interleukin 6.
Anti-inflammatory Effect of Scaffold-Supported hUCMSCs Following Their Transplantation Into the Rat Brain
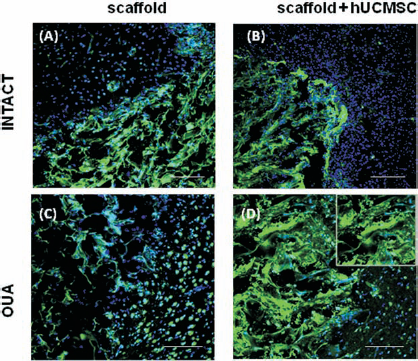
Studies were performed to reveal if GL scaffolds promoted the survival of transplanted hUCMSCs and what their influence on the brain tissue was. Two different transplantation materials were used: (1) hUCMSCs and (2) hUCMSCs encapsulated within GL scaffolds. Transplantation of hUCMSCs within the GL scaffolds improved cell survival significantly after their transplantation (Fig. 5). Most hUCMSCs remained undifferentiated 14 days after their transplantation into the ouabain-injured rat brain. A few hUCMSCs present inside the scaffolds expressed the astrocyte marker GFAP or A2B5 and O4 markers characteristic for oligodendrocyte precursor cells (Fig. 5H, K, L). Implanted scaffolds filled with hUCMSCs modified the local brain environment. There was a significant decrease in activated microglia/macrophages that infiltrated the graft site, as indicated by ED1 staining in comparison with empty scaffold transplantation. Additionally, we observed a distinct zone around the transplanted bioscaffolds, in which activated microglia/macrophages appeared to be excluded (Fig. 5A–C, G, H). However, peri-infarct reactive astrocytosis, revealed by increased GFAP immunostaining, was similar following transplantation of empty scaffolds or those filled with hUCMSCs (Fig. 5D, F). In addition, we evaluated the proteolytic activity of MMP-9 and MMP-2 enzymes by means of in situ zymography in both intact and ouabain-injured rat brains following grafting of GL scaffolds versus GL scaffolds filled with hUCMSCs (Fig. 6). In the injured rat tissue, MMP-connected activities were additionally enhanced by the transplantation of hUCMSCs encapsulated within GL scaffolds. Microphotographs showed activation of MMP-9/-2 in the injured cerebral hemisphere mainly between the cells surrounding scaffolds as well as inside the scaffold (Fig. 6D).

Microglia and astrocytic activation in the rat brain following transplantation of GL empty scaffolds or encapsulated with hUCMSCs. Empty scaffolds (A, D), hUCMSCs [labeled with 5-chloromethyl-fluorescein-diacetate cell tracker (CMFDA)] in suspension (B, E), or 3D aggregates with hUCMSCs (C, F) were transplanted into the intact rat brain. A significant immune (ED1) and glia (GFAP) response was observed mainly from the hUCMSCs injected in suspension (labeled with CMFDA and stained with NuMa, green) leading to cell death (yellow; B, E). This reaction decreased when the hUCMSCs were within transplanted scaffolds (C, F). The transplanted cells occupy the scaffold as well as neighboring host tissue. In the next step, the activation of microglia and astroglia in the injured rat brain after transplantation of GL empty (G, I) or hUCMSC (stained green with NuMa)-encapsulated scaffolds (H, J) is shown. Immunostaining analyses of injured rat brain tissue 7 days after hUCMSC scaffold transplantation revealed a reduced number of ED1 and GFAP-positive cells (red) in comparison with the effect of empty scaffold. Additionally, GL skeletons provided the structural support for the survival of transplanted hUCMSCs and their further differentiation toward microglia: astrocytes (J) and oligodendrocytes (K, L). Cell nuclei were counterstained with Hoechst (blue). Scale bar: 100 μm.
Activation of MMP-9/MMP-2 in the rat brain following transplantation of GL empty (A, C) or hUCMSC-encapsulated (B, D) scaffolds. The activity of matrix metalloproteinase-9 (MMP-9) and MMP-2 (green) was evaluated by in situ zymography 7 days after scaffold transplantation. In the injured rat tissue (C, D), the activation of metalloproteinase was greater than in the intact tissue (A, B). This activation was further enhanced by scaffold-hUCMSCs grafts in surrounding rat brain tissue (B, D). Cell nuclei were stained with Hoechst (blue). Scale bar: 200 μm.
