
Abstract
Stem cell transplantation therapy has emerged as a potential treatment for ischemic stroke and other neurodegenerative diseases. Effective delivery of exogenous cells and homing of these cells to the lesion region, however, have been challenging issues that hinder the efficacy and efficiency of cell-based therapy. In the present investigation, we tested a delayed treatment of noninvasive and brain-targeted intranasal delivery of bone marrow mesenchymal stem cells (BMSCs) in a mouse focal cerebral ischemia model. The investigation tested the feasibility and effectiveness of intranasal delivery of BMSCs to the ischemic cortex. Hypoxia preconditioning (HP) of BMSCs was performed before transplantation in order to promote their survival, migration, and homing to the ischemic brain region after intranasal transplantation. Hoechst dye-labeled normoxic- or hypoxic-pretreated BMSCs (1 × 106 cells/animal) were delivered intranasally 24 h after stroke. Cells reached the ischemic cortex and deposited outside of vasculatures as early as 1.5 h after administration. HP-treated BMSCs (HP-BMSCs) showed a higher level of expression of proteins associated with migration, including CXC chemokine receptor type 4 (CXCR4), matrix metalloproteinase 2 (MMP-2), and MMP-9. HP-BMSCs exhibited enhanced migratory capacities in vitro and dramatically enhanced homing efficiency to the infarct cortex when compared with normoxic cultured BMSCs (N-BMSCs). Three days after transplantation and 4 days after stroke, both N-BMSCs and HP-BMSCs decreased cell death in the peri-infarct region; significant neuroprotection of reduced infarct volume was seen in mice that received HP-BMSCs. In adhesive removal test of sensorimotor functional assay performed 3 days after transplantation, HP-BMSC-treated mice performed significantly better than N-BMSC- and vehicle-treated animals. These data suggest that delayed intranasal administration of stem cells is feasible in the treatment of stroke and hypoxic preconditioning of transplanted cells, significantly enhances cell's homing to the ischemic region, and optimizes the therapeutic efficacy.
Keywords
Introduction
Cellular therapy has emerged as a potential novel treatment for ischemic stroke (40). Promising progress in the safety and efficacy of the therapy has been made in preclinical and clinical studies using bone marrow mesenchymal stem cells (BMSCs) and other cell types (8, 34, 56). The possibility of allogenic transplantation of BMSCs and other benefits has driven the application of these cells toward clinical trials (3, 19, 27, 45). It is commonly recognized that a successful cell-based therapy for stroke depends largely on cell homing and engraftment to the brain ischemic region. Current experimental and clinical research on BMSC stroke therapy utilizes two approaches: local delivery and systemic administration. Local delivery directly targets cells in the injured brain region. However, this approach requires general anesthesia and neurosurgery procedures that hinder clinical applications, and the high degree of invasiveness may cause additional brain damage. Systemic administration can be achieved by intravenous (IV) or intra-arterial (IA) injection, which is less invasive. However, IV delivery of stem cells results in massive cell entrapment in capillary beds of other organs such as lungs, liver, spleen, and kidneys (1, 4, 14, 18, 26) with only a small amount of cells being able to reach the brain (17, 31, 54). Even for those cells that arrive at targeted organs, many of them become passively entrapped inside the capillaries or microvessels (24, 37, 54). Injection of stem cells via the IA route could result in a reduction of cerebral blood flow and micro-embolization within the cerebrovasculature (4, 40, 54). The trapped cells in the vasculature need time (from 24 h to as long as 10 days) to transmigrate through endothelium into tissue parenchyma (24, 37, 54). Another direct result of the shortcomings for systemic delivery is that large numbers of cells have to be injected into patients, which may increase potential side effects and the cost of the treatment (52). Nonneurosurgical alternatives that can effectively deliver cells to the ischemic brain region with reduced systemic exposure would undoubtedly be beneficial for enhancing therapeutic potential and the safety of cell transplantation therapies (52).
Intranasal administration is a noninvasive and convenient drug delivery method that bypasses the blood–brain barrier (BBB) and directly guides therapeutics to the central nervous system (CNS) (12, 49). Although there are multiple barriers to absorption, in healthy rodents intranasal administered BMSCs can migrate to different brain regions utilizing pathways along olfactory and trigeminal nerves innervating the nasal passages (10). The cells delivered by intranasal administration accumulated with the highest number in the brain compared to other analyzed organs including the heart, kidney, lung, liver, spleen, and stomach (9). There are very little to no known reports about intranasal administration of growth factors (15, 30, 55) and two reports on cell therapy for Parkinson's disease (9) or neonatal hypoxia-ischemia injury (51). Nevertheless, there has been no information on intranasal cell delivery for the treatment of ischemic stroke.
Recent reports from our group demonstrate that hypoxic preconditioning (HP) pretreatment of stem cell-derived neural progenitor cells and BMSCs can enhance survival of these cells after transplantation into the ischemic brain and heart. We show that systemic injection of HP-treated cells exhibit improved directional migration and homing to the lesion site either in vitro or following IV injection into the ischemic animals (20, 21, 48). In the present investigation, we examined the brain distribution of intranasally delivered BMSCs in a focal cerebral ischemia model of mice. We specifically tested the hypothesis that HP pretreatment and intranasal delivery of BMSCs are two feasible and effective strategies for improving the clinical potential of cell transplantation therapy.
