
Abstract
Human bone marrow stem cell populations have been applied for cardiac regeneration purposes within different clinical settings in the recent past. The migratory capacity of applied stem cell populations towards injured tissue, after undergoing specific peri-interventional harvesting and isolation procedures, represents a key factor limiting therapeutic efficacy. We therefore aimed at analyzing the migratory capacity of human cluster of differentiation (CD) 133+ bone marrow stem cells in vivo after intraoperative harvesting from the sternal bone marrow. Human CD133+ bone marrow stem cells were isolated from the sternal bone marrow of patients undergoing cardiac surgery at our institution. Migratory capacity towards stromal cell-derived factor-1α (SDF-1α) gradients was tested in vitro and in vivo by intravital fluoresecence microscopy, utilizing the cremaster muscle model in severe combined immunodeficient (SCID) mice and analyzing CD133+ cell interaction with the local endothelium. Furthermore, the role of a local inflammatory stimulus for CD133+ cell interaction with the endothelium was studied. In order to describe endothelial response upon chemokine stimulation laser scanning microscopy of histological cremaster muscle samples was performed. SDF-1α alone was capable to induce relevant early CD133+ cell interaction with the endothelium, indicated by the percentage of rolling CD133+ cells (45.9 ± 1.8% in “SDF-1” vs. 17.7 ± 2.7% in “control,” p < 0.001) and the significantly reduced rolling velocity after SDF-1α treatment. Furthermore, SDF-1α induced firm endothelial adhesion of CD133+ cells in vivo. Firm endothelial adhesion, however, was significantly enhanced by additional inflammatory stimulation with tumor necrosis factor-α (TNF-α) (27.9 ± 4.3 cells/mm2 in “SDF-1 + TNF” vs. 2.2 ± 1.1 cells/mm2 in “control,” p < 0.001). CD133+ bone marrow stem cells exhibit sufficient in vivo homing towards SDF-1α gradients in an inflammatory microenvironment after undergoing standardized intraoperative harvesting and isolation from the sternal bone marrow.
Keywords
Introduction
The application of bone marrow stem cells for cardiac regeneration purposes has been introduced within clinical trial protocols in the last decade. Catheter-based interventional (2) as well as surgical intramyocardial (5) delivery routes have been described. However, despite comparably elaborated clinical cell therapy protocols, underlying mechanisms responsible for the reported beneficial functional effects of cardiac stem cell application remain unclear. Especially, the molecular interplay, regulating stem cell trafficking to injured tissue, still needs further clarification. Although the importance of chemokines and local inflammation for successful targeted cell migration has been shown in the recent past for murine stem cell populations (6,9,16), few data describe the in vivo migratory properties of human bone marrow stem cell subpopulations already applied within running clinical trials. A better understanding of these properties, however, seems crucial, since the capability to migrate towards specific chemokine gradients is likely to represent one key factor limiting regenerative capacity of bone marrow cell subpopulations isolated for therapeutic applications (8). Intramyocardial application of autologous cluster of differentiation (CD) 133+ bone marrow stem cells (BMSCs) in combination with coronary artery bypass grafting (CABG) has been established at our institution for the treatment of chronic ischemic heart disease (20). In the current study, we aimed to characterize this specific subpopulation regarding its migratory capacity towards chemokine gradients in vivo. Based on the crucial relevance of stromal cell-derived factor-1α (SDF-1α)/C-X-C chemokine receptor type 4 (CXCR-4) axis for human bone marrow stem cells trafficking (24), we analyzed CXCR-4 expression of human CD133+ stem cells isolated from sternal bone marrow and tested their migratory potential in vitro. Intravital fluroscence microscopy in a xenogeneic mouse model enabled us to investigate the CD133+ cell's capability to interact with peripheral endothelial cells in vivo upon local SDF-1α stimulation. Since our previous studies have demonstrated the importance of local tumor necrosis factor-α (TNF-α)-induced inflammation for SDF-1α-mediated peripheral endothelial adhesion of c-kit+ bone marrow stem cells (9), we tested the hypothesis that human CD133+ also relay on a local inflammatory stimulus for sufficient interaction with the endothelium. Owing to the known relevance of CXCR-4 for SDF-1α-mediated stem cell migration, we quantified its endothelial expression at basal level as well as upon chemokine stimulation.
