
Abstract
Embryonic stem cells (ESCs) have the potential to be used as an unlimited cell source for cell transplantation therapy, as well as for studying mechanisms of disease and early mammalian development. However, applications involving ESCs have been limited by the lack of reliable differentiation methods in many cases. Mesenchymal stem cells (MSCs) have also emerged as a promising cell source, but as suggested in recent studies, these cells display limited potential for proliferation and differentiation, thereby limiting their usefulness in the clinic and in the laboratory. Unfortunately, effective methods for induction of MSCs from pluripotent stem cells have not been established, and the development of such methods remains a major challenge facing stem cell biologists. Oxygen concentration is one of the most important factors regulating tissue development. It has profound effects on cell metabolism and physiology and can strongly influence stem cell fate. Here we demonstrate that severe low O2 concentrations (1%) can function as a selective pressure for removing undifferentiated pluripotent cells during the induction of MSCs from rabbit ESCs (rESCs) and that MSCs induced under severe hypoxic conditions function as normal MSCs; that is, they repopulate after cloning, express specific markers (vimentin, CD29, CD90, CD105, and CD140a) and differentiate into adipocytes, osteoblasts, and chondrocytes. Furthermore, we demonstrate that these cells can contribute to cartilage regeneration in an in vivo rabbit model for joint cartilage injury. These results support the notion that exposing ESCs to severe hypoxic conditions during differentiation can be used as a strategy for the preparation of functional MSCs from ESCs.
Introduction
Pluripotent stem cells are undifferentiated cells that can self-renew indefinitely. Pluripotent stem cells have been established from a number of sources including embryonic stem cells (ESCs) derived from mammalian preimplantation embryos, embryonic germ cells derived from primordial germ cells from the fetus, multipotent germline stem cells derived from mouse neonatal testes, as well as induced pluripotent stem cells produced by transfecting reprogramming factors into somatic cells (24). These cells are of particular interest for both regenerative medicine and basic research since they can give rise to many types of cells that derive from a single common genetic background. A number of studies have reported the ability to direct the differentiation of pluripotent cells in vitro, yet for some target cell types, there remains a need for reliable and efficacious protocols to control differentiation.
Mesenchymal stem cells (MSCs) are multipotent progenitor cells found in bone marrow, fat, synovium, and other tissues that give rise to connective tissues (13, 48). These cells are capable of differentiating into adipocytes, chondrocytes, myocytes, and osteoblasts and have thus been proposed as a source of cells for regenerative medicine (63). In addition to their potential for in vitro expansion and directed differentiation, these cells are capable of homing to injury sites where they release cytokines and promote tissue repair. In contrast to other sources of transplanted cells, MSCs express very low levels of cell surface immunogenic proteins from the major histocompatibility complex (MHC) and secrete immunosuppressive cytokines after interaction with host tissues (63, 84). For these reasons, they can be used for either autogenic or allogeneic transplantation and thus have emerged as a promising cell source for articular cartilage transplantation therapy (50).
Unfortunately, there are limits to the proliferative capacity, differentiation potential, and secretion of key molecular factors during in vitro expansion that represent barriers to the use of MSCs for clinical therapy. Normally, MSCs are obtained by extracting adult tissue and expanding cells in culture. However, aging significantly impairs MSC multipotency as well as the total number of cells that can be differentiated (67, 74). There is therefore emerging interest in the development of alternative cell sources for MSCs. Recently, some protocols have been reported for inducing MSCs from human embryonic stem cells (hESCs) and induced pluripotent stem cells (iPSCs) (6, 29, 39, 42, 46, 61). However, a safe and effective method for preparing clinically usable MSCs from pluripotent stem cells has not been realized, and numerous technical problems associated with differentiation of these cells remain to be resolved.
Oxygen (O2) concentration is one of the most important factors in the stem cell niche (15, 49). Direct measurement of O2 concentration has revealed that the bone marrow environment is very hypoxic, with some regions containing as low as ~1–2% O2 (49). It has also been demonstrated that oxygen concentration plays a fundamental role in maintaining the potency, proliferation, and plasticity of various stem cells (33, 51, 53). MSCs cultured in low oxygen display greater colony forming potential, enhanced cell proliferation and better maintain their undifferentiated characteristics (14, 23, 28, 30, 57, 79). These facts support the notion that MSCs are tolerant to hypoxic conditions. In contrast, some reports suggest that low O2 concentration impedes pluripotent self-renewal of ESCs. Niebruegge et al. and Prado-Lopez et al. demonstrated that the expression of lineage commitment-related genes is upregulated in human ESCs (hESCs) in response to hypoxic conditions (59, 64). Furthermore, Prado-Lopez et al. demonstrated that severe hypoxic conditions (1%) induce high levels of cell death in hESCs (64). Lee et al. reported the hypoxia induced cell death was also observed in the mouse embryoid body (EB), a structure comprised of ESCs in an early stage of differentiation (41). On the other hand, Abaci et al. showed that hypoxic culture could bring about cell cycle arrest in hESCs and hiPSCs (1).
The detailed mechanism of how ESC self-renewal is obstructed by hypoxic conditions is yet to be revealed. However, these findings raise the possibility that exposing ESC cultures to hypoxic condition during differentiation can be used as an effective and selective method for objective differentiated cells from ESCs. Here, we demonstrate that hypoxic (1% O2) treatment functions as a selective pressure to differentiate rabbit ESCs (rESCs) that possess the hESC-like characteristics referred to as “primed-form pluripotency” (25, 26, 58) into functional MSCs. We analyzed the properties of the rESC-derived MSCs (rESMSCs) to show that they displayed multiple differentiation properties following clonal expansion.
Materials and Methods
Animal Use and Care
The use of animals complied with the regulations of the Institutional Animal Use and Care Committee of the Department of Biology-Oriented Science and Technology at Kinki University and the Kinki University Faculty of Medicine. Each rabbit was housed in an individual pen with food and water ad libitum. Animals were exposed to an artificially controlled light–dark regime with 14-h lighting and 10-h darkness. Temperature was maintained between 20°C and 25°C in a ventilated room. All operations were performed under anesthesia induced by intravenous injection of 45 mg/kg sodium pentobarbital and local injection of 2% Xylocaine with epinephrine.
Derivation and Culture of Rabbit Embryonic Stem Cells
Dutch Belted females (Kitayama Labes, Nagano, Japan) were superovulated by injecting them intraperitoneally with pregnant mare serum gonadotrophin (PMSG, Sumitomo Pharmaceutical, Inc., Osaka, Japan) and human chorionic gonadotrophin (hCG, Sumitomo Pharmaceutical, Inc.) and then mated with Dutch Belted males. Two days after mating, fertilized four- to eight-cell stage embryos were collected by flushing out the oviduct and were then cultured in vitro to the expanded blastocyst stage.
For derivation of ESCs, the blastocysts were transferred onto mitomycin-C-treated (10 μg/ml in medium for 90 min, Invitrogen Corporation, Carlsbad, CA, USA) mouse embryonic fibroblast (MEF) feeder cells prepared from 13.5-day-old ICR mouse fetuses (CLEA Japan, Tokyo, Japan) and cultured in rabbit ESC medium (rESM) consisting of 20% knockout serum replacement (KSR; Invitrogen), Dulbecco's modified Eagle's medium (DMEM)/F12 (Invitrogen), 2 mM l-glutamine (Wako Pure Chemical Industries, Tokyo, Japan), 1% nonessential amino acids (Invitrogen), 0.1 mM β-mercaptoethanol (Invitrogen), and 8 ng/ml human recombinant basic fibroblast growth factor (bFGF, Wako) for 10 days. These rESCs were passaged using CTK colony dissociation solution (0.25% trypsin, 0.1% collagenase IV, 20% KSR, and 1 mM CaCl2 in PBS) as previously reported (75). The multiple differentiation potentials of these cells were examined both in vitro and in vivo. For in vitro differentiation, the rESC colonies were collected with CTK solution, washed once with fresh rESM, and then transferred into suspension culture in 10% fetal calf serum (FCS)-supplemented DMEM (Invitrogen). After 6 days in suspension culture, embryoid bodies that formed from the rESCs were collected and replated onto gelatin-coated culture dishes. The cells derived from the EBs were evaluated by immunofluorescent staining with specific antibodies following 6 days in culture.