Materials and Methods
Isolation and Culture of BMSCs From Rats
BMSCs were isolated and harvested as previously described (21). In brief, BMSCs were flushed from the fibias of postnatal day 21 Wistar rats (Charles River, Wilmington, MA, USA) using a 25-gauge needle. Mononuclear cells were suspended in Dulbecco's modified Eagle's medium (Cellgro, Manassas, VA, USA) supplemented with 15% fetal bovine serum (Sigma, St. Louis, MO, USA) and plated into dishes. Cultures were maintained at 37°C in a humidified atmosphere containing 5% carbon dioxide. After 24 h, nonadherent cells were discarded, and adherent cells were washed four times with phosphate-buffered saline solution (PBS; Sigma). Fresh complete medium was added and replaced every 2 days. Each primary culture was subcultured 1:3 when MSCs grew to approximately 80% confluent. We performed fluorescence-activated cell sorting to characterize MSC population using CD105, CD73, CD34, and CD45 markers (eBioscience, San Diego, CA, USA). All cells used in this study were freshly isolated within five passages and harvested for analysis when they were around 80–90% confluent.
In Vitro Hypoxic Preconditioning
Cells were incubated under normoxic conditions or in a finely controlled ProOx C-chamber system (Biospherix, Redfield, NY, USA). For hypoxia preconditioning, the oxygen concentration in the chamber was maintained at 0.1–0.3% with a residual gas mixture composed of 5% carbon dioxide balanced with nitrogen for 24 h followed by various lengths of reoxygenation time before analyses. For in vitro migration assays and in vivo experiments, cells with 70–80% confluence were incubated under normoxic or hypoxic conditions for 24 h followed by 1 h reoxygenation before testing or harvesting.
Cell Viability Assay
Cell viability at the time of transplantation was determined using a flow cytometry-based Annexin V/propidum iodide (PI; Sigma) assay and Scepter cell counter. The Annexin V/PI assay detects early apoptotic cells, while the Scepter cell counter provides quantitative information of cell health by inspecting cell size/volume at submicron and subpicoliter resolution. Flow cytometry was performed using an Annexin V-fluorescein isothiocyanate (FITC) antibody (BD Pharmingen, San Jose, CA, USA) according to the manufacturer's instruction. Briefly, cells were harvested, resuspended in binding buffer, incubated in dark with Annexin V-FITC and/or PI for 15 min, and then analyzed with flow cytometry. A Scepter cell counter (Millipore, Billerica, MA, USA) was used to count cells and check their healthy states according to manufacturer's instruction. The percentages of defined nonviable cells and debris (6–12 μm) and viable cells (12–26 μm) were detected by gating the two distinct histogram peaks using the Sceptor software version 1.2.
Western Blotting
The expression of migration-related factors in BMSCs was examined by Western blot. Three dishes of cells in each experimental group were examined at 1, 3, and 24 h after reoxygenation. Cultured cells were lysed with modified BCIP/NBT solution (Sigma; 50 mmol/L HEPES, pH 7.3, 1% sodium deoxycholate, 1% Triton X-100, 0.1% sodium dodecyl sulfate, 150 mmol/L NaCl, 1 mmol/L ethylenediaminetetraacetic acid, 1 mmol/L Na3VO4, 1 mmol/L NaF) and protease inhibitor cocktail (Sigma) for 30 min, followed by centrifugation at 17,000 × g for 15 min. Protein concentration of each sample was determined using the bicinchoninic acid assay (Sigma). Samples of 30 μg proteins were electrophoresed on a 6–20% sodium dodecyl sulfate–polyacrylamide gradient gel (BioRad, Hercules, CA, USA) in a Hoefer Mini-Gel system (Amersham Biosciences, Piscataway, NJ, USA) and transferred in the Hoefer Transfer Tank (Amersham Biosciences) to a polyvinylidene difluoride membrane (BioRad). Membranes were blocked with buffer [Tris-buffered saline (Sigma) containing 0.1% Tween-20 (Bio-Rad), pH 7.6%, 7% milk (Carnation, Wilkes-Barre, PA, USA)] at room temperature for 2 h and incubated overnight at 4°C with one of the following antibodies: anti-human CXC chemokine receptor type 4 (CXCR4) monoclonal mouse IgG2B (clone 44716) (1:1,000, R&D Systems, Minneapolis, MN, USA), anti-matrix metalloproteinase 9 (MMP-9) rabbit polyclonal antibody (1:1,000; Millipore, Billerica, MA, USA), or anti-MMP-2 rabbit polycolonal antibody (1:500; Millipore). Mouse β-tubulin (1:2,000; Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used as the protein loading control. The blots were washed in 0.5% Tris-buffered saline containing 0.1% Tween-20 (TBST) and incubated with alkaline phosphatase-conjugated anti-rabbit or anti-mouse IgG (Promega, Madison, WI, USA) for 2 h at room temperature. Finally, membranes were washed with TBST followed by three washes with Tris-buffered saline. Signal was detected by the addition of 5-bromo-4-chloro-3-indolyl-phosphate/nitroblue tetrazolium (BCIP/NBT) solution (Sigma). Data were quantified and analyzed using the NIH Image J program (NIH, Bethesda, MD, USA). The expression level of each protein was corrected against the loading control of β-tubulin.
Directed Migration Assay In Vitro
BMSC migration ability in vitro was measured with scratch wound healing assay and transwell assay as described before (20, 21, 28). In the wound healing assay, BMSCs were grown to 70–80% confluence in 35-mm dishes (Cellstar/Sigma). The cell layer in both the normoxic group and hypoxic cells was scratched along the central vertical line using a 200-μl pipette tip. The debris was removed, and the edges of the scratch was smoothed by washing the cells once with 1 ml serum-free medium and then replacing with 5 ml of 2% serum medium in order to slow down the cell proliferation rate. Pictures at the same location were taken immediately at time 0 and 24 h after the scratch. The distances that cells migrated from the edge of the scratch to the “wounded” central area were measured (28). The migration ability of cells was assessed as a function of how far from the scratch line the cells had progressed within the 24-h period. Migration and chemotaxis assays were further confirmed and quantified using transwell polycarbonate inserts (8-μm pore size, 24-well plate, BD Biosciences, San Jose, CA, USA) coated with 200 μg/ml Matrigel (BD Biosciences). Normoxic-cultured BMSCs (N-BMSCs) or HP-BMSCs (1 × 105 cells) resuspended in DMEM containing 0.4% FBS were loaded into the upper inserts, and stromal cell-derived factor (SDF)-1α (200 ng/ml; ProSpec, East Brunswick, NJ, USA) was added to the lower chambers as a chemoattractant. The cells were allowed to transmigrate for 18 h. Nonmigrating cells on the upper surface of the memberane were gently removed using cotton swabs. Cells migrating to the lower surface were fixed with 4% paraformaldehyde, then stained with Hoechst 33342 (Molecular Probes, Carlsbad, CA, USA), and photographed in seven random, nonoverlapping areas using 100 × magnification field. The number of cell nuclei was counted using the NIH Image J software. In some experiments, the CXCR4 antagonist ADM3100 (25 μg/ml, Sigma) or a monoclonal antibody against MMP-2 or MMP-9 (10 μg/ml, Millipore) was added into the upper chamber to block the specific signaling during the test. All experiments were carried out in triplicate.