Materials and Methods
Bone Marrow Aspiration
Bone marrow was harvested by sternal aspiration from informed donors giving written consent after approval by the local authorities and in conformity with the Declaration of Helsinki. All donors were patients undergoing cardiac surgery (age 67 ± 8 years, 69% male) including median sternotomy at Rostock University Hospital, Rostock, Germany. The incidence of major cardiovascular risk factors is described in Table 1 and does not show any unexpected distribution for the major risk factors. Sternal aspiration was performed immediately before median sternotomy using a 14-gauge needle (Zamar, Suzzara, Italy).
Baseline Cardiovascular Risk Factors Within the Cell Donor Cohort

Data are expressed as percentage of all patients. NYHA, New York Heart Association score.
Isolation of CD133+ Cells
Mononuclear cells were isolated from bone marrow by overlayering onto lymphocyte separation medium (LSM) 1077 (PAA, Linz, Austria) and density gradient centrifugation (Eppendorf, Hamburg, Germany). The magnetic-activated cell sorting (MACS) cell separation system (Miltenyi Biotec, Bergisch Gladbach, Germany) was employed to purify CD133+ cells via positive magnetic selection.
Fluorescence-Activated Cell Sorting Analysis (FACS)
Reagents used for FACS analysis were obtained from Miltenyi Biotec [anti-CD133-phycoerythrin (PE) (293C2), anti-CD34-fluorescein isothiocyanate (FITC)], and BD Biosciences [Schwechat, Austria; anti-CD45-allophycocyanin-H7 (APC-H7), anti-CXCR-4-APC, and 7-aminoactinomycin (7-AAD)].
Cells were suspended in buffer containing 0.5% bovine serum albumin. To reduce unspecific binding, FcR blocking reagent (Miltenyi Biotec) was added to all samples. Cells were incubated with the antibodies for 10 min in the dark at 4°C and analyzed by LSR-II flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA) employing FACSDiva software (Becton Dickinson). Compensation was established using single stained controls and gating was performed with matched isotype/fluorescence minus one (FMO) controls. The Boolean gating strategy was modeled on the International Society of Hematotherapy and Graft Engineering (ISHAGE) guidelines for CD34+ cell analysis.
In Vitro Migration of Isolated CD133+ Cells Against SDF-1 (Boyden Chamber)
In vitro migration assays were performed in transwell plates (Boyden chamber; pore size 5.0 μm, 96-well plate) (Corning Inc., Life Sciences, Lowell, USA). The transwell membrane was coated with 100 ng/ml fibronectin (Sigma-Aldrich, Munich, Germany) in phosphate-buffered saline (PBS; PAN Biotech) for 1 h at room temperature, then washed thoroughly with PBS. CD133+ cells (5×104) were seeded in 30 μl StemSpan medium (Stem Cell Technologies, Grenoble, France) into the upper compartment, whereas the lower compartment was filled with 300 μl of medium containing 100 ng/ml human recombinant SDF-1 (PAN Biotech, Aidenbach, Germany) or with medium alone. After incubation at 37°C for 15 h, migrated cells were counted microscopically. Assays were performed in duplicates, six independent experiments were performed.
Intravital Fluorescence Microscopy
All animal experiments were carried out according to the US National Institutes of Health guidelines for the care and use of and were permitted by the local animal care and use committee. Male severe combined immunodeficient (SCID) mice (C.B.–17 Icr-Prkdcscid/IcrIco Crl, Charles River, Sulzfeld, Germany) weighing 20–25 g (n = 18) were assigned to intravital fluorescence microscopy analysis. The mice underwent surgical procedure and topical administration of either 200 μl SDF-1α (200 ng/ml in PBS; PAN Biotech) in the “SDF-1α” group (n = 6), TNF-α (2,000 units; R&D Systems, Minneapolis, MN, USA) in the “TNF-α” group (n = 6), or 200 μl of SDF-1α (200 ng/ml in PBS) and TNF-α (2000 units) in the “SDF-1α + TNFα” group (n = 6). Control animals underwent topical treatment with 200 μl of PBS in the “Control” group (n = 6). All in vivo experiments were performed on the day of intraoperative CD133+ harvesting.
