
Abstract
Effects of leukemia inhibitory factor (LIF) and fibroblast growth factor 2 (FGF2) on establishment and maintenance of rabbit embryonic stem cell (rESC) lines were assessed. When grown on MEF feeders, rESC lines derived from fertilized embryos were established and maintained in medium containing paracrine factors LIF (via STAT3) and/or FGF2 (via MEK-ERK1/2 and PI3K-AKT). However, high levels of ERK1/2 and AKT activities in rESCs were crucial for maintaining their undifferentiated proliferation. Although rESCs under the influence of either LIF (500, 1,000, and 2,000 U/ml) or FGF2 (5, 10, and 20 ng/ml) alone had enhanced expression of pluripotency markers, peak expression occurred when both LIF (1,000 U/ml) and FGF2 (10 ng/ml) were applied. Induced dephosphorylation of STAT3, ERK1/2, and AKT by specific inhibitors limited growth of rESCs and caused remarkable losses of self-renewal capacity; therefore, we inferred that STAT3, ERK, and AKT had essential roles in maintaining rESC proliferation and self-renewal. We concluded that LIF and FGF2 jointly maintained the undifferentiated state and self-renewal of rESCs through an integrative signaling module.
Keywords
Introduction
Human embryos, as a primitive form of human life, are ethically restricted with regards to their availability for establishment of human embryonic stem cell (hESC) lines and related therapeutic applications. Presently, several model animals are potential alternatives for biomedical research and clinical applications. Among them, rabbits (Oryctolagus cuniculus) are classical laboratory animals with many advantages over other species, making them one of the most common laboratory species for research on human diseases, including hypertension (2,35) myocardial infarction (36,45,71), bone and cartilage disorders (14,34), arteriosclerosis (18,44,68), and diabetes (30,33,64), as well as an ideal bioreactor for production of pharmaceutical proteins (20,46). The earliest isolation of fertilized embryo-derived rabbit ESC (rESC) lines was reported by Graves and Moreadith (19), followed by Schoonjans et al. (49). Recently, many more rESC lines, including those derived from parthenotes (24), have been established (9,12,16,22,25,59).
The mitotically inactivated mouse embryonic fibroblast (MEF) (9,12,15,16,22,25,54,59) and Sandos inbred mouse embryo-derived thioquanine and ouabain-resistant cells are two types of feeder cells most commonly used in establishing and maintaining ESC lines derived from several species, including mice (50), cattle (54), and humans (42,43). In hESCs, paracrine factors, such as fibroblast growth factor 2 (FGF2), wingless-related mouse mammary tumor virus integration site (Wnt) and/or Activin/Nodal, have been implicated in maintaining cell self-renewal and stemness (1,13,27,54–57,67). Undifferentiated hESCs expressed high levels of FGF ligands and their cognate receptors (7,48,51). In addition, extracellular regulated kinases 1/2 (ERK1/2) and phosphatidylinositol-3 kinase (PI3K)-AKT downstream of the FGF pathway are responsible for the survival, proliferation, and maintenance of pluripotent stem cell lines (6,28,32). It has also been well established that leukemia inhibitory factor (LIF) is required for mouse ESC (mESC) self-renewal (39,50,63,70), and that signal transducers and activators of transcription 3 (STAT3) functions as the key downstream transcription factor in the LIF/glycoprotein 130 (gp130) pathway (37,39). Bypassing the LIF signaling pathway may still maintain the pluripotency of mESCs (17,31,38,66,70) and rESCs (16,24). Similarly, addition of integrins (21), heparan sulfate (47), vitamin A (10,11), or small molecules such as SC1 (pluripotin) to the culture medium without LIF maintains mESC pluripotency (10,11,21,47).
Nevertheless, the demand for LIF to keep ESCs undifferentiated in culture seems to be species-dependent. In our previous studies, LIF by itself was sufficient to sustain undifferentiation and self-renewal of rESCs on MEF feeders (24). These rESCs expressed pluripotency markers and retained the capacity to differentiate into cell lineages of all germ layers (9,16,19,22,24,25,49,59). However, we could not exclude the possibility that other signaling pathways also contributed to maintenance of stemness in rESCs.