Scaffold Degradation Following Transplantation Into the Injured Rat Brain
One of the most important properties of GL scaffolds is their spontaneous biodegradation. At day 7 following transplantation of GL scaffolds with hUCMSCs into the injured rat brain, we observed slow scaffold degradation. What is particularly important is that this was without any additional immune response or exacerbation or cytotoxic effect (Fig. 5). Ten days after transplantation, in the place of the graft, only transplanted cells coexpressing CD73and CD90 (markers characteristic for immature mesenchymal stem cells) were found. The degraded scaffold structure was replaced by extracellular fibronectin (Fig. 7).
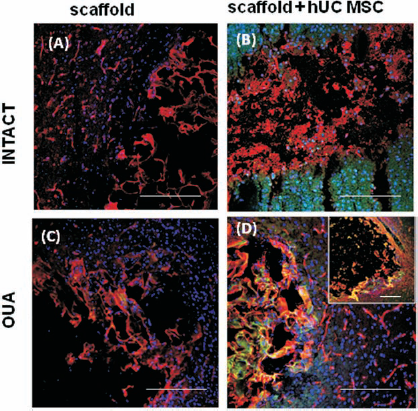
Scaffold degradation following their transplantation into intact (A, B) or injured (C, D) rat brain. Ten days after transplantation in the place of scaffold grafting, a cavity filled by amorphic debris staining with fibronectin (A, B; red) was observed. Few hUCMSCs coexpressing CD73 and CD90 (green) were found (B, D inset) in this area. Cell nuclei were stained with Hoechst (blue). Scale bar: 200 μm.
Discussion
Tissue-engineered implants made of 3D bioactive scaffolds and stem cells are very promising tools for treating many neurodegenerative diseases, given their success in preclinical studies (39,41,42,50). Biocompatibility of the implantable devices with brain tissue and stem cells used for therapy is the main challenge of tissue engineering. Here we describe the brain-compatible GL bioactive cryogels seeded with hUCMSCs derived from Wharton jelly tissue. The cryogels used contained gelatin (collagen) and LM, the components characteristic for the brain extracellular matrix. Such implants are biocompatible with the central nervous system microenvironment, are neutral immunologically, and share advantages of both the supportive and integrative types of scaffolds described in the introduction (1,26). Clinical compliance of tissue engineering therapy requires a minimal manipulation of biological material in vitro. To meet this criterion, we introduced naive hUCMSCs into the scaffolds by centrifugation or direct injection just prior to transplantation into the brain tissue. This technique reduces the potential risk of contamination and cost of GL tissue-engineered cultures significantly. We have observed that hUCMSCs within GL scaffolds were viable following transplantation into the OHC or rat brain tissue, due to the protective effect of the gelatin skeleton. It is worth stressing here that transplantation of neuronally induced cells may be necessary in some cases to obtain the desired therapeutic effect. In that case, in vitro neuronal induction of encapsulated hUCMSCs will be necessary in static or bioreactor systems. We have shown here that hUCMSCs seeded into the GL scaffold survived and created a 3D cellular network in vitro and in vivo. That was possible due to optimal pore size for human stem cells in the range of 20–160 μm. The hUCMSCs seeded into the scaffold penetrated deep into the core of the sponge structure through the large pore network and became anchored in the small pore niches. Previous results showed that an optimal range of scaffold porosity should resemble the average size of an adult human stem cell (approximately 20 μm). In the case of large mesenchymal stem cells, this range is 30–50 μm (30,37). Such scaffolds with pore sizes of 30–60 μm were shown to promote neuron differentiation and enhance cell proliferation (27).
Functional regeneration of damaged brain tissue requires delivery of stem cells capable of time-locked proliferation, migration, and ultimately neuronal differentiation (23). We have shown here a high proliferation for cells transplanted into bioactive scaffolds. Moreover, a proportion of these cells differentiated, over a relatively short period of time (7 days), into potentially young neural cells (oligoprecursors, A2B5 or neuroblasts, NF200) in vitro. This neurogenic effect correlated with the neuroepithelial characteristic of mesenchymal stem cells described by Takashima and colleagues (51). To support neural differentiation of hUCMSCs, the scaffolds were additionally covered with LM. This extracellular matrix molecule was described to increase neuronal cell proliferation and to reduce in vitro apoptosis among fetal neuronal stem cells and adult neurons (19). Moreover, the scaffolds used in our experiments created a mechanical barrier protecting cells from immune cell afflux. We observed an accumulation of inflammatory cells around the graft, both in vitro and in vivo, in the intact and injured tissue. It is worth noting that only a few microglia/macrophages infiltrated inside the scaffold, suggesting a strong inhibitory effect of the GL bioactive scaffolds. The shield provided by the GL scaffold against the immune system resulted in better survival of transplanted cells, as opposed to unsupported cells, which were destroyed within 72 h of transplantation. Similar observations have been published recently by the group of Delcroix (10).