Focal Ischemia Stroke Model of Mice
All animal experiments and surgical procedures were approved by the University Animal Research Committee and met NIH standards. Adult male C57BL/6 mice (National Cancer Institute, NCI, Bethesda, MD, USA) weighing 20–25 g and in the age range of 2–2.5 months were used in this study. The focal cortex ischemic stroke was induced as previously described (57). Briefly, animals were anesthesized with intraperitoneal (IP) injection of 4% chloral hydrate (100 mg/kg; Sigma). Focal cortex ischemia was induced by permanent occlusion of two to three branches of the middle cerebral artery (MCA). This was accompanied by 7-min bilateral common carotid artery (CCA) ligation followed by reperfusion. During surgery and recovery periods, body temperature was monitored using a rectal probe and maintained at 37.0°C using a heating pad and a temperature-controlled, ventilated incubator.
Cell Labeling
BMSCs were labeled with Hoechst 33342 in order to track and count these cells in the brain. Sterile 1 mM Hoechst 33342 was added, and the culture was incubated for 1 h before transplantation. The cells were rinsed six times with PBS to remove unbound Hoechst 33342 and harvested by trypsinization for intranasal delivery.
Intranasal Administration of BMSCs
Intranasal delivery was performed as previously described (10) with minor modifications. Twenty-four hours after ischemia, the mice were anesthetized with 4% chloral hydrate (100 mg/kg, IP) and placed on a heating pad, with the head in the upright position. All animals received 100 U hyaluronidase (Sigma) dissolved in sterile PBS 30 min prior to the administration of cells. By catalyzing the hydrolysis of hyaluronan, hyaluronidase can increase tissue permeability. Hyaluronidase was used here to disrupt the barrier function of the nasopharyngeal mucosa and facilitate the cell entry to the brain (10). Five-microliter drops containing the cell suspension or vehicle were carefully placed on one nostril, allowing it to be snorted, alternating the nostrils with 1-min intervals. A total volume of 100 μl of cell suspension (1 × 106 cells) or vehicle was used. In total, the procedure took about 30 min. Animals were scarified under isoflurane (Piramal Healthcare, LBS Marg, Vikhroli, Mumbai) either 1.5 h or 3 days from the initial cell delivery.
Assessment of Transplanted BMSCs
At 1.5 h after delivery, the brain was immediately removed and mounted in optimal cutting temperature (OCT) compound (Sakura Finetek USA, Inc., Torrance, CA, USA) at −80°C, and 20-μm frozen sections were cut from the olfactory bulb caudally toward bregma −2.50. Brain coronal sections were counterstained with PI (red) to reveal the whole cell population. Brain sections were analyzed by fluorescence microscopy (BX51, Olympus, Tokyo, Japan). We checked sections starting from the olfactory bulb (OB, bregma 5.5–3.5, 20-μm thickness of 20 sections, 80 μm apart) and other regions of the brain (e.g., forebrain: bregma 3.0–1.0) for migration, distribution, and localization of intranasally delivered BMSCs. To examine homing capacity, the PI and Hoechst doublelabeled cells (magenta) were quantified from bregma 1.10 to −2.10 mm. Among the total 150 brain sections per animal, which covered most of ischemic lesion of our stroke model, every fifth brain section across the entire region of interest was counted. Thus, 30 brain sections of 80 μm apart among the total 150 sections were counted. Cell count was performed on the entire area of interest. The counted number from the 30 sections was then multiplied by 5 to reflect the total population of counted cells in the surveyed brain volume.
The lesion region was defined as the ischemic core and penumbra area as described before (39, 57). The results were presented as cell number in different brain regions, as well as percentage of cells reaching certain brain regions compared to all cells found in analyzed brain regions (bregma 1.10 to −2.10 mm) or to the number of transplanted cells (1 × 106 cells).
Immunohistochemical Staining
Immunofluorescence staining was used in order to determine the location of prelabeled cells in brain tissue with the respect to brain vessels. Brain tissues were fixed with 10% buffered formalin for 10 min, permeabilized with 0.2% Triton X-100 for 5 min, and blocked with 1% fish gelatin (Sigma) for 1 h at room temperature. Specimens were then incubated with primary antibodies overnight at 4°C (goat anti-collagen type IV, 1:400; Millipore), then washed with PBS three times, and incubated with Alexa Fluor 488 anti-goat immunoglobulin G (1:200; Molecular Probes) for 1 h at room temperature. Slides were mounted with ProLong Antifade mounting medium (Molecular Probes) and analyzed under a florescent microscope (BX51).