Based on their origin, it is not surprising that rESCs share many, albeit not all, biological or physiological characteristics with mESC and hESC lines, for example, in morphology and surface marker expressions and in signaling molecules for maintaining pluripotency (9,16,19,22–25,58,59). Therefore, it is likely that an adequate microenvironment supported by LIF and/or FGF2 with other paracrine factors may be advantageous in maintaining pluripotency and self-renewal of rESCs. In this study, we tested LIF and FGF2, the two key signaling ligands for mESC and/or hESC self-renewal, and we hypothesized that these two factors have a cooperative effect on rESC identity in culture.
Materials and Methods
Reagents and Animals
The care and management of all animals used for recovering embryos complied with the guidelines and was approved by the Institutional Animal Care and Use Committee (IACUC) of National Chung Hsing University, Taiwan, ROC (IACUC Permit No. 96–72). Nearly all chemicals and reagents used were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA) unless otherwise mentioned. The severe combined immunodeficient (SCID) mice were purchased from BioLasco Taiwan Co., Ltd. (Taipei, Taiwan), and raised in accordance with the Guide of the IACUC of National Chung Hsing University, Taiwan, ROC. When animals had to be euthanized, all efforts were made to minimize suffering.
Generation and Culture of rESC Lines
Derivation of rESCs
Blastocyst embryos were flushed and recovered from the uterus of New Zealand White rabbits (Livestock Research Institute, Tainan, Taiwan) 4 days after mating. The flushing medium was M199 (31100–027; Gibco, Grand Island, NY, USA) supplemented with 3.36 g/L NaHCO3 (S5761; Sigma), 10% fetal calf serum (26140–079; Gibco) or 20% knockout serum replacement (KSR; Gibco), 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (H3375), and 1% antimycotics (15240; Gibco). Blastocyst or isolated inner cell mass (ICM) plating, recipes for culture media, outgrowth selection, or passage and culturing of ESCs were all based on previous protocols (12,24,25).
Karyotyping of ESC Lines
The ESCs in the log growth phase were incubated with 2 μg/ml of colcemid (KaryoMAX® Colcemid™; Gibco) at 37°C in a 5% CO2 incubator for 6 h. Karyotypic analyses of cultured rESCs at passage 15 were performed as previously described (24).
Labeling of Alkaline Phosphatase (AP) and Pluripotency Markers
The ESC lines for marker detection were grown on sixwell dishes and rinsed with Dulbecco's phosphate-buffered saline (DPBS) prior to fixation in 4% paraformaldehyde (P-6148; Sigma-Aldrich) solution. At 15 min after fixation at room temperature (RT), ESCs were rinsed with DPBS (three times) prior to AP staining, as previously described (12,24,25). For specific protein marker expressions, ESC colonies were rinsed with DPBS and then fixed in 4% paraformaldehyde 2–4 days after culture. After washing with DPBS for 10 min, cells were stained as previously described (24,25). Briefly, ESC lines were first incubated with primary antibodies [anti-stage-specific embryonic antigen 4 (SSEA-4), 1:15, MAB 4304; Chemicon, Temecula, CA, USA; anti-octamer binding transcription factor 4 (Oct4) 1:500, SC8628; Santa Cruz, Santa Cruz, CA, USA; anti-keratan sulfate antigens (TRA)-1–60, 1:100, ab16288 or anti-TRA-1–81, 1:100, ab16289; Abcam, Cambridge, MA, USA], and then were washed three times with DPBS and 0.05% Tween-20 (DPBST, 20605; Amersham Life Science, Arlington Heights, IL, USA) prior to incubation with secondary antibodies [SSEA-4, fluorescein isothiocyanate (FITC)-conjugated rabbit anti-rat IgG + IgM + IgA, 1:200, ab520; Abcam; Oct4, FITC-conjugated rabbit antigoat IgG, 1:400, F736 and TRA-1–60 or TRA-1–81, goat anti-mouse IgG, 1:200, F2012] for 60 min. For nuclear labeling, 4′,6-diamidino-2-phenylindole (DAPI; 1 μg/ml) in DPBST was used for staining, followed by two DPBST rinses before examination with epifluorescence microscopy (24,25).