Mice were anesthetized with ketamine (75 mg/kg; Pfizer, Berlin, Germany) and xylazine (25 mg/kg; Bayer, Leverkusen, Germany). An arterial catheter was inserted retrograde into the left femoral artery to establish the injection route for fluorescent reagents and labeled CD133+ cells. The right cremaster muscle was dissected and prepared for intravital fluorescence microscopic analysis as previously described (3,22). For intravital microscopy, human CD133+ cells obtained from sternal bone marrow were fluorescently labeled with the carboxy-fluorescein diacetate, succinimidyl ester (CFDA SE) (Molecular Probes, Eugene, OR, USA) according to the manufacturer's protocol. Rhodamine 6G (Sigma-Aldrich) was used for background visualization. Intravital microscopy was performed using an Axiotech fluorescence microscope (Carl Zeiss, Jena, Germany), modified for epi-illumination and connected to a charge-coupled device (CCD) video camera. Six postcapillary venules were randomly chosen before cell injection to define the areas of cellular adherence analysis. With respect to the groups, the cremaster muscles were exposed to SDF-1α, SDF-1α + TNF-α, or PBS 15 min prior to the first CD133+ cell injection. Human CD133+ bone marrow stem cells were applied with a total number of 0.4 × 106 CD133+ cells using five consecutive injections. CD133+ cell behavior along the endothelial lining was considered as “rolling” when a more than 50% reduction of cell velocity in combination with the typical cellular “stick and release” motion was present. The rolling cells were expressed as percentage of all passing cells along a predefined venular distance during 1 min of observation. All the cells that showed random brief tethering phenomena were not considered as rolling cells. Furthermore, specific CD133+ rolling velocity was measured upon SDF-1α stimulation. Firm adhesion was recorded when cells did not move for more than 30 s. The number of firmly adherent c-kit+ cells was calculated in relation to the endothelial surface of the predefined venules (diameter × length × p) and was expressed as adherent cells/mm2 endothelial surface.
In order to provide similar microcirculatory conditions within the experimental groups, several parameters were measured and taken into careful consideration. Red blood cell velocity profile was verified using the line shift method on intravital microscopy recordings (CapImage Software, Zeintl, Heidelberg, Germany). The analyses of micro circulation also included the measurement of vessel diameter and wall shear rates based on the Newtonian definition γ = 8 × v/d, where v represents the red blood cell velocity divided by 1.6 according to the Baker–Wayland factor (4) and d represents the single vessel diameter.
Following intravital microscopic analysis, mice were sacrificed, and their cremaster muscles were snap-frozen in liquid nitrogen for histological examination.
Laser Scanning Microscopy and CXCR-4 Expression Quantification
Frozen transverse tissue sections of cremaster muscles were cut at 5–10 μm thickness.
For visualization and subsequent signal intensity quantification of CXCR-4 cremaster muscle sections were incubated with a primary antibody [rat anti-mouse CD184 (CXCR-4) Clone 2B11, eBioscience, San Diego, CA, USA; 1:100] and a secondary antibody (donkey anti-rat IgG, Molecular Probes; Alexa Fluor 488, 1:1,000) according to manufacturer's protocol. For nuclear staining, samples were incubated with 4,6-diamidino-2phenylindol- dihydrochloride (DAPI; Molecular Probes) for 10 min, washed intensively and mounted in FluorSave” (Calbiochem, Torrey Pines, CA, USA). For signal intensity quantification a Laser scanning microscope (LSM) (780 ELYRA PS.1 microscope, Carl Zeiss) was applied, using a specific algorithm for intensity calculations. In LSM mode Alexa Fluor 488 was excited using 488 nm laser and recorded with the 510–550 nm emission filter [main dichroic beam splitter (MBS) 488]. The excitation of DAPI was achieved using 405 nm laser and recorded with 415–470 nm emission filter (MBS 405). Transmitted light images were collected simultaneously with the excitation of DAPI, using 405 nm laser and photomultiplier for transmitted light. All experiments were performed utilizing 63× Plan Apochromat objective [numerical aperture (NA) = 1.4] with immersion oil (Immersol” 518F, Carl Zeiss). For reliable quantification, all acquisition settings were kept constant for all analyzed groups of samples.