Infarct Volume Assessment
A separate group of mice was subjected to stroke surgery and 24 h later received N-BMSCs, HP-BMSCs, or PBS as described above. On day 4 after stroke, the brain was removed and sliced into 1-mm coronal sections using a mouse brain matrix (Harvard Bioscience, South Natick, MA, USA) and incubated in 2% 2,3-5-triphenyl-tetrazolium chloride (TTC; Sigma) solution at 37°C for 5 min. Brain sections were scanned, and the unstained versus stained area was determined using NIH Image J on the ventral side of six brain slides per animal. Then the infarct area (mm2) of staining in each slice was multiplied by the slice thickness (1 mm) to get the infarct volume (mm3). The indirect infarct volume was calculated by the difference between the volume of contralateral cortex and the volume of the TTC-stained portion (nonischemic) of ipsilateral cortex of each mouse, following the equation, contralateral volume – (ipsilateral volume – infarct volume). This method helps to correct edema in the ipsilateral hemisphere in order to achieve accurate assessments of infarct volume (46). The brain infarct volume was the summation of six individual section volumes.
Terminal Deoxynucleotidyl Transferase Biotin-dUPT Nick End Labeling Staining
Brain sections were cut into 10 μm thickness, and terminal deoxynucleotidyl transferase biotin-dUPT nick end labeling (TUNEL) staining kit (DeadEnd Fluorometric TUNEL System, Promega, Madison, WI, USA) was used to visualize DNA fragmentation and cell death in those brain sections according to the manufacturer's instructions. The slides were counterstained with Hoechst 33342 (1:20,000, Molecular Probes) for 5 min to reveal the nucleus of all cells before mounting with ProLong Antifade mounting medium (Molecular Probes). For systematic sampling in design-based stereological cell counting, every fifth brain section (50 μm apart) across the entire region of interest was counted. For multistage random sampling, six fields per brain section were randomly chosen in the ischemic boundary region using a florescent microscope (BX51). This was repeated in six separate sections per brain. The result was presented as percentage of TUNEL-positive cells compared with all nuclei within 400 × magnification fields.
Evaluation of Neurological Function Deficits
Adhesive-removal test, which is a sensitive method to assess sensorimotor deficits in focal cerebral ischemic mice, was used to evaluate the neurological dysfunction after stroke as previously described (6). Briefly, a training session (for 3 days, one to two trials per day) was performed until the mice could take off the sticky dots on their paws within 12 s before surgical procedures. Animals were tested before, 1 day (before cell application), and 4 days after ischemia by an investigator who was blind to the experimental groups. The mean time (seconds, averaged from four to five trials) required to remove sticky dot from the left paw (time to remove) was recorded. All testing trials were conducted during the day.
Statistical Analysis
Student's two-tailed t test was used for comparison of two experimental groups. Multiple comparisons were performed using one-way analysis of variance followed by Tukey's test for multiple pair examinations. Changes were identified as significant if value of p < 0.05. Data were expressed as mean ± SEM.
Results
Characteristics and Cell Viability of BMSCs
Cell surface markers and multipotency of rat's BMSCs were verified (Fig. 1) and published in our previous investigations (20, 21). Flow cytometry analysis confirmed that these cells expressed CD73, CD90, and CD105 surface markers but were CD34 and CD45 negative, consistent with characteristic surface markers of undifferentiated BMSCs (Fig. 1A–D). The lack of expression of CD34 and CD45 suggested that the cell population was depleted of hematopoietic stem cells (20). The multipotency of these cells further verified the cellular nature of BMSCs (13, 20).
We examined the cell viability in both normoxic and hypoxic culture conditions using a flow cytometry-based Annexin V/PI assay and a cell volume assessment using a Scepter cell counter. In the AnnexinV/PI assay, the percentage of viable BMSCs cultured in 0.1% O2 for 24 h was comparable to that of cells cultured under normoxic condition (Fig. 1E, G). Both cultures showed at least 80–85% of viable cells. Given that cell shrinkage and swelling are morphological landmarks of apoptosis and necrosis, respectively, the distribution of cell size/volume was checked as indication of cell's health status. The percentage of normal healthy BMSCs, defined in the range of diameter between 12 and 26 μm, was consistent with the flow cytometry data with around 90% of healthy cells in both normoxic and hypoxic cultures (Fig. 1F, H). These viability assessments confirmed the healthy status of our transplanted cells.
Characterization and viability of isolated bone marrow cells. Bone marrow cells were collected from postnatal day 21 rats. After 24 h in our culture condition, adhesive cells were harvested and characterized for cell surface markers specific for bone marrow mesenchymal stem cells (BMSCs). (A–D) Fluorescence-activated cell sorting using flow cytometry identified cell populations that are positive to CD105, CD73 (A, B) but not to the hematopoietic cell marker CD45, CD34 (C, D), which are consistent with the characteristics of BMSCs. We had also demonstrated multipotency of these cells, being able to differentiate into osteocyte phenotype cells, fat cells, and chondrogenic cells. Hypoxia preconditioning did not change the phenotypes of cells. (E) Cell viability under normoxic (N) and hypoxic culture (HP) conditions was tested by flow cytometry with Annexin V and propidium iodide staining. (F) Cell viability before transplantation was also checked by cell size/volume assessment using Scepter cell counter. (G, H) Quantification of flow cytometry data (G) and Scepter cell counting (H) demonstrated the healthy state of our BMSCs in control and experimental cell groups (n = 3 for flow cytometry and n = 6 for Scepter cell counting).
Migration Properties of BMSCs
Western blot analysis was performed at 1, 3, and 24 h after termination of the sublethal 24-h hypoxia; the three time points were selected to estimate acute, subacute, and chronic regulations of related proteins after the hypoxic exposure. The HP treatment and reoxygenation significantly upregulated the expression level of some migration-related proteins, including MMP-2, MMP-9, and the SDF-1 receptor CXCR4 (Fig. 2). At 1 h after reoxygenation, the upregulation reached their peak levels, increasing these factors by over 50% to twofolds (Fig. 2). Expression of MMP-2 and active MMP-9 in HP-BMSCs remained elevated up to 24 h after reoxygenation (Fig. 2).