Embryoid Body (EB) Formation and Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
rESC colonies were lifted from feeders with dispase (1 mg/ml, 17105041; Gibco) at 37°C and split into small clumps by gentle pipetting. Procedures for EB formation and RNA extraction were performed as previously described (24,25). Total RNAs of ESC lines (F1, F3, F8, and F10) were extracted using a total RNA extraction kit (RT050; Geneaid, New Taipei City, Taiwan) as previously described (24,25). The primer sequences including sense and antisense, annealing temperatures used in the PCR reactions, and the expected product sizes are shown (Table 1).
Primer Sequences of Target Genes and Conditions for PCR Analyses

Oct4, octamer binding transcription factor; Sox2, sex determining region Y box 2; Fgfr2, fibroblast growth factor receptor 2; Pax6, paired box 6; Gata4, guanine-adenine-thymine-adenine binding protein 4; Gapdh, glyceraldehyde 3-phosphate dehydrogenase.
Quantitative Real-Time PCR
Quantitative PCR was performed under the following conditions: 10 min at 95°C, and 40 cycles of 15 s at 95°C, 1 min at 60°C using 2× Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA) with 200 nM of forward and reverse primers. Each assay was run on an Applied Biosystems 7300 Real-Time PCR system in triplicates and expression fold changes were derived using the comparative CT method. Primer sequences and expected product sizes for real-time PCR analyses are shown (Table 2).
Primer Sequences and Expected Product Sizes for Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR) Analysis

Maintenance of ESCs in a Feeder-Free Culture System
rESC lines (passage 17) were treated with dispase (1 mg/ml, 17105041; Gibco) and split into small clumps by gentle pipetting. Cells were maintained on feeder-free and gelatin-coated plates in culture media consisting of 81.5% Dulbecco's modified Eagle's medium/F12 (12400–024; Gibco), 15% fetal bovine serum (FBS, 10437–028; Gibco), 4 mM l-glutamine (G8540), 0.5% nonessential amino acids, 0.1 mM β-mercaptoethanol (M7522; Sigma-Aldrich), and various concentrations (0, 500, 1,000, or 2,000 U/ml) of murine LIF (ESG 1107; Chemicon, Billerica, MA, USA) or human FGF2 (0, 5, 10, or 20 ng/ml, CYT-218; Prospec, East Brunswick, NJ, USA). The ESCs were analyzed for expression of pluripotency markers 4 days after seeding.
Joint Effects of LIF and/or FGF2 Treatment and Signaling Inhibitors on ESCs
In addition to adding LIF and FGF2 to the culture medium, Janus kinase 3 (JAK3) inhibitor I (JAKi, PHZ1084; Biosource, Camarillo, CA, USA) was used to suppress JAK kinase signaling, which would otherwise activate STAT3 phosphorylation via LIF stimulation. Inhibitors U0126 and LY294002 (9903 and 9901; Cell Signaling Technology, Boston, MA, USA) were used to block mitogen-activated protein kinase kinase (MEK)-ERK1/2 and PI3K-AKT pathways, respectively, of ESCs, as previously described (24). JAK3-inhibitor I (10 mM), 10 mM U0126, 10 mM LY294002, or dimethyl sulfoxide (DMSO, Sigma-Aldrich) were added to the medium to culture rESCs for 4 days in order to block the STAT3, ERK1/2, or AKT pathways. After that, the cells were subjected to AP and immunocytochemical staining or Western blotting to detect the expression of pluripotency markers.