Differentiation properties of the rESCs were evaluated in vivo via teratoma formation assay. Cell suspensions containing 5 × 106 rESCs were subcutaneously injected into the femora of male severe combined immunodeficient (SCID) mice at 6 weeks of age (CLEA Japan, Tokyo Japan). After 8 weeks from the time of cell injection, teratomas were collected, stained by hematoxylin and eosin, and observed histologically.
To observe the effect of inhibitors on the pluripotency of rESCs, the ESCs were passaged onto Matrigel (BD Falcon, Bedford, MA, USA) and cultured in MEF-conditioned rESM. After 24 h of matrigel culture, 8 ng/ml bFGF, 1,000 U/ml leukemia inhibitory factor (LIF; Millipore Billerica, MA, USA), 1 μM Janus kinase (JAK) inhibitor I (Merck, Darmstadt, Germany), 1 μM specific mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (Erk) inhibitor PD0325901 (Stemgent, Cambridge, MA, USA), or 0.5 μM a specific activin-like receptor 4/5/7 (ALK4/5/7) inhibitor A83-01 (Stemgent) were added to the culture medium. After 48 h, cultures were collected and analyzed for the pluripotent marker genes Nanog and POU5f1 [pituitary-specific, octamer-binding transcription factor (OCT) Unc-86 domain family class 5 homeobox 1] using quantitative RT-PCR (qRT-PCR).
To further demonstrate that these cells exhibited primed form pluripotency, we performed a single-cell digestion assay by treating the rESCs with 0.25% trypsin (Invitrogen) and 0.04% EDTA (Sigma-Aldrich, St. Louis, MO, USA) (trypsin-EDTA), seeding the dissociated rESCs onto gelatin-coated tissue culture dishes, and culturing for 24 h in rESM with/without 10 μM of a Rho-associated, coiled-coil containing protein kinase (ROCK) inhibitor Y27632 (Wako). Cell death was then examined by flow cytometry (fluorescence-activated cell sorting; FACS) with the dead cell marker propidium iodide (PI, BD Pharmingen, San Diego, CA, USA) and by Western blot analysis for caspase 3, which is a key protein mediating apoptosis. In the qRT-PCR and FACS analysis, three independent experiments were performed.
Establishment and Culture of Rabbit Bone Marrow Mesenchymal Stem Cells (rBMMSCs)
The control MSC line was established from rabbit bone marrow tissue. Establishment of a stem cell line was performed following a previously published protocol with slight modifications (71, 81). Briefly, bone marrow cells were isolated from a male Japanese white (JW) rabbit (3.0 kg in weight; Hamaguchi Animals, Osaka, Japan) by flushing out the femoral and tibial cavities with phosphate-buffered saline and plating the flushed material onto 10-cm dishes in 10% FCS-α-modified minimum essential medium (αMEM). Three days after plating, nonadherent cells were washed away in PBS, and adherent cells were further propagated. Once confluent, these cells were dissociated by trypsin-EDTA, washed, diluted to 200 cells/35-mm dish, and cultured. These cells were determined to be positive for the MSC markers vimentin, CD29, and CD105 by Western blot analysis prior to further experiments. The differentiation potentials of the established rBMMSCs were comparable to standard MSCs, as established by induced differentiation to adipocytes, osteocytes, and chondrocytes (data not shown).
Hypoxic Treatment of Undifferentiated rESCs
Rabbit ESCs were cultured on Matrigel (BD Falcon) in MEF-conditioned rESM medium under normoxic (20% O2 with 5% CO2) culture conditions for 24 h and then transferred into either 20% O2 with 5% CO2, 5% O2 with 5% CO2, or 1% O2 with 5% CO2 for 48 h. Hypoxic atmospheres were balanced with nitrogen. All conditions were controlled by a multigas incubator MCO-5M (SANYO Electric Co. Ltd., Osaka, Japan). Actual O2 concentrations were monitored by a paperless recorder DXAdvanced DX1000 (Yokogawa Electric Corporation, Tokyo, Japan) throughout the culture period (data not shown).
Quantitative RT-PCR (qRT-PCR) Analysis
Total RNA was extracted with the TRIzol reagent (Invitrogen) and reverse transcribed with the high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA, USA). QRT-PCR with total cDNA was performed using Perfect Real-Time SYBR Green II (Takara Bio, Inc., Shiga, Japan) with rabbit specific primers (Table 1) in the Thermal Cycler Dice® Real-Time System (Takara Bio, Inc.) at 95°C for 10 s followed by 40 cycles of 95°C for 5 s, 60°C for 30 s. Primers for putative sex-determining region Y box 2 (Sox2), Krüppel-like factor 4 (Klf4), and platelet-derived growth factor receptor a (PDGFRα) were designed in the sequences originally obtained by TA-cloning and BigDye terminator sequencing (data not shown). Briefly, partial sequences of the rabbit putative Sox2, Klf4, and PDGFRα were amplified using universal primers designed on conserved sequences in mouse and human total cDNA from the rESCs or rabbit bone marrow tissues and the Platinum Taq PCRx DNA polymerase (Invitrogen). The amplicons were then ligated into the pGEM-T easy vector (Promega Corporation, Madison, WI, USA), transformed into E. coli JM109 (Takara Bio), purified by a QIAprep Spin mini-prep kit (QIAGEN, Valencia, CA, USA), and sequenced using the Big Dye Terminator v3.1 cycle sequencing ready reaction kit (Applied Biosystems) and the ABI3730 capillary sequencer (Applied Biosystems). To quantify the relative expression of each gene, the Ct (threshold cycle) values were normalized to an endogenous reference (ΔCt = Cttarget - Ctreference) and compared with a calibrator (control), using the “ΔΔCt method (ΔΔCt = ΔCtsample - ΔCtcalibrator)” (16). For the reference genes, we used 28s rRNA to evaluate pluripotent or mesenchymal gene expression during hypoxic experiments and used glyceraldehyde 3-phosphate dehydrogenase (GAPDH) to observe chondrocyte-specific gene expression in vitro and in vivo.
Primer Sequences Used in qRT-PCR

Pou5f1, Pituitary-specific, Octamer-binding transcription factor (OCT) Unc-86 domain family class 5 homeobox 1; Sox2, sex-determining region Y box 2; Klf4, Krüppel-like factor 4; PDFGRα, platelet-derived growth factor receptor α; Col2A1, collagen type II α1; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Immunofluorescence of Cultured Cells
The cultures were fixed with Mildform 10N (Wako) at room temperature for 1 h. Fixed cells were washed with PBS, blocked with Block Ace (Dainippon Sumitomo Pharma, Osaka, Japan) for 1 h, washed twice more, and incubated with each primary antibody overnight at 4°C. The samples were washed twice and then incubated with secondary antibodies [fluorescein-isothiocyanate (FITC) or Texas red-conjugated rabbit, goat, donkey, or bovine polyclonal antibodies; all were purchased from Santa Cruz Biotechnology] (Table 2). Samples were then counter stained with DAPI (1 μg/ml in PBS; Wako) prior to direct observation. Negative controls were prepared omitting the primary antibodies (data not shown).
Primary Antibodies Used in Immunofluorescent Analysis and/or WB Analyses

SSEA-4, stage specific embryonic antigen-4; Cdx2, caudal type homeobox 2.