Effect of in vitro hypoxic precondition on expression of CXCR4, MMP-2, and MMP-9 in BMSCs. Western blot was performed to detect protein levels of CXC chemokine receptor type 4 (CXCR4), matrix metalloproteinase 2 (MMP-2), and MMP-9 in BMSCs pre-exposed to normaxia (21% O2) or sublethal hypoxia (0.1–0.3% O2, 24 h) and reoxygenation. (A) Hypoxia followed by 1 h reoxygenation significantly enhanced the expression of CXCR4 in BMSCs. The upper panel above the bar graph shows representative Western blotting. (B) The expression of MMP-2 was enhanced during 1–24 h posthypoxia reoxygenation. (C) Western blotting of the expression levels of pro-MMP-9 and active MMP-9 in BMSCs at different times after hypoxia–reoxygenation. (D, E) Hypoxia and reoxygenation increased expression of pro-MMP-9 (D) as well as active MMP-9 (E) in BMSCs. n = 6–9; *p < 0.05 versus normoxia controls.
An in vitro scratch-stimulated cell migration assay was applied to test whether HP exposure could enhance the migration activity of BMSCs. Compared with N-BMSCs, HP-BMSCs moved significantly further into the central wounded area from the scratch edges over a 24-h period (Fig. 3A, B). Thus, the directed migration in vitro test suggested that HP-BMSCs acquired enhanced movement activity toward injury-induced migration cues. To verify that CXCR4 receptor and MMP-2/MMP-9 played key roles in the directional migration, transwell migration assay was performed using SDF-1 as the chemoattractant in the lower chamber of the transwell. Consistent with the wound healing test, greater numbers of HP-BMSCs migrated into the lower chamber where SDF-1 was placed (Fig. 3C, D). In the presence of the CXCR4 inhibitor AMD3100 (25 mg/ml) in the upper chamber, the increased migration activity was largely blocked, supporting that the SDF-1/CXCR4 signaling system largely mediated the directional migration (Fig. 3E). The SDF-1-induced migration was also inhibited by the MMP-9 blocking mAb (10 μg/ml) but not the MMP-2 blocking mAb (10 μg/ml), respectively (Fig. 3F).
Hypoxic preconditioning promoted directional migration of BMSCs in vitro. The directional migration activity of cultured BMSCs in responding to wound injury signals or chemoattractants was tested using the scratch induced wound healing test and transwell migration test. (A) Wound healing test was performed to test the migration ability of N-BMSCs and HP-BMSCs into the lesion area. Representative phase contrast photos of BMSC cultures show cell locations immediately and 24 h after a scratch damage in the middle of the culture dish. (B) Quantification of distance that cells moved from the scratch edge into the empty/wounded area over a 24-h period. More HP-BMSCs moved into the central area than N-BMSCs. n = 12; *p < 0.001. (C–E) Transwell test measured cell migration from the upper chamber toward the chemoattractant stromal cell-derived factor-1 (SDF-1) (200 mg/ml) placed in the lower chamber. The experiments with HP-BMSCs were performed in the absence or presence of the CXCR4 inhibitor ADM3100 (25 μg/ml) or a monoclonal antibody against MMP-2 or MMP-9 (not shown). (F) Quantified data of results from experiments in (C) to (E). Compared with N-BMSCs, significantly more HP-BMSCs moved from the upper chamber to the lower chamber, indicating their greater activity of directional migration. AMD3100 added into the upper chamber significantly prevented the increased migration of HP-BMSCs. The MMP-9 blocking mAb (10 μg/ml) also showed inhibitory effects on the directional migration of HP-BMSCs, although MMP-2 mAb only showed a trend of inhibition on the HP-BMSC migration. n = 6, *p < 0.05 versus N-BMSCs; #p < 0.05 versus HP-BMSCs in PBS control.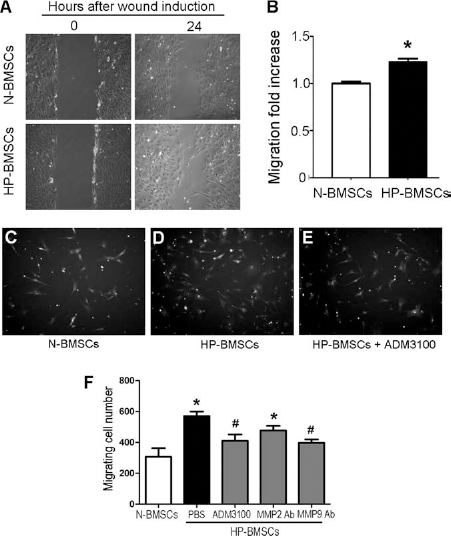
Detection and Distribution of BMSCs in the Ischemic Brain After Intranasal Delivery
Twenty-four hours after the onset of ischemia, a total volume of 100 μl sterile PBS containing N-BMSCs (1 × 106 cells), HP-BMSCs (1 × 106 cells), or PBS alone was administered alternatively into both nostrils. At 1.5 h after the first drop, some animals were sacrificed and brain sections were surveyed for distribution of transplanted cells (Fig. 4). Hoechst 33342 fluorescence-positive N-BMSCs and HP-BMSCs were detected in the OB and ipsilateral cortex around the ischemic region at this early time point (Fig. 4A, C). Transplanted cells were also observed in the contralateral hemisphere (Fig. 4D). Hoechst 33342-positive cells were immediately deposited around the vasculature, both inside and lining outside of the brain vessels (Fig. 4B).