Images were acquired as z-stacks, consisting of six optical sections with ~1 μm each. Afterwards, maximum intensity projections of acquired z-stacks were formed and analyzed using the histogram view of ZEN 2010D software (Carl Zeiss). CXCR-4 expression was analyzed regarding to the intensity of Alexa Fluor 488 signal. To this end, several elements were defined first: part of background area, outer border of endothelium, and inner area of endothelium. The background area was used to detect the threshold. Total intensity of the endothelial layer normalized to its area was calculated using the following formula:
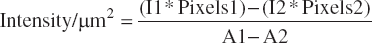
A1 = area, defined by the outer border of endothelium, μm2
A2 = area, defined by the inner border of the endothelium, μm2
I1 = mean intensity of the area 1
I2 = mean intensity of the area 2
Pixels 1 = total number of pixels in the area 1
Pixels 2 = total number of pixels in the area 2
LSM images were collected and analyzed as described above for 15 different vessels of each treatment group.
Statistical Analysis
All statistical analyses were performed using Sigma Stat software version 3.0 (SPSS Inc., Chicago, IL, USA). Results are given as mean ± SEM. Comparisons of the groups were executed using one-way analysis of variance (ANOVA) with subsequent post hoc multiple Holm– Sidak analysis. In case the data failed normality testing, the nonparametric Kruskal–Wallis or post hoc multiple Dunn tests were employed. Values of p < 0.05 were considered statistically significant.
Results
Viability and CXCR-4 Expression Level After CD133+ Stem Cell Isolation From the Sternal Bone Marrow
After the isolation procedure from donor patients' bone marrow, FACS analysis was performed to confirm quality of the isolated cells. Mean purity, expressed as content of CD133+ CD34+ cells, was 83.6 ± 2.2%, whereas the mean viability of isolated cells was 95.0 ± 1.2%. CXCR-4 protein, representing the main receptor of SDF-1α, was expressed on 71.7 ± 3.49% of the CD133+ cells.
CD133+ Cells Migrate Towards SDF-1α In Vitro
In order to quantify the general chemoattractant capacity of SDF-1α on CD133+ cells after intraoperative sternal harvesting, we tested the effect of SDF-1α on the migration of CD133+ cells in the Boyden Chamber.
SDF-1α caused a significant migration of isolated CD133+ cells in vitro. The number of migrated cells increased almost fivefold compared to medium alone (p = 0.030) (Fig. 1).

CD133+ cell migration towards SDF-1α gradients in vitro. Quantitative analysis of cluster of differentiation 133 positive (CD133+) cell migration in Boyden Chamber assays. CD133+ cells from sternal bone marrow were seeded at the upper compartment of Boyden chamber assays with and without stromal cell-derived factor (SDF)-1α in the lower compartment. The number of migrated cells increased fivefold when SDF-1α was added to the assay “SDF-1” (n = 6) compared to medium alone “Control” (n = 6) (*p < 0.05 vs. “Control”).
In Vivo Interaction of CD133+ Cells with the Endothelium Occurs Predominantly in Postcapillary Venules
Overall, interaction between injected CD133+ cells and the peripheral vascular endothelium was a rare event and occurred almost exclusively in postcapillary venules. CD133+ cell contact with the endothelial lining varied to a different extent in the respective experimental groups. The microcirculatory conditions, represented by the red blood cell velocity and the wall shear rate, did not differ significantly between the experimental groups (Table 2). In the observed venules, rolling and firm adhesion of CD133+ cells in response to superfusion with the respective reagent could be clearly quantified utilizing intravital fluorescence microscopy (Fig. 2).

Intravital fluorescence microscopy. Representative intravital fluorescence microscopy images of rolling and adherent CD133+ cells in a murine cremaster muscle preparation. Intravascular background is visualized by rhodamine. White arrows indicate rolling CD133+ cells; black arrows indicate adherent CD133+ cells.