Western Blot Analyses: Protein Expressions and Phosphorylation
For Oct4 and Nanog, ESCs were recovered for Western blotting. Protein extracts were prepared by resuspending ESCs with 400 μl of lysis buffer [0.15 mM NaCl, 5 mM ethylenediaminetetraacetic acid (EDTA), 1% Triton X-100, 10 mM Tris-HCl, and 5 mM dithiothreitol; Sigma] containing 0.1 mM protease inhibitor cocktail (P8340; Sigma) on ice for 10 min, and then centrifuged (18,600 × g for 10 min at 4°C). The supernatant was stored at −80°C until use. Prior to electrophoresis, extracted proteins were boiled for 5 min and then loaded onto 10% (v/v) sodium dodecyl sulfate (SDS) polyacrylamide gels. Proteins were resolved and analyzed as previously described (24,25). In brief, the extracted protein was boiled for 5 min and subjected to electrophoresis in 10 % (v/v) SDS polyacrylamide gels. The resolved proteins were transferred to polyvinylidene difluoride membranes. After blotting, the membrane was incubated for 1 h at room temperature in blocking buffer (5% skim milk powder in DPBS) and then incubated with primary antibodies at 4°C overnight. The nitrocellulose membrane was washed with 1× TBST (blocking solution; 200 mM Tris-HCl, 25 mM NaCl, 0.05% Tween-20, pH 7.4) and then subjected to the treatment of horseradish peroxidase (HRP)-conjugated secondary antibodies (28169; Anaspec, Fremont, CA, USA) for 1 h for visualization with a Super-Signal West chemiluminescent substrate kit (Thermo Scientific, West Palm Beach, FL, USA). The intensity of protein signals in three duplicates was determined by using ImageJ software (National Institutes of Health, Bethesda, MD, USA) and by Java software (Version 1.42 for Windows; Oracle Corp., Redwood City, CA, USA). β-Actin served as a loading control to normalize expressions of Oct4 or Nanog, and total STAT3, ERK1/2, and AKT were used to normalize expression of phospho-STAT3, phospho-MEK kinase (MEKK)/ERK1/2, and phospho-AKT, respectively.
Phosphorylation of STAT3, ERK1/2, and AKT of ESCs were detected as previously described (24). Briefly, ESCs were washed with DPBS and harvested with 0.05% trypsin-EDTA (1 mM) for protein extraction. After centrifugation, protein concentrations of the supernatants were determined by 10% SDS-PAGE. For Western blotting, anti-STAT3 (1:1,000, 9132; Cell Signaling, Boston, MA, USA), antiphospho- STAT3 (serine 727, 1:1,000, 9134; Cell Signaling), anti-ERK1 (1:1,000, SC94; Santa Cruz), anti-phospho-MEKK/ERK1/2 (1:1,000, 9101; Cell Signaling), anti-AKT (1:1,000, 9272; Cell Signaling), anti-phospho-AKT (Ser 473, 1:1,000; Cell Signaling), and anti-β-actin (1:1,000, 4967; Cell Signaling) were used. Similarly, primary antibodies were incubated overnight with the blot and then reacted with HRP-conjugated secondary antibodies (KLP, 474–1516; for anti-p-AKT: 7067; Cell Signaling) for 1 h. Protein signals on the blots were visualized and analyzed as aforementioned.
Statistical Analysis
Percentile data were normalized by arcsine transformation prior to statistical analysis by ANOVA procedure in SAS (Version 9; SAS Institute, Cary NC, USA), followed by Tukey's test. The results are presented as mean ± SEM. Statistical significance was considered at p < 0.05.