Distribution and neurovasculature location of BMSCs after intranasal delivery into the ischemic brain. Immunohistochemical staining was applied to reveal the distribution of transplanted cells after intranasal delivery of BMSCs. (A) BMSCs (blue color with Hoechst 33342 staining) were seen in the area of olfactory bulb. (B) Collagen IV was used to stain the brain vessels (green). Transplanted cells (blue) were found inside a vessel (arrowhead), but many were lining vessels (dash arrows) or were deposited outside the vessels (arrows). (C) Many BMSCs moved to the ipsilateral brain cortex in and around the ischemic core. (D) Some Hoechstpositive cells can also be found in contralateral cortex. The morphology of prelabeled cells can be better seen under higher magnification (400 ×, magenta cells, inset images).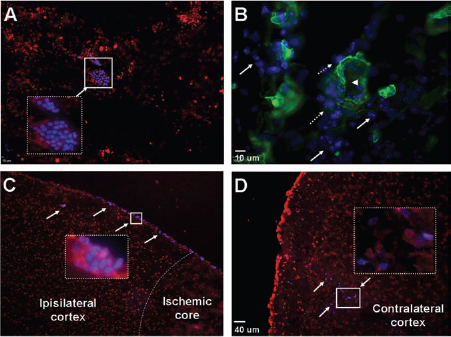
Hypoxic Preconditioning Increased BMSC Homing to the Ischemic Region
Although both N-BMSCs and HP-BMSCs were visible in the ischemic region 1.5 h after intranasal delivery, the majority of N-BMSCs were distributed along the olfactory passage route, including olfactory tubercle, piriform cortex, and amygdala (Fig. 5). N-BMSCs were also seen in the contralateral cortex, upper midline or lower midline brain region, and there was no significant difference in their distribution between ipsilateral and contralateral cortex (Fig. 5A, D). On the other hand, a drastically higher number and percentage of HP-BMSCs were identified in the ischemic region compared to all other analyzed brain areas (Fig. 5A–C, E). This was true even when compared with the transplanted cells in the olfactory passage regions (Fig. 5E). In cell counting assays on 30 brain sections obtained from the brain location of bregma 1.1 to −2.1 mm range (80-mm interval between counted sections), approximately 50 ± 7% of counted HP-BMSCs located to the infarct region compared with 23 ± 8% of N-BMSCs that mostly distributed in other brain areas (n = 6, p < 0.05) (Fig. 5F). When comparing with the total number of transplanted cells (1 × 106 cells), only 0.05 ± 0.01% of N-BMSCs were counted in the ischemic core and penumbra regions (region 4 in Fig. 5A; bregma 1.1 to −2.1 mm) while three times as many (0.16 ± 0.05%) HP-BMSCs homed to the ischemic region.
Distribution and homing of intranasally delivered BMSCs in the ischemic brain. Distribution of BMSCs in different brain regions was measured and quantified by counting Hoechst 33342 dye-positive cells in different brain regions. (A) Ischemic damage to the right cortex is shown as a negative/white area of TTC staining 24 h after ischemia. The dotted line areas with numbers show the brain regions where we quantified the distribution of intranasally delivered cells: 1, Motor cortex; 2, Upper midline; 3, Lower midline; 4, Ischemic cortex; 5, Contralateral cortex; and 6, Olfactory pathway (including olfactory tuvercle, piriform cortex and amygdala). (B, C) N-BMSCs or HP-BMSCs (1 × 106) were intranasally delivered 24 h after the onset of focal ischemia. One and half hours after the intranasal administration, animals were sacrificed and brain sections were counterstained with propidium iodide (PI) (red) to visualize the nuclei of the host brain cells and transplanted BMSCs (blue). Hoechst 33342-labeled HP-BMSCs (blue) were identified in the ischemia region (*). In image (C), HP-BMSCs counterstained with PI (magenta, arrows) were found in ipsilateral cortex. Inset image is enlarged from the framed area (original magnification: 400 ×). (D, E) Summary of distribution of N-BMSCs (D) and HP-BMSCs (E) in different brain regions as marked in (A). HP-BMSCs were heavily located to the ischemic cortex (area 4 in A). (F) The bar graphs summarized the percentage of cells that reached the ischemic brain region 1.5 h after intranasal delivery compared to all the cells found in the other brain region from bregma 1.10 to −2.10 mm. There is no significant difference in the distribution percentage of N-BMSCs between ipsilateral (ipsilat.) and contralateral (contralat.) cortex region. Significantly higher percentage of HP-BMSCs was seen in the ispilateral side of the brain. n = 6, *p < 0.05 versus HP-BMSCs in contralateral brain, #p < 0.05 versus N-BMSCs in the ispilateral brain.
Delayed Intranasal Delivery of HP-BMSCs Reduced Brain Infarct Volume and Cell Death After Ischemic Stroke
In this investigation, we focused on delayed postischemia treatment. BMSCs were administrated 24 h after ischemia, targeting a highly feasible therapeutic window for stroke patients. In TTC assays of brain sections 3 days after intranasal delivery, stroke animals that received HP-BMSCs showed significantly smaller infarct volume (12.51 ± 1.15 mm3; n = 13) when compared with those in the vehicle-treated group (17.25 ± 1.24 mm3; n = 18; p < 0.05; one-way ANOVA with Tukey's multiple comparison test). In mice receiving N-BMSCs, the infarct volume (12.93 ± 1.47 mm3, n = 14) was also significantly reduced when examined by ANOVA with Bonferroni correction (although no significance in ANOVA analysis with Tukey's test) (Fig. 6A, B).
TUNEL staining was performed to detect the cell death in the ischemic peri-infarct region 3 days after BMSC transplantation (i.e., 4 days after ischemia). The percentage of TUNEL-positive cells among the total cells was significantly decreased in both N-BMSC- and HP-BMSC-treated mice when comparing with that of control mice (Fig. 6C–F).