Western Blot Analysis
Cells were collected by scraping, homogenized in sodium dodecyl sulfate (SDS) buffer (4% SDS, 125 mM tris-glycine, 10% 2-mercaptoethanol, 2% bromophenol blue in 30% glycerol), and then subjected to polyacrylamide gel electrophoresis (PAGE) in the presence of SDS (SDS/PAGE) followed by electrotransfer onto polyvinylidene difluoride (PVDF) membranes (Hybond-P; Amersham Pharmacia Biotech, Buckinghamshire, UK). The blotted membranes were blocked overnight with Block Ace (Dainippon Pharmaceutical, Osaka, Japan) and treated with each primary antibody (Tables 2 and 3) overnight at 4°C. Detection was realized by enhanced chemiluminescence with an ECL plus Western blotting detection system (Amersham Pharmacia Biotech, Buckinghamshire, UK) and horseradish peroxidase (HRP)-conjugated secondary antibodies (all were purchased from Santa Cruz Biotechnology, Santa Cruz, CA, USA) corresponding to each primary antibody. The lumino-labeled membranes were analyzed by the CCD-based chemiluminescent analyzer LAS 4000 (Fujifilm, Toyko, Japan).
Primary Antibodies Used in WB Analysis

Mdm2, murine double minute 2 oncogene; pRb, retinoblastoma protein.
Cell Cycle Analysis
To observe the effect of hypoxic culture on the rESC cell cycle, the rESC-1 cell line was cultured on Matrigel in MEF-conditioned rESM under 20% O2 with 5% CO2, 5% O2 with 5% CO2, or 1% O2 with 5% CO2 for 48 h. Cells were then dissociated to single cells with trypsin-EDTA, washed twice with PBS containing 2% FCS, and fixed with chilled 99.5 % ethanol (final concentration 70%) overnight at −20°C. After fixation, cells were washed once with PBS and resuspended in PBS containing 2% FCS and incubated with 50 U DNase-free RNase A (Calbiochem, San Diego, CA, USA) for 30 min at 37°C. After incubation, cells were stained with PI for 15 min at room temperature. Flow cytometry analysis was performed using a FACS Calibur (BD Biosciences, San Diego, CA, USA), and cell cycle analysis was performed using CELLQUEST software (BD Biosciences). rESCs cultured with 1 μg/ml Colcemid (Invitrogen) or 1 μg/ml Nocodazole (Sigma) for 6 h were used as control samples.
Embryoid Body (EB) Formation, Adhesion, and Hypoxic Treatment of Differentiated Cells
Suspension cultures of embryoid bodies were initiated by resuspending 100 colonies per milliliter in 10% FCS-DMEM. EBs were maintained for 4 days in suspension with medium refreshment performed every 2 days by allowing EBs to sediment in 15-ml centrifuge tubes, aspirating the old medium, and resuspending in fresh medium. EBs were then transferred onto gelatin-coated dishes and cultured in 10% FCS-αMEM for 6 days. During adherent culture, oxygen concentrations were changed to either 20% O2, 5% O2, or 1% O2 (Step 1). After 6 days of oxygen-controlled culture, EB-derived cells were treated with trypsin-EDTA, washed once, resuspended in 10% FCS-αMEM, and cultured for another 3 days in each O2 concentration (Step 2). Colony formation assays were performed by culturing the EB derivatives at Step 1 or Step 2 for 14 days after trypsin dissociation and dilution of the treated cells to 200 cells/35-mm dish. To prevent single-cell digestion-related cell death of human-type ESCs, suspension medium was supplemented with 10 μM of Y27632. After 14 days, the cultures were fixed with ice-cold ethanol, washed twice with PBS, and used for analysis of alkaline phosphatase (ALP) activity with Leukocyte Alkaline Phosphatase Kit (Sigma) or colony counting after crystal violet staining. Reproducibility was confirmed by three independent experiments.
Cloning and Reexpansion of Induced MSCs
Fibroblastic colonies were scraped from their substrates with a glass capillary, transferred into TrypLE Express (Invitrogen) in a 96-well multiplate to be dissociated, and plated onto gelatin-coated plates in 10% FCS-αMEM supplemented with 10 ng/ml bFGF. The medium was changed after the cells attached and every 2 days thereafter. The cells were passaged every 3–4 days with trypsin-EDTA (Invitrogen) and maintained for further experiments in 10% FCS-αMEM supplemented with 10 ng/ml bFGF.
In Vitro Differentiation Assay of rESC-Derived MSCs
Cells were plated at a density of either 2 × 105 cells/35 mm dish or 1 × 104 cells/96-well multiplate dish and cultured to confluence. To promote adipogenesis, the medium was switched to adipocyte differentiation medium consisting of 10% FCS-αMEM supplemented with 10−7 M dexamethasone (Sigma), 0.5 mM isobutylmethylxanthine (Sigma), and 100 μM indomethacin (Sigma). After 10 days, the adipogenic cultures were fixed with 10% neutral formalin and stained with Oil Red-O solution (Primary Cell Co., Ltd., Sapporo, Japan). To promote osteoblast differentiation, the medium was switched to osteoblast differentiation medium consisting of 10% FCS-αMEM supplemented with 10−8 M dexamethasone, 10 mM 2-glycerophosphate (Sigma), and 50 μg/ml ascorbate (Sigma) for 14 days. After differentiation, the dishes were fixed with 10% neutral formalin and stained with 0.5% Alizarin Red S (Sigma) solution. To promote chondrogenesis, 2.5 × 105 cells were cultured in a 15-ml polypropylene tube (BD Falcon) as a pellet or 1 × 105 cells were plated in 96-well plates and culture in chondrocyte differentiation medium (Invitrogen) for 21 days. After chondrocyte differentiation, cells were treated with TRIzol solution; RNA was purified and used for further studies.
Preparation of Green Fluorescent Protein (GFP)-Expressing rESMSCs
GFP-expressing rESCs were produced by introducing pCAG-GFP-IRES-Puro vector into the rESCs also used above. To create the pCAG-GFP-IRES-Puro vector, the GFP sequence obtained from the pCAG-GFP vector (Addgene, #11150, distributed by Dr. Connie Cepko; Harvard Medical School, Boston, MA) was cloned into the NotI/EcoRI site of the pCAG-IRES-Puro plasmid, which was kindly gifted from Dr. Hirofumi Suemori (Kyoto University., Japan). The pCAG-GFP-IRES-Puro vector was linearized by SspI and electroporated into 3 × 106 rESCs using a Gene Pulser II (BioRad Laboratories, Hercules, CA, USA) at 250 V, 950 μF. The rESC cultures were selected in rESM supplemented with 10 μM Y27632 and 7 ng/ml puromycin (Invitrogen) for 4 days. The puromycin-selected cells were treated with trypsin, passed through a cell strainer (BD Biosciences), washed once with rESM, and resuspended in rESM supplemented with 10 μM Y27632. The GFP-expressing cells were further purified by FACS vantage (BD Biosciences). The sorted cells were seeded onto mitomycin-C-treated MEF feeder cells and cultured in Y27632-supplemented rESM for 24 h. The medium was changed with fresh rESM every 2 days until colonies were observed. MSC induction was performed with the procedures described in this manuscript. Induced MSCs from the GFP-expressing rESC line (rgESMSC-1) were used for transplantation studies after cloning, determination of differentiation to osteoblasts and adipocytes, and confirmation of MSC marker expression.
Transplantation Assays with Rabbit Articular Cartilage Defects
Cells were transplanted as sheets (31). Transplantation as cell sheets is rapid and convenient since they stably and reproducibly adhere to defect sites after only short periods of time. The cell line rESMSC-1 was seeded directly at a density of 2 × 106 cells per temperature-responsive cell culture dish (35-mm diameter, Cell Seed, Inc., Tokyo, Japan). Cells were maintained in 10% FCS-αMEM supplemented with 10 ng/ml bFGF until they reached confluence and formed a cell sheet. On the day of transplantation, viable cell sheets were harvested by reducing temperature of the culture to room temperature (approximately 24°C) for 30 min, following a previously described protocol. For cell transplantation, skeletally mature JW male rabbits, weighing an average of 3.0 kg, were used. The rabbits were anesthetized, the right knee joint was approached through a medial parapatellar incision, and the patella was dislocated laterally. Full thickness osteochondral defects (5 mm diameter, 3 mm deep) were created in the trochlear groove of the femur as reported previously (35), and then the defects were filled with the cell sheet. All rabbits were returned to their cages after the operation and were allowed to move freely. Animals were euthanized with an overdose of sodium pentobarbital at 2 and 4 weeks after the operation, and the articular tissues were examined by histological staining, immunofluorescence, or FACS. For the transplantation experiment, we prepared three animals for histological analysis and three animals for FACS analysis (a total of six animals).