Results
Derivation of rESC Lines From Fertilized Embryos
Effects of FGF2 on the Establishment of ESC Lines
In our previous studies, adding 5 ng/ml FGF2 helped to maintain rESC lines (R3 and R4). In this study, various FGF2 treatments were applied to evaluate their effects on derivation of ESC lines from fertilized embryos (Table 3). A total of 100 blastocysts were randomly allocated to five treatment groups (0, 5, 10, or 20 ng/ml of FGF2 in FBS and 5 ng/ml FGF2 in KSR) and cultured on MEF feeders. When treated with FGF2, 55–65% of embryos formed primary ICM colonies. A low concentration of FGF2 (5 ng/ml) worked as effectively as a high concentration (e.g., 20 ng/ml) in establishing rESC lines (35% vs. 40%). The established rESC lines with normal morphology were easily expanded beyond 25 passages in either FBS or KSR medium supplemented with FGF. However, in the control (without FGF2) group, colonies lost their defined boundary within four passages (Fig. 1A). The rESC lines (F1, F3, F10, and F8) isolated with various concentrations of FGF2 (5, 10, or 20 ng/ml of FGF2 in FBS or 5 ng/ml FGF2 in KSR, respectively) all evidently expressed pluripotency markers (Fig. 1B), including AP, Oct4, TRA-1–60, TRA-1–81, and SSEA-4. These rESC lines have been continuously cultured and passaged for more than 30 generations with normal karyotypes (>80% with 44 chromosomes, n = 12). Expression of pluripotency genes of Oct4, Nanog, and Sox2 after treating the rESCs line (F1) with various concentrations of FGF2 was analyzed by qPCR (Fig. 1C). When cultured under the feeder-free condition with supplemented FGF2 (5, 10, or 20 ng/ml in FBS or KSR), rESCs (F1) expressed higher levels of pluripotency marker genes than the untreated group and fibroblast cells (Fig. 1C). In particular, when cells were treated with 20 ng/ml FGF2 in FBS, expression levels of Oct4, Nanog, and Sox2 were significantly increased (p < 0.05) (Fig. 1C). Rabbit EBs derived from the four rESC lines in the presence of serum by suspension culture (Fig. 1D) contained three germ layers, as evidenced by expression of the three marker genes, Pax6 (ectoderm), Desmin (mesoderm), and Gata4 (endoderm) (Fig. 1E).
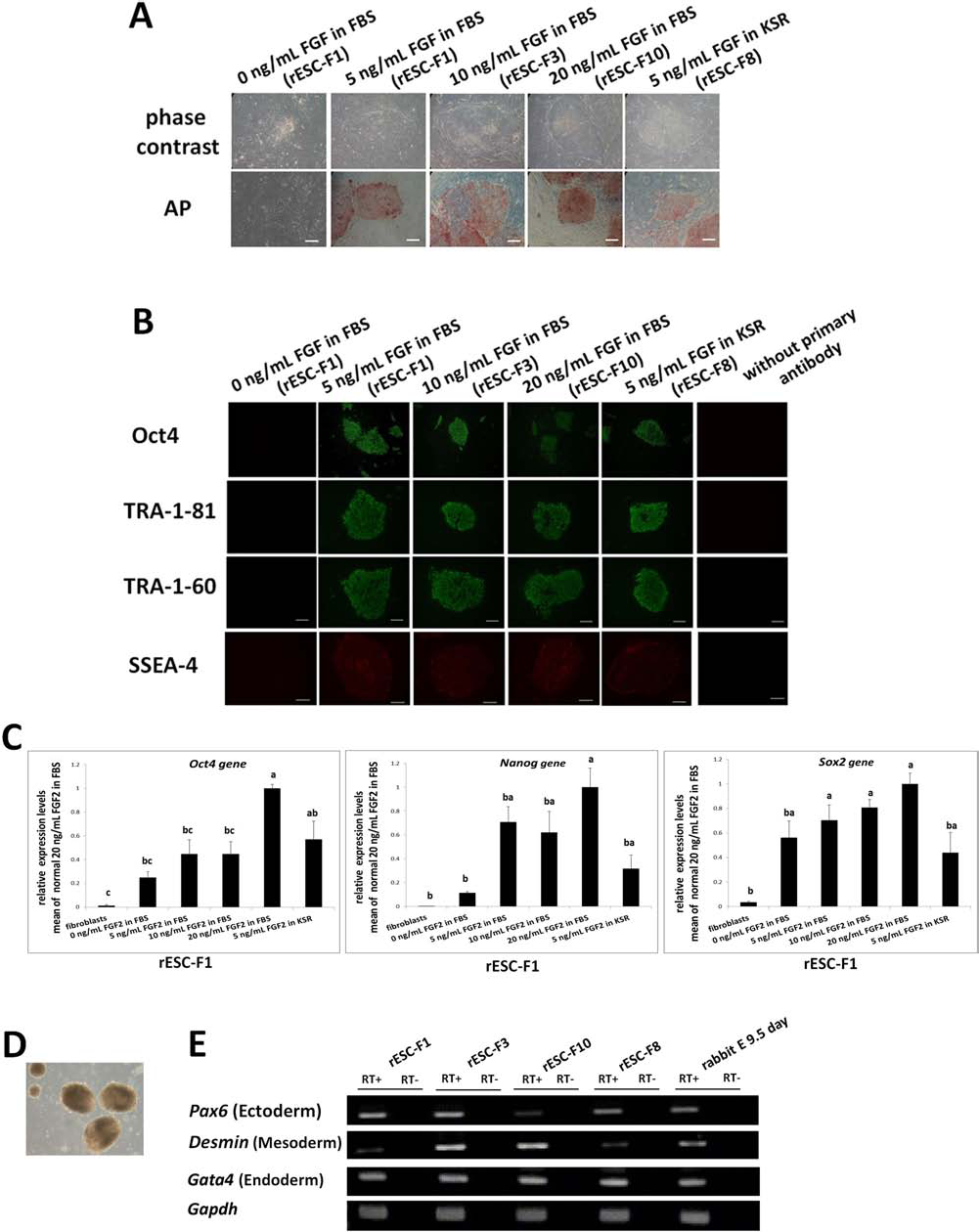
Effects of FGF2 on the establishment of rESC lines. (A) The ESC colonies at passage 12 after being seeded on MEF feeders in the medium containing FBS or KSR plus varying concentrations of FGF2 (0–20 ng/ml). The four rESC lines examined had AP activity and the ESC pluripotency markers as detected by immunocytochemistry. Untreated cell control (0 ng/ml FGF2) and secondary antibody-minus groups served as negative controls (B). (C) Gene expressions of Oct4, Nanog, and sex determining region Y box 2 (Sox2) after treating the rESC line (LF1) with varying concentrations of FGF2 were analyzed by qPCR. Fibroblasts