Intranasal BMSC treatment reduced cell death and brain damage after stroke. (A) Representative TTC staining of the mice brain 4 days after permanent middle cerebral artery (MCA) occlusion plus 7-min common carotid artery (CCA) ligation. (B) Quantification of 2, 3-5-triphenyl-tetrazolium chloride (TTC) staining shows a significant reduction in infarct ratio in N-BMSC-(n = 14) and HP-BMSC-treated animals (n = 13) compared with stroke vehicle controls (n = 18, *p < 0.05 by one-way ANOVA with Tukey's test or Bonferroni correction). (C–E) Images of terminal deoxynucleotidyl transferase biotin-dUPT nick end labeling (TUNEL)-positive cells in ischemic peri-infarct area of animals received vehicle, N-BMSCs and HP-BMSCs, respectively. (F) Quantified data from experiments of (C) to (E). Both N-BMSCs and HP-BMSCs showed a significant reduction in the number of TUNEL-positive cells in the peri-infarct area of the postischemic brain. n = 8; *p < 0.05 versus stroke vehicle controls.
Delayed Intranasal Delivery of HP-BMSCs Attenuated Ischemia-Induced Neurological Deficits
Adhesive removal test was used to assess sensorimotor deficits in mice 1 day after focal cerebral ischemia and 3 days after intranasal delivery of BMSCs. The time needed for stroke animals to feel and remove the sticky dot from the left paw (time to remove) was markedly prolonged after stroke due to the central damage in the sensorimotor cortex (29). This functional impairment was significantly attenuated in mice receiving HP-BMSCs (n = 12) compared with vehicle-treated mice (Fig. 7). In N-BMSC-treated mice, the time to remove was improved but the difference was not statistically significant compared to stroke controls (n = 10) (Fig. 7).
Intranasal BMSC treatment promoted functional recovery after stroke. Adhesive removal test was performed to evaluate sensorimotor deficits of the mice 4 days after focal cerebral ischemia (3 days after intranasal delivery of cells). The time to remove the sticky dot from the left paw was significantly reduced in mice receiving HP-BMSCs (n = 12) compared with vehicle-treated mice (n = 15; *p < 0.05). The time-to-remove value in N-BMSC-treated mice (n = 10) showed a trend of shortening but it was not statistically significant (p > 0.05).
Discussion
The present investigation demonstrates that intranasally administered BMSCs can survive and migrate into various regions of the brain. Importantly, many cells moved into the ischemic region and deposit outside of cerebrovasculature as early as 1.5 h after administration. This is a sharp contrast to systemic administration of BMSCs. Previous studies showed that, although cerebral local implanted (25, 50) or systemic injected (8, 43, 59) BMSCs could home to damaged brain tissues, the efficiency of the homing ability is extremely low and the precise mechanism underlying the directional migration remains largely unknown. Moreover, systemically administered cells are often trapped inside the vessels as long as 10 days after transplantation (24, 54). Strategies that could promote BMSC migration activity and homing to the ischemic brain region have been extensively explored, but significant improvement has been absent with the methods of systemic transplantation. Here, we provide the first evidence showing the distribution of intranasally delivered BMSCs in a focal cerebral ischemic model and their rapid homing ability and deposition outside of the brain vasculature. The intranasal delivery method of BMSCs is significantly improved by applying hypoxic preconditioning to the transplanted cells.
BMSCs have been a favorable cell source for cell transplantation therapy because of the accessibility for expansion in culture, their immusuppression property, multipotent potential, and the allogeneic transplantation potential that eliminates ethical considerations (1, 38, 52). Growing evidence suggests that BMSCs provide therapeutic benefits for ischemic stroke and other CNS diseases (2, 8, 34, 43) with increased trophic support and/or paracrine activity (24, 43). Clinical trials with BMSCs show absence of major adverse side effects (3, 19, 27, 45). Current clinical trials of BMSCs utilize either systemic administration or local injections. The disadvantages of these methods such as the inefficient brain delivery and high deposition of transplanted cells inside other organs with intraarterial/intravenial injections, invasive and time-consuming nature of cerebral injections, have hindered the efficacy of the cell-based therapy for acute stroke patients (1, 4, 17, 24, 54). Optimal cell delivery for large-scale clinical applications of BMSC therapies is essentially needed.
We show here that intranasal transplantation can be performed as late as 24 h after ischemia and still shows marked neuroprotective effects in the postischemic brain. This is a highly practical therapeutic window for most of stroke patients. Given the observations that some of the systemically administrated BMSCs may become passively trapped in the brain vessels (24, 54), we examined the localization of cells in respect to vessels following intranasal administration by counterstaining brain sections with a vessel marker. Our results provide the evidence that this delivery method leads to cells being directly deposited outside of the brain vessels. The observations also suggest a more efficient way of using transplanted cells to deliver therapeutic factors (i.e., regenerative genes and neurotrophines) to brain tissues.
Although intranasal administration of drugs has been used for decades, application of this method in cell-based therapy has only recently been tested in a Parkinson's disease model (9), a neonatal hypoxic-ischemic stroke model (51), and in normal rats and mice (10). In the neonatal study, 9-day-old mice were subjected to cerebral hypoxia-ischemia (HI) insults, and mouse BMSCs were transplanted intranasally 10 days after HI. At 28 days after HI, BMSCs were still present in the affected hemisphere although no differentiation was observed. Intranasal BMSC treatment decreased gray and white matter area loss at 28 days after HI and significantly improved sensorimotor function in the cylinder rearing. To the best of our knowledge, no study has been performed in an ischemia-only model or adult stroke models. The present investigation focused on brain delivery of BMSCs, we did not evaluate differentiation of transplanted cells. BMSCs are able to differentiate into neuronal and nonneuronal cells both in vitro and after systemic transplantation (32, 33, 58). Demonstration of neuronal differentiation and engraftment of cells into the host brain structures are key issues for the goal of a cell replacement therapy. Systematical investigations will be needed to explore this possibility with intranasal delivered BMSCs. After the transnasal process, migrating cells can move into the olfactory bulb and to other parts of the brain, enter into the cerebrospinal fluid (CSF) with movement along the surface of the cortex followed by entrance into the brain parenchyma (10). This is consistent with our observation that large portions of cells appeared along the olfactory pathway and on the surface of the brain. Ischemic insult and hypoxic preconditioning markedly shifted cell migration to the ischemic cortex. We saw more cells depositing inside the brain parenchyma both inside and around stroke core at 3, 6, and 12 h post delivery, suggesting that migration pathways other than the olfactory pathway exist in the ischemic brain. A close analysis of the BMSC distribution in the brain reveals that many cells inside brain parenchyma distributed outside along tube-like structures. Further immuno-assaying revealed that these structures are vessels. These findings provide evidence that intranasally administered cells might migrate through perivascular spaces (10), which provide rapid transport of particles/molecules within the CNS (16). Many labeled cells were found in perivascular area of the large vessels at the ventral side of the brain, which also suggests the possible perivascular migration pathway. More quantitative data are needed for evaluating migration routes of the cells after intranasal administration into the postischemic brain.