Histology and Fluorescence Microscopy
The knee joints including regeneration sites were fixed in Mildform 10N and decalcified with 85% formic acid and 20% aqueous sodium citrate. For histological observation, the samples were dehydrated and embedded in paraffin. The sections were then stained with Safranin-O (Sigma) or double-stained with Alcian blue (Wako) and Alizarin red. For fluorescent observation of GFP-expressing cells in the regenerated cartilage, deparaffinized and rehydrated paraffin sections were blocked with Block Ace for 1 h, washed twice with PBS, and incubated with 1/200 diluted anti-GFP rabbit polyclonal antibody (Santa Cruz Biotechnology, sc-8334) at 4°C overnight. The specimens were then washed twice with PBS containing 10% Block Ace and incubated with 1/1,000 diluted FITC-conjugated anti-rabbit IgG bovine secondary antibody. After two washes, FITC-labeled specimens were stained with 1/1,000 diluted DAPI and observed using a fluorescence microscope (BZ-9000, Keyence, Osaka, Japan).
FACS Sorting of the GFP-Positive Transplanted Cells From Recipient Cartilage Tissues
Regenerated sites were collected using a scalpel, washed twice with PBS, and dissociated enzymatically for 3 h in 300 U/mg collagenase (Wako) in DMEM/F12 supplemented with 0.3% bovine serum albumin (Sigma). The dissociated cells were collected, filtered through a 40-μm cell strainer (BD Falcon), washed twice with PBS, and sorted by FACS vantage. To avoid contamination with false-positive cells, we examined the samples with two bandpass filters [530 nm for FITC/GFP and 585nm for phycoerythrin (PE)] and displayed the data as a FL-1 (GFP) versus FL-2 (PE) density plot as suggested previously (45). The FL-1 cells were selected, and the GFP-expressing transplanted cells were further analyzed.
Statistical Analysis of the Data
Significant difference was detected by Tukey–Kramer HSD test or Student's t test. A value of p < 0.05 was considered significant.
Results
Derivation and Characterizations of Rabbit ESCs
Eighteen rESC lines were successfully established from 71 blastocysts. In this study, we selected two cell lines and used them for subsequent studies. Both rESC-lines expressed ALP, POU5f1, and stage-specific embryonic antigen-4 (SSEA-4) (Fig. 1A). When the rESCs were transferred to nonadherent environments, they aggregated and formed EBs. Several types of lineage-committed cells were identified after the EBs adhered; class III β-tubulin-expressing neural cells, Desmin-expressing mesodermal cells, and albumin-expressing endodermal cells were observed. Furthermore, expression of caudal type homeobox 2 (Cdx2), a differentiation marker for extra-embryonic lineages, was prominent in ESCs (Fig. 1A). The rESCs generated teratomas in SCID mice after 2 months of cell injection. Hematoxylin–eosin staining of teratoma sections showed tissues representative of the three different germ layers (Fig. 1A).

Generation and characterization of rabbit embryonic stem cells (rESCs). (A) Characterization of the rESC-1. Pluripotency of the rESCs were determined by alkaline phosphatase activity (ALP), gene expression, and immunofluorescence labeling for pluripotent cell markers pituitary-specific, octamer-binding transcription factor (OCT) Unc-86 domain family class 5 homeobox 1(POU5f1), and stage-specific embryonic antigen-4 (SSEA-4). Scale bars: 50 μm. These images were presented without DAPI to clearly show their localization. Multiple differentiation properties in vitro were evaluated by immunofluorescence with anti-neuron-specific class III β-tubulin antibody for neural cells, anti-Desmin antibody for cardiac cells, anti-albumin antibody for hepatocytes, and anti-caudal type homeobox 2 (Cdx2) antibody for trophoblastic cells. Scale bars: 50 μm. Sections of teratoma were analyzed for structures that developed from three germ cell layers after hematoxylin and eosin (H&E) staining. Scale bars: 250 μm. (B) RT-PCR and WB-based characterization of the rESCs in their undifferentiated state. Fb, fibroblast cells; DW, distilled water; Sox2, sex-determining region Y box 2; GAPDH, glyceraldehyde 3-phosphate dehydrogenase. (C) Specific inhibition of the signals required for maintenance of pluripotency. The x-axis show relative expression ratios [rESCs in the basic fibroblast growth factor (bFGF) added condition with no inhibitors = 1.0]. Two cell lines (shown as #1 and #2 in the figure) were used in this study. Bars show the mean score of three independent experiments and bars depict SD. ∗,∗∗Statistically significant differences of p < 0.05 from the standard culture condition (i.e., the bFGF added condition with no inhibitors, shown uppermost in the graph). ∗Significant differences in the rESC-1 line; ∗∗significant differences in the rESC-2 line. LIF, leukemia inhibitory factor; JAKi, Janus kinase inhibitor; MEKi, mitogen-activated protein kinase kinase; ALKi, activin-like receptor inhibitor. (D) Single cell digestion caused rESCs cell death in 24-h cultures after seeding. Y27632 supplementation improved the survival rate. PI, propidium iodide.
The rESCs displayed pluripotent gene expression as evidenced by Nanog, POU5f1, and Sox2 detection using RT-PCR. ESC marker proteins POU5f1 and E-cadherin were also observed by Western blot analysis (Fig. 1B). To confirm that the rESC lines possessed hESC-like characteristics as previously reported (26), we observed the response of the rESCs to administration of specific inhibitors, which block pluripotency-related signaling cascades. In accordance with other reports involving rESCs (26), maintaining an undifferentiated cell state is dependent on the bFGF and Activin/Nodal signaling pathways, and is not affected by LIF addition. In our experience, rESCs could not maintain an undifferentiated status in the presence of PD0325901, a selective inhibitor of MEK/ERK, which are downstream targets of the FGF pathway, or in the presence of the Activin/Nodal signaling inhibitor A83-01, which selectively blocks ALK4/5/7. On the other hand, inhibition of LIF/signal transducer and activator of transcription 3 (STAT3) signaling with a Janus kinase (JAK) inhibitor did not have a significant effect on rESC differentiation status (Fig. 1C). These data demonstrate that both Activin/Nodal signaling and FGF signaling are important for maintaining undifferentiated cell states and that LIF/STAT3 signaling is not essential for maintenance of undifferentiated cell states for rESCs.
Another specific characteristic of primed-form pluripotent stem cells is their sensitivity to enzymatic digestion, which can be reduced by inhibition of ROCK activity (77, 82). Thus, we examined the effect of Y27632 administration during single-cell digestion and culture. After trypsin digestion, rESCs clearly proceeded to cell death. Without treatment with Y27632, Western blot analysis showed the appearance of a band at 20-kDa, representing one of the activated domains of caspase 3. Y27632 supplementation dramatically improved the survival rates from approximately 15–20% to 80%, and in this condition, the band showing activated caspase 3 was dramatically diminished (Fig. 1D). These data clearly demonstrated that the cell lines we selected possessed characteristics of pluripotent ESCs and indicated that the rESCs possessed hESC-like primed-form characteristics, corresponding to the previous reports (26, 82).