served as a negative control. Relative quantification in qPCR was determined using glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as an endogenous control, and sample “fibroblasts” were used as a calibrator. The EBs derived from the ESCs (D) all expressed markers of the three germ layers (E). The 9.5-day embryos were sources of differentiated cells for positive control. RT- means RT reactions lacking transcriptase and served as negative controls. Scale bar: 100 μm.
Effects of Fibroblast Growth Factor 2 on Establishment of rESC Lines

Three replicates.
Within a column, means without a common superscript differed (p < 0.05).
FGF2 Pathway Sustains Self-Renewal of rESCs on MEF Feeders
In our previous study, we determined that STAT3 was phosphorylated in rESCs cultured in medium supplemented with LIF (24). In this study, we further examined the roles of ERK1/2, AKT, and downstream regulators of the FGF pathway in sustaining self-renewal. The four f-rESC lines (F1, F3, F8, F10) all expressed FGF receptor 2 (Fgfr2) (Fig. 2A). Western blot analyses confirmed that activations of ERK1/2 and AKT were key events of rESC signaling. Both ERK1/2 and AKT were highly phosphorylated in undifferentiated rESC lines under various FGF2 treatments (5, 10, or 20 ng/ml in FBS or 5 ng/ml FGF2 in KSR) for 10 or 30 min (Fig. 2B). Phosphorylation status of ERK1/2 and AKT was both dose and time dependent on FGF2 treatment. In addition to Western blotting, highly phosphorylated ERK1/2 and AKT in rESCs were further confirmed by immunocytochemical staining (Fig. 2C).

MEK/ERK and PI3K/AKT signaling pathways maintained pluripotency of rESCs. The rESC lines expressed Fgfr2 gene as detected by RT-PCR (A). Fibroblasts were used as negative controls. RT means that RT reactions were performed without adding transcriptase, which also served as a negative control. Transient FGF2 stimulation induced phosphorylation of ERK1/2 and AKT in FBS-starved ESCs. The ESC line F1 was first deprived of FGF2 in FBS or KSR overnight and then restimulated with FGF2 (5, 10, or 20 ng/ml) for 10 or 30 min (B). (C) Cells were treated with FGF2 (20 ng/ml) for 30 min and subjected to immunocytochemical analyses to determine phosphorylation status of ERK1/2 and AKT (scale bar: 100 μm). Nuclei were counterstained with DAPI. (D, E) Cells going through FBS starvation overnight received various treatments: (a) without LIF and FGF2; (b) with 2,000 U/ml LIF; (c) 20 ng/ml FGF; and (d) LIF (2,000 U/ml) and FGF2 (20 ng/ml), in a feeder-free system. For phosphorylation status, whole-cell extracts were blotted with phospho-specific antibodies against phospho-signal transducers and activators of transcription 3 (STAT3), ERK1/2, and AKT (Ser-473). β-Actin served as a loading control (three replicates). Bars without a common superscript differed (p < 0.05).