Vessel Diameter, Red Blood Cell Velocities, and Wall Share Rates in Venules of Murine Cremaster Muscles at Baselines in Control (n = 6), SDF-1α (n = 6), TNF-α (n = 6), and SDF-1α + TNF-α (n = 6) Groups

Analysis was performed using CapImage Software (Zeintl, Heidelberg, Germany). Data are expressed as mean ± SEM. Variation between the individual groups was not significant. SDF-1α, stromal cell-derived factor-1α; TNF-α, tumor necrosis factor-α.
SDF-1α Alone Enhances Venular Rolling of CD133+ Cells In Vivo
In the PBS-superfused cremaster muscle (“Control”), the fraction of rolling CD133+ cells was 17.7 ± 2.7%. Following local administration of SDF-1α, a significant increase in CD133+ cell rolling was detected (45.9 ± 1.8% in “SDF-1α” vs. 17.7 ± 2.7% in “Control,” p < 0.001). Local application of TNF-α alone induced an increase in CD133+ cell rolling, however not in a significant extent (23.9 ± 2.1% in “TNF-α” vs. 17.7 ± 2.7% in “Control,” p > 0.05). In response to combined treatment with SDF-1α and TNF-α, the fraction of rolling CD133+ cells was not significantly different compared to CD133+ cell rolling after exposure to SDF-1α alone but significantly exceeded the fraction of rolling CD133+ cells following treatment with TNF-α alone (41.7 ± 3.3% in “SDF-1α + TNF-α” vs. 45.9 ± 1.8% in “SDF-1α” and 23.9 ± 2.1% in “TNF-α,” p > 0.05 and p < 0.001, respectively) (Fig. 3A). Interestingly, the average rolling velocity was also significantly reduced after SDF-1α and SDF-1α + TNF-α treatment in comparison to control animals (102.2 ± 11.7 μm/s in “Control” vs. 47.3 ± 3.8 μm/s in “SDF” and 31.1 ± 2.1 μm/s in “SDF-1α + TNF-α,” p < 0.001), indicating a relevant enhancement of early interaction between applied CD133+ cells and the local endothelium upon stimulation with SDF-1α and SDF-1α + TNF-α, respectively (Fig. 3B).

SDF-1α- and TNF-α-mediated rolling of CD133+ cells in cremaster muscle microcirculation. Quantitative analysis of CD133+ cells exhibiting endothelial rolling based on intravital fluorescence microscopy. (A) Bars indicate the percentage of rolling CD133+ cells in “Control” (n = 6), “SDF-1α” (n = 6), “tumor necrosis factor (TNF)-α” (n = 6), and “SDF-1α + TNF-α” (n = 6) groups. (B) Bars indicate the velocity of CD133+ cells exhibiting endothelial rolling in “Control” (n = 6), “SDF-1α” (n = 6), and “SDF-1α + TNF-α” (n = 6) groups (*p < 0.05 vs. “Control”).
Local Stimulation with TNF-α in Addition to SDF-1α Treatment Enhances Firm Endothelial Adhesion of CD133+ Cells In Vivo
In order to investigate the capability of SDF-1α on induction of firm CD133+ cell–endothelial cell interaction, we quantified the number of firm adherent CD133+ cells on predefined postcapillary venules following local application of SDF-1α. Furthermore, the role of additional inflammatory stimulation by TNF-α was analyzed.
In PBS-superfused cremaster muscle (“Control”) the number of firmly adherent CD133+ was 2.2 ± 1.1 cells/mm2. In animals treated with SDF-1α, the number of firmly adherent CD133+ cells was significantly increased (9.8 ± 1.9 cells/mm2 in “SDF-1α” vs. 2.2 ± 1.1 cells/mm2 in “Control,” p < 0.001). Local administration of TNF-α did not show a significant difference as compared to treatment with SDF-1α alone (11.3 ± 2.0 cells/mm2 in “TNF-α” vs. 9.8 ± 1.9 cells/mm2 in “SDF-1α,” p > 0.05). In response to combined treatment with TNF-α and SDF-1α, however, the number of firmly adherent CD133+ cells exceeded that of untreated control more than 13-fold (27.9 ± 4.3 cells/mm2 in “SDF-1α + TNF-α” vs. 2.2 ± 1.1 cells/mm2 in “Control,” p < 0.001) (Fig. 4).