Severe Hypoxia-Induced Cell Death in rESCs
In agreement with previous studies, pluripotent gene expression levels were maintained in hypoxic conditions at both 5% O2 and 1% O2 conditions (data not shown). However, the colony morphology of the rESCs cultured in 1% O2 condition was dramatically different with many dead cells and spaces between cells observed (Fig. 2). FACS analysis showed that approximately 50% of cells were dead (Fig. 2A). To determine if hypoxia-induced cell death resulted from apoptosis, we performed Western blot analysis for caspase 3. The activated (cleaved) form of caspase 3 was not detected in any of the rBMMSC experimental groups but was detected in all groups of rESCs. The levels of cleaved caspase 3 increased in response to severe hypoxia (Fig. 3A), suggesting that hypoxia induces apoptosis in the rESCs.

Effect of hypoxic treatment on rESCs. (A) Fluorescence activated cell sorting (FACS) analysis for cell survival rate in each oxygen condition. (a) FACS images of PI-stained rESCs in each condition. The areas enclosed by the black lines show PI-negative cell fractions. (b) Cell survival rate obtained by calculation from the PI-negative fractions. Two different rES cell lines (shown as #1 and #2 in the figure) were used in this study. Bars show the mean scores from three independent experiments. Error bars depict SD. (B) Cell cycle analysis of the rESCs and rabbit bone marrow mesenchymal stem cells (rBMMSCs) maintained in each condition. The leftmost image shows the gate used to sort only single cells for use in the present analysis. Colcemid-treated rESCs and Nocodazole-treated rESCs were examined as controls for cell cycle transition. The y axis represents cell numbers. Rates of each phase were represented as means of three independent experiments. The rESC-1 line was used for this study. (C) Numbers of rESCs after 48 h of culture in each condition. Two different rES cell lines (shown as #1 and #2 in the figure) were used in this study. Bars represent the mean scores from three independent experiments. Error bars depict SD.
When we observed cell cycle status, a larger proportion of cells cultured in the 1% O2 condition were in G0/G1 phase than for the 20% or 5% O2 culture conditions (Fig. 2B). This suggests that the cell cycle was arrested in the 1% O2 condition. These trends carried over to the total cell numbers observed after hypoxic treatment, as total cell numbers after 48 h of culture in the 1% O2 condition decreased by approximately 3- to 4-fold relative to the 20% or 5% O2 conditions (Fig. 2C).
In previous studies using tumor cell lines, hypoxia has been shown to induce accumulation of p53 and hypophosphorylated retinoblastoma protein (pRb) (12), which prevent cell growth by inhibiting cell cycle progression. We examined whether hypoxia-induced apoptosis or cell cycle arrest were triggered by participate with these molecules. The quantity of murine double minute 2 oncogene (Mdm2), which is an important negative regulator of p53, decreased after 1% O2 treatment. However, no differences were observed in the expression levels of p53 and POU5f1. It is known that p53 translocates to the nucleus and induces cell growth arrest and apoptosis in response to various stresses (9, 66). We observed the localization of p53 after hypoxic treatment and noticed that treatment with 1% O2 caused p53 to translocate to the nucleus in most cells (Fig. 3C). These results suggest that the cell death induced in severe hypoxia may be mediated by p53 and that this process is possibly regulated by interaction with Mdm2. Furthermore, in the 1% O2 condition, the phosphorylated form of retinoblastoma protein (p-pRb), which indicates progression through the cell cycle, decreased. This result might support a shift to cell quiescence rather than active progression through the cell cycle shown in the FACS analysis.

Analysis for the effects of the hypoxic treatment in rESCs. (A) Caspase 3 activity in rBMMSCs and rESCs cultured in each oxygen concentration for 48 h. The band at 19 kDa shows activated caspase 3. Two different rES cell lines (shown as #1 and #2 in the figure) were used in this study. (B) Western blot analysis of murine double minute 2 oncogene (Mdm2), p53, phosphorylated retinoblastoma protein (p-pRb), and POU5f1 in the rESCs cultured in each condition for 48 h. (C) Localization of p53 protein in rESCs cultured in each oxygen condition was shown by FITC. Nuclear localization of p53 was only observed in the 1% O2 culture condition. These images show the result of an experiment using the rESC-1. Scale bar: 20 μm.
Hypoxic Treatment Inhibits Pluripotent Cell Expansion During Differentiation
We determined if hypoxic treatment decreases the number of undifferentiated cells during in vitro differentiation to MSCs. After 10 days from the onset of differentiation (i.e., 6 days after EB attachment), fibroblastic cells appeared in peripheral regions of the EBs (Fig. 4A). The mesenchymal lineage marker vimentin was expressed in these cells, indicating that they were committed to a lineage capable of producing MSCs. When the differentiation process was performed in 20%, 5%, and 1% O2 conditions, significant decreases in Nanog, POU5f1, Sox2, and Klf4 gene expressions were detected by qRT-PCR. Statistically significant differences were only observed in cells derivative from rESC-2s (Fig. 4B). Immunoblotting showed diminished levels of two undifferentiated ESC markers POU5f1 and E-cadherin in response to severe hypoxia (Fig. 4C). These results suggested that the number of undifferentiated cells was effectively decreased by culture in the 1% O2 environment.

Selective deletion of the undifferentiated cells in hypoxic condition. (A) Immunofluorescence for vimentin in rabbit embryoid body (rEB)-derivatives. (a) Phase contrast image, (b) DAPI, (c) vimentin expression detected with anti-vimentin antibody and fluorescein isothiocyanate (FITC)-conjugated secondary antibody, (d) merged image. Scale bar: 200 μm. (e) Magnified image of the vimentin expressing fibroblastic cells that emerged in the peripheral regions of the EBs. Scale bar: 100 μm. (B) Alteration of marker gene expressions of EB derivatives in each oxygen condition. Two different rES cell lines (white bars show the rESC-1, and gray bar shows the rESC-2) were used in this study. ∗,∗∗Statistically significant differences (p < 0.05) from the normoxic conditions. ∗Significant differences in the rESC-1 line, and ∗∗significant differences in the rESC-2 line. KLF4, Krüppel-like factor 4; PDFGRα, platelet-derived growth factor receptor a. (C) Western blot analysis for the undifferentiated ESC markers E-cadherin and POU5f1 and the mesenchymal cell marker vimentin. These images show the result of an experiment using the rESC-1. (D) Proliferation assays of the EB derivatives of the rESC-1 in each oxygen condition. Each dot shows the mean scores of three independent experiments. EBC, the EB derivative cells. (E) Teratomas formed from the EB-derivative of the rESC-1 prepared in each oxygen concentration. As a control, teratoma developed from the undifferentiated rESC-1 for same experimental period is shown.
Next, we observed proliferation of cells from the primary cultures through five-time passages and compared proliferation rates. In agreement with other studies in humans (14, 57), hypoxic cultures slightly improved proliferation of the rBMMSCs, and proliferation rates of the rBMMSCs were apparently slower than that of the EB-derived cells. It is likely that the EB-derived cells contained many undifferentiated or partially differentiated cells, which can proliferate rapidly and might explain the increased cell numbers in the EB-derived cells. The proliferation rate of the EB-derived cells was slower for the 1% O2 condition than for the 5% and 20% conditions but was comparable to that of the control rBMMSCs cultured in 1% O2 (Fig. 4D). We transplanted the EB-derived cells from each condition and examined alterations in tumorigenicity. Interestingly, the EB derivative prepared and passaged in 1% O2 lost the ability to form teratomas even after 3 months of injection in SCID mice (no teratoma formation in three transplantation cases). The cells obtained from 5% O2 and 20% O2 formed teratomas (teratoma formations were determined in all cases of three transplantations) (Fig. 4E).