The exact mechanisms underlying BMSC migration capability to injury site are only partly understood. Adhesive interactions and chemokines released from injured tissue or endothelial cells may mediate BMSCs homing to specific sites (24). The interactions between the CXC chemokine SDF-1 and its receptor CXCR4 have been shown to play a critical role in mediating the ischemia-induced or other damages-induced recruitment of stem/progenitor cells (23, 41, 42, 47). We show in this investigation that blocking the interaction between SDF-1 and CXCR4 largely prevents the directional migration of BMSCs. This observation suggests that an upregulation of CXCR4 in transplanted cells may be a target for improved homing of transplanted cells. In addition to chemokines, BMSCs often secrete proteases to degrade the extracellular matrix (ECM), which is an essential step for their migration and invasion toward chemotactic factors (24). Knocking down MMP-2 with antibodies or siRNA substantially impaired BMSCs invasion (11, 35). Upregulating MMP-2 and/or MMP-9 activity in BMSCs augmented their invasive capacity (35). We have previously shown that HP promotes cell migration by a novel mechanism of upregulating the formation of the potassium voltage-gated channel (Kv2.1) and focal adhesion kinase-1 (FAK) complex that enhances FAK phosphorylation and activation (20). Together with upregulated CXCR4, MMP-2, and MMP-9 shown in this investigation, HP-BMSCs gain a significantly enforced ability to move across vascular/tissue barriers and into the injured site that is essential for the cell-based therapy.
Intranasal administration of BMSCs shows marked neuroprotective effect even 24 h after stroke. Significant protection of reduced infarct volume was seen with HP-BMSCs and N-BMSCs. More and more investigators have postulated that attenuated brain injury by transplantation of BMSCs is related to their trophic support and induction of neurotrophic factors in the host brain (53) as well as their modulation of inflammatory and immune responses (7, 34), which promotes survival of neurons in the stressful environment of penumbra. To this end, hypoxic preconditioning selectively increases neurotrophic and prosurvival trophic support of BMSCs that should contribute to the therapeutic effect of BMSCs (21, 48).
Very low cell survival, insufficient cell migration, and poor ability of homing to the injured site have been the major dilemmas in BMSC therapy for stroke and other CNS disorders such as traumatic brain injury (17, 31, 40, 54). Hypoxic preconditioning is a well-known endogenous mechanism that stimulates multiple prosurvival pathways, regenerative pathways, and suppression of inflammatory activities (5, 23, 44); all are particularly beneficial for increasing therapeutic efficacy and efficiency of stem cell therapy (36, 47). Our previous work showed that hypoxic preconditioning markedly increases survival of transplanted embryonic stem cell-derived neural progenitor cells and BMSCs in the ischemic brain and heart following IV injection (21, 48). Transplantation of HP-BMSCs promotes functional recovery after ischemic stroke and heart ischemia (20, 21, 48). In recent investigations, we demonstrate that HP enhances directed migration of BMSCs toward injured areas in cell culture studies and ischemic region after intravenous injection in animal experiments (20). The present investigation shows the synergic effects of hypoxic preconditioning and intranasal delivery of cells. The combination approach shows significant improvement of the cell-based therapy.
The tracking method of labeling transplanted cells with Hoechst dye in this investigation has its limitations. A major concern is that the dye might leak from prelabeled cells when they die and be picked up by neighboring cells that give false results. We quantified BMSC distribution as early as 1.5 h after delivery. According to previous knowledge of cell death time course of transplanted cells, cells can start to show apoptotic features 3 h after delivery (7). Our assessment thus was performed much earlier than cell deterioration or cell death. However, we cannot completely rule out that some leakage of Hoechst dye may occur. Optimized labeling method of BMSCs such as those with superparamagnetic iron oxide nanoparticles (22) or use of green fluorescent protein (GFP) transgenic BMSCs is preferred in further investigations. In the current study, we mainly focus on the HP treatment on brain delivery of BMSCs and the protective effect of intransally delivered cells acutely or subacutely after ischemic stroke. Long-term recovery experiments are needed to validate the therapeutic benefits of using HP-treated BMSCs. Further work is also needed to determine the optimal therapeutic time window, dosages, and the underlying mechanisms involved in intranasal delivery of BMSC treatment after ischemic stroke.
Footnotes
Acknowledgments
This work was supported by NIH grants NS057255 (S.P.Y.), NS073378 (S.P.Y.), NS058710 (L.W.), NS062097 (L.W.), NS075338 (L.W.), and American Heart Association Established Investigator Award (L.W.). This work was also supported by the NIH grant C06 RR015455 from the Extramural Research Facilities Program of the National Center for Research Resources. We gratefully acknowledge the technical support from Dongdong Chen in flow cytometry and Denise Song for assistance in immunohistochemical staining. The authors declare no conflicts of interest.