SDF-1α- and TNF-α-mediated firm endothelial adhesion of CD133+ cells in cremaster muscle microcirculation. Quantitative analysis of CD133+ exhibiting firm endothelial adhesion based on intravital fluorescence microscopy. Bars indicate the total amount of firm adherent CD133+ cells per mm2 endothelial lining in “Control” (n = 6), “SDF-1α” (n = 6), “TNF-α,” and “SDF-1α + TNF-α” (n = 6) groups (*p < 0.05 vs. “Control”).
Stimulation with SDF-1α and TNF-α Induces CXCR-4 Expression on Cremaster Muscle Endothelium
In order to analyze the effect of SDF-1α and TNF-α treatment on the endothelial expression of CXCR-4 in the cremaster muscle venules, laser scanning microscopy with consecutive CXCR-4 signal intensity quantification was performed. In SDF-1α and TNF-α treated tissue, we detected a clear induction of CXCR-4 receptor on the endothelial lining when compared with untreated control animals and tissue treated with SDF-1α only (3,316 ± 596 I/μm2 in “SDF-1α + TNF-α” vs. 1171 ± 181 I/μm2 in “SDF-1α,” p = 0.02 and 308 ± 52 I/μm2 in “Control,” p < 0.001) (Figs. 5 and 6).

Representative laser laser scanning microscopy image, illustrating endothelial CXCR-4 intensity calculation in cremaster muscle samples. Green line indicates the background area used to detect the fluorescence threshold. Yellow line indicates the area defined by the outer border of the endothelium and light blue line indicates the area defined by the inner border of the endothelium. Total fluorescence intensity of the endothelial layer normalized to its area was calculated according to the formula mentioned within the materials and methods part. CXCR-4, chemokine (C-X-C motif) receptor-4.

SDF-1α- and TNF-α-mediated induction of endothelial CXCR-4 receptor expression. (A) The left panel shows representative laser scanning microscopy images detecting CXCR-4 signal intensity (light green fluorescence) on the cremaster muscle endothelium within the respective treatment groups. Double staining additionally shows all nuclei (blue). The right panel shows respective negative controls. (B) Bars indicate the quantification of CXCR-4 signal intensity detected by laser scanning microscopy in “Control” (n = 6), “SDF-1α” (n = 6), and “SDF-1α + TNF-α” (n = 6) groups (*p < 0.05 vs. “Control”).
Discussion
Efficient migration towards ischemic myocardium forms a key factor limiting the therapeutic efficacy of bone marrow stem cells administered for cardiac regeneration purposes (25). Although a series of chemokines, upregulated in the context of myocardial ischemia, are known to promote targeted stem cell migration (21), bone marrow stem cell subpopulations, applied within clinical protocols, have not been described conclusively regarding their in vivo migratory capacities towards chemokine gradients in a peri-interventional setting. However, consequent determination of these properties seems crucial, since, for example, clinical cell harvesting procedures and patients' morbidity (15) might alter specific migratory capacities, thereby influencing the possible therapeutic efficacy of respective cell populations. The current study aimed at analyzing the migratory properties of human CD133+ BMSCs towards SDF-1α in vivo after isolation from the sternal bone marrow.
Our results suggest that SDF-1α is capable to induce both, transient CD133+ cell–endothelial cell interaction and firm endothelial adhesion of CD133+ BMSCs in vivo. However, in terms of firm endothelial adhesion, which is required for subsequent stem cell migration across the endothelial layer, additional inflammatory stimulation with TNF-α enhanced the CD133+ cell–endothelial cell interaction in a highly significant manner.