One of the important features of MSCs is the ability of these cells to form colonies from single cells after limiting dilution. Therefore, we observed colony formation properties following limiting dilution of the EB-derived cells cultured in 20%, 5%, and 1% O2 conditions. After 2 weeks of limiting dilution culture, three types of colonies were observed: (1) round and compact colonies that stained intensely with crystal violet, (2) round colonies with spaces between cells that stained slightly less, and (3) fibroblastic colonies stained at comparable levels to type 2 (Fig. 5A). We also examined ALP activity to detect undifferentiated cell colonies. Since the round and compact colonies displayed high levels of ALP activity, it was thought that these colonies formed from remaining undifferentiated rESCs. When the same experiment was performed using primary cultures of EBs, that is, the EB-derived cells were directly used; rates of fibroblastic colony formation were approximately 40% in the 20% O2 condition, 30% in the 5% O2 condition and 85% in the 1% O2 condition. About half of the total colonies formed in 20% or 5% O2 were ALP-positive ESC-like colonies. Only 10% of colonies were ALP-positive ESC-like colonies in the 1% O2 condition. These trends were more pronounced when the same experiments were performed using passaged EB-derived cells. In this case, the rate of fibroblastic colony formation was approximately 90% in the 1% O2 condition. In contrast, almost all colonies formed from the EB-derivative cells cultured in 20% or 5% O2 showed ESC-like undifferentiated features (Fig. 5B).
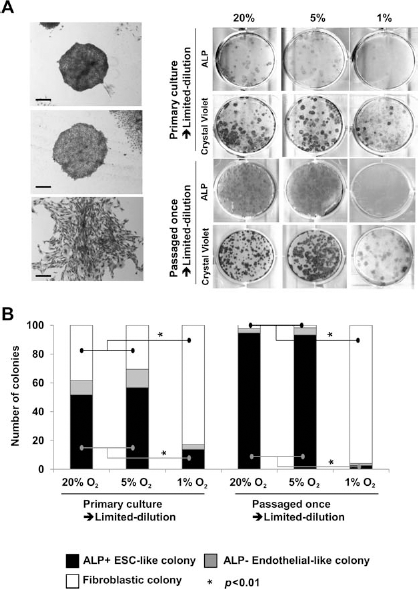
Colony formation assay of the differentiated cells in each oxygen condition. (A) Limiting dilution of differentiated cells formed three types of colonies; compact round ESC-like colony strongly stained by crystal violet (upper), sparse and round epithelial-like colony weakly stained by crystal violet (middle), and fibroblastic colonies very weakly stained by crystal violet (lower). Scale bars: 100 μm. (Right) Colonies obtained from primary (directly obtained from the EB derivatives) or passaged (passaged the primary cultures once and cultured again) cultures in each oxygen condition. (B) Abundance ratios of the three types colonies obtained from primary or passaged cultures in each condition. ∗Conditions where significant differences (p < 0.05) were observed.
Mesenchymal Stem Cell Characteristics of Differentiated Cells in Hypoxic Condition
We confirmed whether the MSC-like cells induced in 1% O2 possessed MSC-specific characteristics. We isolated 25 fibroblastic colonies formed from EB derivatives cultured and passaged in 1% O2, expanded them, and obtained 12 expandable clones. Eleven clones showed differentiation potentials to both osteoblast and adipocyte fates (data not shown). Then, we randomly selected five clones for Western blot analysis. All clones expressed the MSC markers vimentin, CD29, CD90, CD105, and CD140a and lost the undifferentiated cell markers POU5f1 and E-cadherin (Fig. 6A). When one clone of these (rESMSC-1) was randomly selected and observed for its proliferation capacity, it possessed colony-forming properties from a single cell and proliferated logarithmically in normoxic culture conditions (Fig. 6B, C). In accordance with the result in the BMMSCs, the rESMSC-1 more effectively proliferated under hypoxic than normoxic conditions (Table 4).

Characterization of the rESC-derived MSCs (rESMSCs). (A) Marker expression analysis for five independent rESMSC-clones. The undifferentiated rESC-1 line was used as a negative control and rBMMSCs were used as a positive control. (B) Colony formation assay using the rESMSC-1. The clones reformed colonies after limiting dilution. Scale bar: 200 μm. (C) Long-phase cell proliferation analysis of cloned rESMSCs (rESMSC-1) in normoxic conditions. Both cell lines maintained logarithmic proliferation.
Cell Proliferation Analysis of rESMSC-1 and rBMMSCs in Each O2 Condition

rESMSC-1, rabbit embryonic stem cell derived mesenchymal stem cell line; rBMMSCs, rabbit bone marrow mesenchymal stem cells.
The rESMSC-1 line could differentiate to osteoblasts and adipocytes after at least six passages, and these cells could also differentiate to chondrocytes. We examined chondorogenetic properties in vitro by comparing expression levels of Aggrecan and collagen type II α1 (Col2A1) with rBMMSCs and rabbit cultured chondrocytes after induction of differentiation. In this study, cultured chondrocytes were used as a control and were also stimulated to chondrocyte differentiation since the differentiated phenotype of chondrocytes is rapidly lost during in vitro culture through a process of dedifferentiation (4, 85). In all experimental groups, expression levels of both marker genes dramatically increased after differentiation (approximately 100-fold increase in Aggrecan and approximately 160-fold increase in Col2A1 between predifferentiation and postdifferentiation), and statistically significant differences between each of the two groups were not observed (Fig. 7B). To determine the differentiation potential of the rESMSCs in vivo, we produced a GFP-expressing rESC-line by introducing CAG-GFP constructs to the rESC-1 line and induced its differentiation to MSCs using our hypoxia protocol, that is, EB formation, adherent culture of EBs in 1% O2 condition for 6 days, further culture in 1% O2 condition after passage of the EB-derivatives, colony isolation after limiting dilution, and reexpansion as a clonal cell line ubiquitously expressed GFP (Fig. 8A). The induced GFP-expressing rESCs (rgESMSC-1) expressed the MSC markers vimentin, CD29, CD90, CD105, and CD140a (data not shown) and possessed multipotent differentiation to osteoblasts and adipocytes (Fig. 8B). By propagation of the rgESMSC-1 to confluence on temperature-responsive cell culture plates, GFP-expressed cell sheets were successfully prepared (Fig. 8C). Then, we examined whether the rESMSC cell sheets could contribute to cartilage regeneration and stably engraft into the transplanted sites. A histological examination 4 weeks posttransplantation suggested that full-thickness articular cartilage defects could be repaired by transplantation of the cell sheets; the transplanted area covered by cartilage layers well stained by Alcian blue or Safranin-O (Fig. 8D). When observed by immunofluorescence with an anti-GFP antibody, GFP-expressing cells were located mainly in the middle to deep regions of cartilage in the regenerated areas (Fig. 8E). To reject the possibilities of background signal accounting for this observation of transplanted cells, we collected the cartilage from the joints of the transplant recipients at 4 weeks and analyzed GFP-positive cells by FACS. We dissected three transplanted animals and determined that the GFP-positive cells existed at 1.3%, 2.0%, and 8.7% in the regenerated cartilage by FACS (one instance was shown in Fig. 8F). To confirm that the GFP-positive cells acquired chondrocyte phenotype, we sorted GFP-positive fraction and performed qRT-PCR for chondrocyte-specific markers Col2A1 and Aggrecan. GFP-expressing fractions expressed the chondrocyte-specific genes Aggrecan and Col2A1, and the expression quantities were comparable to that of the GFP-negative (i.e., native chondrocytes of the recipients) or normal chondrocyte collected from the contralateral knee (Fig. 8G). Teratoma formation was not observed in any of the treated animals (total of six animals). Therefore, we concluded that the transplanted rESMSCs could successfully engraft and differentiate at the transplanted sites.

Differentiation properties of the rESMSCs in vitro. (A) Alizarin Red staining for osteogenesis (upper left). Scale bar: 200 μm. Oil Red O staining for adipogenesis (upper right). Scale bar: 100 μm. The rightmost picture shows magnified images of the area marked by the white rectangle. Scale bar: 30 μm. (B) Chondrocyte pellet and gene expression analysis for chondrogenesis, significant differences (p < 0.01) were detected between undifferentiated cells and differentiated cells for both genes. Dif, differentiation; rBM, rabbit bone marrow MSCs.