As important as the modeling of optimal scaffolds is the proper choice of transplanted stem cells. hUCMSCs derived from Wharton jelly were chosen because of their unique features and possible future autologous transplantation and also, as mentioned before, for overcoming the availability, immunological-related, and ethical concerns around the use of stem cells (54). Moreover, these cells may favor less tumor or teratoma progression compared with embryonic stem cells or induced pluripotent stem cells. hMSCs derived from Wharton's jelly are considered a potential alternative to cells from bone marrow, on account of their ease of isolation, higher cell expansion potential, and the greater accessibility of noninvasive clinical sampling. It is worth mentioning that alternative MSC sources can be found in adult humans in adipose (fat) tissue and dental pulp (55). hUCMSCs would also seem optimal for clinical practice because of their low immunogenic and anti-inflammatory properties. Previous studies in animal models of different organ injuries have suggested MSCs' ability to modulate inflammatory response of damaged tissue (4,20,47). In our studies, we have shown the reduction of neuroinflammatory response in the injured brain tissue following grafting of the scaffolds seeded with cells compared with empty skeleton transplantation. It is worth stressing that the strong anti-inflammatory effect of MSCs was also increased as a result of the low immunogenic properties of the bioactive materials used. We managed to match here the unique properties of the biomaterial and cellular components of a tissue-engineered implant to maximize the immunomodulatory effect in vivo. Owing to such a strong property of immune modulation and neuroepithelial commitment, the hUCMSCs ought to be considered a double-edged sword where brain injuries are concerned, not only bringing about potential neural tissue repair but also attenuating inflammation or adverse inflammatory reactions in an allogenic setting. Our study additionally infers that, following hUCMSC/GL scaffold transplantation into the rat brain, hUCMSCs and surrounding tissue are characterized by significant activation of MMP-2 and MMP-9. A similar observation was also described by the Blouch group, which connected the degradation of the cell basement membrane via activation of MMPs in MSCs with their migration properties. Recently, De Becker et al. demonstrated a functional involvement of MMP-2 in MSC homing through bone marrow endothelium (9). The marked capacity for MMP activation evoked in MSCs could also be responsible for their fate after transplantation. MMPs are described to regulate cell proliferation, facilitating the interaction between growth-suppressive or mitogenic factors with their cell surface receptors (7,36). Modifying the extracellular matrix directly, MMPs can obstruct or bring about the exposure of adhesion sites, therefore influencing cell attachment and migration, while also being capable of modulating cell migration through the release or inactivation of chemotactic signals (7,36,38). The involvement of MMPs in the differentiation of MSCs into a neural lineage has also been reported (35). The induction of MMP-2/-9 activity is probably also responsible for the fast (after 10 days) biodegradation of transplanted scaffolds as well as the remodeling of tissue at the site of transplantation. Similar observations were made by the Montero-Menei group, who transplanted a cylindrical collagen scaffold seeded with 3 × 106 hMSCs into the lesion cavity 4 days after TBI. The scaffold was readily degradable and was already not detectable 35 days after TBI. At that time, a reduction in lesion volume and enhanced migration of hMSCs within the parenchyma together with improvement of sensory motor function and spatial learning were observed (20). Nevertheless, the fast scaffold degradation observed in our and other experiments did not result in edema or cytotoxity induced by the scaffold-secreted components. Moreover, implanted cells remained viable even after scaffold biodegradation, with no signs of immune system reactivation. In conclusion, a proper combination of tissue engineering and cell therapy seems to be crucial for future therapeutic success in brain injury regeneration where a substantial loss of neural tissue is observed. Nevertheless, there remains a need for better understanding of the early cell death mechanisms after transplantation (lack of oxygen, cell necrosis or apoptosis, shear stress, or deprivation of trophic factors) as well as for deciphering more of the basic phenomena connected with MSC biology.
Footnotes
Acknowledgments
This work was supported by the Novous Sanguis, Jerome Lejeune Foundation Grant, and the National Science Centre for grant No. 05728/B/NZ4/2011/01 and No. 2011/01/B/NZ3/05401. The authors declare no conflict of interest.