The primary intention of this study was to test the migratory potential of CD133+ BMSCs after cell harvesting and isolation procedures as applied within our running clinical cell therapy protocol. In vitro data show that isolated CD133+ cells, obtained from the sternal bone marrow of an unselected patient population undergoing cardiac surgery, express CXCR-4 and are capable of migration towards SDF-1α. The feasibility of a xenogeneic approach for in vivo analysis of homing and engraftment capacity of human bone marrow stem cells has been proven previously (7,18). Herein, a well-established in vivo model (3), allowing for detailed analysis of CD133+ cell–endothelial cell interaction by means of intravital fluroscence microcopy, was used in a xenogeneic setting. Utilizing the same model, we have been able to show before that SDF-1α induces murine c-kit+ cell migration in the presence of TNF-α (9). Since the special relevance of SDF-1α for targeted stem cell migration has been shown also by other groups (19), it was conceivable to test the migratory potential of CD133+ BMSCs in a tissue model upon stimulation with this specific chemokine. Human and murine SDF-1α are cross-reactive and differ in only one amino acid (12). Furthermore, although peak SDF-1α gradients directing the migratory behavior of bone marrow-derived stem cells have primarily been detected following acute ischemia, elevated basal plasma levels seem to be associated with myocardial regeneration and neovascularization also in the setting of chronic ischemic heart disease (11). Our results demonstrate—for the first time—that CD133+ BMSCs, after undergoing sternal harvesting and isolation procedures, exhibit a relevant migratory potential in non-bone marrow tissue. In line with findings for murine progenitor cells, SDF-1α alone is capable to induce transient CD133+ cell interaction with the endothelium (“rolling”) and additional application of TNF-α had no further impact. Significantly reduced rolling velocity after SDF-1α treatment indicates that the observed CD133+ cell rolling represents a relevant interaction, priming endothelial transmigration, rather than an unspecific early tethering phenomenon. It has been demonstrated for endogenous leucocytes that an intraluminal “crawling” process, differing from initial endothelial rolling, takes place immediately prior terminal extravazation (14,23). In terms of firm endothelial adhesion, however, additional TNF-α stimulation significantly enhanced the amount of SDF-1α-induced firm endothelial adhesion. It appears that, as described already by Abbott et al. for c-kit+ cells (1), a certain degree of local inflammation is necessary in order to induce targeted CD133+ cell migration in a relevant extent.
The capability of CD133+ BMSC to display sufficient endothelial adhesion and subsequent migration towards chemokine gradients in a microenvironment similar to ischemic myocardium provides highly relevant information on the therapeutic feasibility of this stem cell subpopulation. Although clinical results of cardiac CD133+ cell transplantation have been promising (10), on-going preclinical analysis regarding the in vivo behavior of applied cell populations is indispensable. Besides established viability testing of the applied cell product, migratory testing protocols could contribute to the therapeutic efficacy of bone marrow stem cell populations in the future. Although complex in vivo models, as performed in the current study, are likely not to be applicable in the clinical routine, in vitro migration assays might complement the peri-interventional testing of clinically applied cell populations in the future.
Results obtained from laser scanning microscopy suggest that the described firm endothelial adhesion of CD133+ BMSCs is likely to depend on both CD133+ BMSC attraction and endothelial activation by an increase in CXCR-4 after SDF-1α and TNF-α stimulation. A clear induction of endothelial CXCR-4 expression observed after combined treatment with SDF-1α and TNF-α indicates that such endothelium-linked mechanisms are crucial for peripheral CD133+ BMSC recruitment, as already described for CD34+ cell homing to the bone marrow (17). After SDF-1α/CXCR-4-mediated endothelial activation subsequent upregulation of endothelial adhesion molecules, such as intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1), is likely to contribute to CD133+ cell adhesion (18). In human cord blood CD34+ cells, SDF-1 upregulates the tetraspanin CD9 via a CXCR-4-dependent mechanism (13). CD9 was demonstrated to be necessary for SDF-1-induced transendothelial migration. However, a detailed analysis of the linkage between the different adhesion molecules contributing to the reported findings was not the aim of this study but should be addressed in upcoming experimental work.
In conclusion, we were able to show that CD133+ BMSCs exhibit sufficient in vivo homing towards SDF-1α gradients after undergoing standardized intraoperative harvesting and isolation from the sternal bone marrow. Therefore, in terms of its migratory capacity in an inflammatory, nonbone marrow microenvironment, this subpopulation qualifies for further clinical use. Migratory testing might add to the per-interventional analysis of stem cell populations isolated for therapeutic application in the future.
Footnotes
Acknowledgment
The authors declare no conflict of interest.