Production of a GFP-expressing rESMSC clone (rgESMSC-1) and transplantation to articular cartilage defects in rabbit. (A) Ubiquitously green fluorescent protein (GFP)-expressing rESMSC-line (rgESMSC-1) induced in the 1% O2 condition from GFP introduced into the rESC-1 line, isolated as a clone and reexpanded for subsequent studies. Scale bar: 100 μm. (B) Differentiation properties of rgESMSC-1 in vitro. Differentiation into osteoblasts (upper) and adipocytes (lower) were determined at day 10 and day 14. GFP expression was also confirmed in the differentiated cells. Scale bars: 100 μm (C) Cell sheets prepared from rgESMSC-1. Scale bar: 1 cm. Panels on the right show magnified brightfield images and a fluorescence image of a cell sheet. Scale bar: 500 μm. (D) Histological observation of the transplanted site at 4 weeks after cell sheet transplantation using the rgESMSC-1 line. (Upper) A section double stained by Alcian blue and Alizarin red. (Lower) A section stained by Safranin O. Scale bar: 500 μm. (E) Immunofluorescence detection of the transplanted rgESMSC-1. Scale bar: 500 μm. (F) FACS analysis of GFP-expressing cells in regenerated cartilage at 4 weeks after transplantation. The area enclosed by the black line in the left shows the putative surviving- and single-cell populations used in the immunofluorescence analysis. The area enclosed by the black line in the right shows the gate for the GFP-positive population used for analysis and sorting. (G) Chondrocyte-specific gene expressions was determined in the cells sorted from regenerated cartilage. Gray bars show the relative quantities of Col2A1 expressions, and white bars shown the relative quantities of Aggrecan expressions. Bars represent the mean scores from three independent experiments. Error bars denote SD. Abbreviation: Contra, contralateral joint cartilage of the recipients.
Discussion
We demonstrated here that (i) severe hypoxic treatment in 1% O2 impeded self-renewal of rESCs by promoting cell death and inhibiting cell proliferation and (ii) MSCs induced from ESCs in this severe hypoxic environment fulfilled the fundamental characteristics of multipotent MSCs.
We established two new rESC lines and determined whether they showed primed-form-specific characteristics and could be used as human stem cell models. Both lines were analyzed and confirmed for pluripotency and possession of multiple differentiation properties to the three different germ layers both in vitro and in vivo. In accordance with results from human ESC studies, our rESC lines showed vulnerability to single cell digestion. This dissociation-induced cell death could be rescued by administration with Y27632, reinforcing our data that these cells show human ESC-like characteristics.
In our first series of experiments, we observed that cell death, as evidenced by detection of activated caspase 3, occurred when cells were cultured under severe hypoxic conditions and that cell proliferation was arrested in rESCs cultured in the 1% O2 condition. Cell death was also observed for the 20% and 5% O2 conditions, albeit far less frequently. It is likely that cell death under the 5% and 20% conditions occurred due to adaptation to feeder-free culture conditions. Enzymatic treatment during preparation of the experimental groups could also have affected the cell viability, since similar tendencies were observed in the cell digestion assays shown in Figure 1.
It is known that accumulation of p53 and hypophosphorylation of pRb occur in cancer cell lines cultured in hypoxic condition (12). However, the role of these molecules in ESCs under hypoxia has not been well examined. In rESCs cultured in 1% O2, decreases in Mdm2 and p-pRB expression were detected. However, significant changes were not determined in the expression levels of p53. In ESCs, it has been reported that p53 expression is ubiquitously maintained at high levels (43, 47, 70). In spite of the presence of high levels of p53, undifferentiated ESCs are neither arrested nor do they undergo apoptosis. It is possible that subtle quantitative alterations in p53, which could result from blocking p53 degradation or induction of de novo expression, were masked by high levels of existing p53 protein.
Alternatively, Sabapathy et al. described that p53 levels may represent a critical determinant of apoptosis and cell cycle arrest; high levels of p53 induce apoptosis, and lower levels results in cell cycle arrest (70). Furthermore, p53 protein levels may be reduced after differentiation (47). Recently, it was reported that the pluripotent status of ESCs is heterogeneous, and even within the same cell line, individual single cells can express different gene expression profiles (17). In other words, if the expression levels of p53 are varied between individual cells, both reactions could occur simultaneously in one culture dish and could therefore mask any net change in p53 expression. In contrast, when p53 function was examined by observing translocation, clear localization into the nucleus was determined in the 1% O2 condition. This fact suggests that hypoxia-induced cell death and inhibition of cell proliferation were the results of p53 translocation. In some cancer cells, epithelial cells and endothelial cells increased apoptosis in response to hypoxia have already been reported (34, 36, 41). Hypoxia is a physiological stabilizer and inducer of a tumor suppressor protein p53, and it can lead to apoptosis in differentiated cell types (3, 37). In pluripotent ESCs, it was suggested that Mdm2, which is a p53-inducible molecule, might contribute to controlling the functions of high levels of p53 expression by repressing p53 function (70). Mdm2 regulates the protein stability of p53 in large part by ubiquitination, which targets p53 for destruction by proteasomes. Importantly, it has been reported that Mdm2 protein levels are downregulated in hypoxic conditions, thereby reducing the level of p53 nuclear export and p53-mediated cell death (2, 37).
On the other hand, decreasing the p-pRB level could also participate in the blocking of ESC proliferation during severe hypoxia. pRB negatively regulates cell cycle progression from G1 to S phase by repressing gene transcription required for G1 to S transition; thus, elevation of pRB activity decreases the S population and is accompanied by increases in the G1 population. pRB is increasingly phosphorylated and inactivated via cyclin-dependent kinases (CDKs) during progression through G1 and is maintained in a hyperphosphorylated state during mitosis (19, 21, 56). In contrast, pRb phosphorylation is inhibited in condition containing CDK inhibitors. Until recently, some researchers thought that hypoxic conditions could act to induce accumulation of the activated form of pRb, which could bring about an arrest in cell proliferation (12, 38). Our results are consistent with these findings, and therefore, our results from immunoblotting and FACS probably do not contradict each other. Of course, hypoxia inducible signaling cascades relating to cell death or arrest in cell proliferation may occur by very complicated mechanisms, so that we must not rule out the possibility that hypoxia-responsive molecules could affect each other.
For example, Uchida et al. and Miwa et al. reported that Mdm2 can regulate p-pRb function (52, 80). Thus, it cannot be ruled out that hypoxia initially affects Mdm2 degradation but then leads to decreasing p-pRB and subsequent cell cycle arrest. Furthermore, the relationship among other signaling cascades or transcription factors might also be important for regulating this process. Hypoxia inducible transcription factors (HIFs), which stabilize and regulate the transcription of various genes during hypoxic conditions (15), are well studied and well supported as participants in regulating the physiological response of cells under hypoxia. In ESCs, HIFs could participate in the regulation of the gene expression both to maintain pluripotency and to direct to differentiation (11, 22, 64, 83). Furthermore, recent reports have shown that HIFs can regulate cell proliferation in hypoxia (40, 72). Importantly, in our study loss of undifferentiated rESCs was observed only in the severe hypoxic conditions and no significant differences were observed between the cells cultured in the 5% O2 condition and the 20% O2 condition. HIF stabilization occurred in both 1% O2 and 5% O2 conditions; thus, it might be reasonable to assume that other mechanisms are also involved in the loss of cells observed under 1% O2 in our study.
Decreases in AKT expression (8) or increases of the inactivated form of glycogen synthase kinase-3β (GSK3β) (55) in severe hypoxic (~1%) conditions are also possible. Phosphatidylinositol 3-kinase (PI3K)/AKT pathway plays an important role in proliferation and maintenance of the undifferentiated state in mESCs (62) and hESCs (5). In contrast, GSK3β negatively regulates self-renewal of ESCs via Wnt/β-catenin signaling (32) or targeting of c-Myc, which promotes the maintenance of ESCs (7).
Now, the effects of hypoxia on ESCs are highly controversial and difficult to interpret. Some researchers reported decreased cell proliferation or increased cell death in severe hypoxic condition (1% O2) in human and mouse ESCs (18, 41, 64, 65), although detailed mechanisms have not been examined. On the contrary, positive effects of hypoxic treatment (3–7% O2 condition) for hESC cultures have also been reported (18, 20, 44). Studying various ESCs in various O2 concentrations and organizing the knowledge obtained from these studies will be helpful to more precisely assign the effect of hypoxia to ESC behavior and may lead to the improvement of methods for engineering pluripotent stem cells.
In the second part of our study, we examined whether severe hypoxic culture conditions would be effective for selective differentiation of MSCs from ESCs. There have been several reports indicating that MSCs are superior to ESCs in terms of safety and ethical aspects; however, we feel that developing the methods for obtaining MSCs from ESCs will be useful for the following reasons. First, MSCs represent a small portion of tissues and require very invasive procedures for collection (73), so in the case of small children, elderly persons or patients with persistent disease states, it may be difficult to obtain sufficient amounts of MSCs for expansion and transplantation in actual clinical practice. Even when MSCs are available in suitable quantities, the ability of expanded MSCs to maintain adequate characteristics including multiple differentiation potentials after excessive numbers of passages is questionable because of cellular senescence (69). On the other hand, ESCs are developmentally pluripotent and able to be expanded infinitely such that MSCs acquired from the ESCs will have higher potency. Second, variability in differentiation potency of MSCs obtained from adult tissue is very high, and it is difficult to collect and selectively expand MSCs that possess high potency. For example, most MSCs lose their homing ability to bone marrow tissue and cannot contribute to the bone marrow niche after ex vivo expansion (54, 68). However, ESCs can provide a source for all types of cells including tissue-specific stem cells possessing very high potentials. Therefore, we think that with the present technology established, it will be possible to easily obtain highly potent MSCs for most clinical needs with minimum to no invasive procedures. Third, the most important point of the present methodology is its applicability to iPSCs. Establishing this protocol will enable us to provide highly potent MSCs to those patients for whom it may be difficult to obtain MSCs, such as the elderly or newborns.
For MSCs, we needed to consider quite different reactions to hypoxia compared to ESCs, since oxygen concentration within bone marrow tissue, which is the principal source of the MSCs, has been measured as very hypoxic (49), and it is plausible to hypothesize that the MSC niche exists within similarly hypoxic conditions. We hypothesized, if the rESMSCs possess similar properties to BMMSCs, they can survive and proliferate better in hypoxic conditions. Following the limiting dilution cultures of the whole EB derivatives in 1% O2, fibroblastic colonies emerged. Repeating the procedure by passaging these cells once in the 1% O2 resulted in colonies that were almost entirely fibroblastic. Cloning of the emergent fibroblastic colonies and subjecting them to differentiation assays demonstrated that at least half of the colonies possessed multiple differentiation potentials. We found that the 1% O2 condition inhibited only undifferentiated cells and non-MSC lineage cells, such as epithelial-like cells, without interfering with the proliferation of the rESMSC population. Moreover, treating the cells with severe hypoxia helped to evade the risk of teratoma formation of the differentiated cells. These results provide important advantages that support the use of our simple strategy for inducing MSCs from ESCs.
Next, we determined whether the induced rESMSCs actually possessed ordinary MSC characteristics; that is, expression of MSC-specific markers, superior proliferation, and maintenance of multiple differentiation potentials to mesenchymal lineages. At first, we randomly selected six clones and confirmed that all cell lines expressed MSC markers and could differentiate to osteoblasts and adipocytes. For more detailed characterization, we compared their proliferation properties with that of BMMSCs and demonstrated BMMSC-like proliferation capacity. The rESMSC clone showed multiple differentiation properties, and the in vitro chondro-differentiation properties of these clones were comparable to that of the rBMMSCs and cultured chondrocytes. The most interesting property of the rESMSC clone was the ability to contribute to cartilage regeneration in vivo. With these demonstrations, we confirmed that rESMSC possessed the cardinal properties of MSCs, including the property to participate in cartilage regeneration and engraftment to the in vivo tissue.
One possible reason for the only partial in vivo contribution of rESMSCs to cartilage repair is conflict with the immune cells of the recipient. In this study, we transferred the rESMSCs as cell sheets to allogeneic recipients to verify their competence as MSCs. In recent studies, MSCs have possessed interesting immunological properties. Some researchers showed that undifferentiated MSCs could evade immune surveillance, induce specific immunological tolerance, and suppress graft-versus-host disease (GVHD) (10). These properties allow the MSCs to survive and contribute to repairing the injured tissues of allogeneic individuals. In other words, if the rESMSCs are provided with the comparable properties to ex vivo isolated MSCs, they can engraft into the transplant site of the allogeneic recipient. Taking this information into consideration, it is possible that there was depletion of the major part of the transplants due to the loss of quality of the MSCs in the cell sheets. However, the cell sheet transplantation method may have clear advantages as a cell transferring strategy for the following reasons. First, it enables the introduction of equal numbers of cells to the transplant site, because the cells contained in the sheets maintain larger amounts of adhesion molecules than those transplanted in the single-cell state. Second, cells transplanted as sheets may also be able to accelerate integration of transplants with the target site (31). Unfortunately, it may be unavoidable that cells contained in sheets are maintained in an overconfluent format that might result in a loss of specific characteristic of MSCs. Recently, Huang et al. reported that differentiated MSCs were recognized by the host immune system and immunologically rejected soon after transplantation using allogeneic MSC transplantation models for cardiac disorder (27). This finding suggests the possible risks that if the MSCs lose undifferentiated status at transplantation, they could be recognized by immune systems and rejected. However, in our study, the residual transplanted cells maintained their differentiated status and expressed chondrocyte specific genes in the repaired cartilages. Presumably, some features of the cartilage, that is, absence of vascular, neural, or lymphatic input enable the rESMSCs to circumvent the immune response even after differentiation to mature chondrocyte in the regenerating cartilage. This is supported by our observation that the rESMSC-derived chondrocytes survived in internal regions, not on the surface, of the cartilage. Indeed, Koga et al. demonstrated that allogenically transplanted MSCs were accepted in rabbit cartilage injury models (35). More versatile studies with various kinds of injury patterns (different wide or depth), animal species, or follow-up periods may be helpful in understanding more detailed characteristics of tissue regeneration after transplantation of ES-derived MSCs.
In conclusion, we describe a novel hypoxia-based differentiation method for deriving MSCs from ESCs. This method of differentiation is mediated by selective cell death and regulation of the cell cycle of undifferentiated cells. Until now, critical factors or molecules to induce MSCs have not been discovered. Retinoic acid (RA) has been reported as an induction factor by Takashima et al. and us (76, 78) previously, but these studies used mice, so that the applicability to humans has not been demonstrated. Furthermore, RA is also known to be a negative factor for expansion of human MSCs (60). Our method of hypoxic induction could avoid the problems encountered in other protocols, because it will not require any growth factors or differentiation induction factors (chemicals) to select cells. Future studies using human cells will be required to evaluate utility and safety of the present strategy. Detailed analysis will lead to further enhancement of transplant safety, efficacy, and resolution of problems associated with pluripotent stem cell transplantation therapy.
Footnotes
Acknowledgments
We gratefully acknowledge Dr. Hirofumi Suemori, Institute for Frontier Medical Science, Kyoto University, for kindly providing pCAG-IRES-Puro plasmid. And we thank Ms. Naomi Backes Kamimura, Department of Biology-Oriented Science and Technology, Kinki University, for the English editing. We also thank Ms. Kanae Shigi and Ms. Naoko Ohoshi for excellent technical assistance. The authors declare no conflicts of interest.