We then wondered whether treatments of rESCs with LIF along with FGF2 would enhance the phosphorylation statuses of STAT3, AKT, and ERK1/2 and in turn fortify cell propagation in preference to LIF or FGF alone. The rESC line F1 was FBS starved overnight and subjected to phosphorylation analyses. Treatment of LIF alone did not seem to consistently affect the phosphorylation of STAT3, AKT, and ERK1/2. However, with FGF2 in the medium, rESCs on MEF feeder cells had higher levels of ERK1/2 phosphorylation than those cultured in medium lacking FGF2 and LIF (Fig. 2D, E). Interestingly, rESCs cultured in the presence of both LIF and FGF2 had consistently increased levels of STAT3, ERK1/2, and AKT phosphorylation compared to the untreated control (p < 0.05), strongly implicating an additive effect of LIF and FGF2 on STAT3, ERK1/2, and AKT phosphorylation statuses.
In addition, we further investigated whether a combination of LIF and FGF2 would help in establishing new rESC lines. A total of 80 blastocysts were randomly allocated to four treatment groups: [1] control, culture medium without LIF and FGF2; [2] medium with 1,000 U/ml LIF, [3] medium with 10 ng/ml FGF, and [4] medium with both LIF (1,000 U/ml) and FGF2 (10 ng/ml). Treatment with the combination of LIF and FGF2 appeared to be acting more effectively in establishing rESC lines than either LIF or FGF2 alone though this was not significant (40% vs. 35%, p > 0.05; data not shown). The rESC line (LF1) was established and maintained in LIF and FGF2 media for more than 20 passages with evidently expressed pluripotency markers including Oct4, Nanog, TRA-1–60, and TRA-1–81 or with the potential of teratoma formation (which occurred in SCID mice injected with 5 × 106 LF-1 cells). All cell types representative of the three germ layers were identified.
LIF or FGF2 Alone Maintained ESC Self-Renewal Under Feeder-Free Condition
To eliminate the effects of paracrine factors secreted by the feeder cells on rESC survival and propagation, a feeder-free culture system was adopted to reexamine the controversial LIF dependency. The rESCs (F1 line) at passage 17 were treated with 0, 500, 1,000, or 2,000 U/ml of LIF, respectively. The ESCs without LIF treatment lost their compact colony morphology shortly thereafter, whereas cells in other groups with LIF treatments sustained normal morphology. Expression levels of the pluripotency markers including AP, TRA-1–60, and TRA-1–81 were upregulated in proportion to LIF supplementation (Fig. 3A). In the rESC line F1, Oct4 and Nanog expressions increased with LIF supplementation, irrespective of the amount added (p < 0.05) (Fig. 3B, C). Similarly, when the rESCs (F1) were cultured under the feeder-free condition with supplemented FGF2 (5, 10, or 20 ng/ml), they remained compact, retained better morphology, and expressed higher levels of TRA-1–60, TRA-1–81, and AP activity than those untreated control (Fig. 4A). In particular, when the added FGF2 exceeded 10 ng/ml, rESCs had higher levels of Oct4 and Nanog expressions than those in the control (p < 0.05) (Fig. 4B, C).

LIF maintained pluripotency of rESC lines. Representative images show the colony morphology of rESC line F1 (passage 17) cultured in the LIF-containing medium, supplemented with or without LIF for 4 days under feeder-free conditions. (A) Expressions of AP, TRA-1–60, and TRA-1–81 were detected in the undifferentiated state and increased with various concentrations of LIF supplementation in ESC media. (B, C) Increased expressions of Oct4 and Nanog proteins were detected in ESC lines (F1, passage 17) cultured in the medium supplemented with LIF for 4 days. β-Actin served as a loading control. Three replicates. Scale bars: 100 μm.
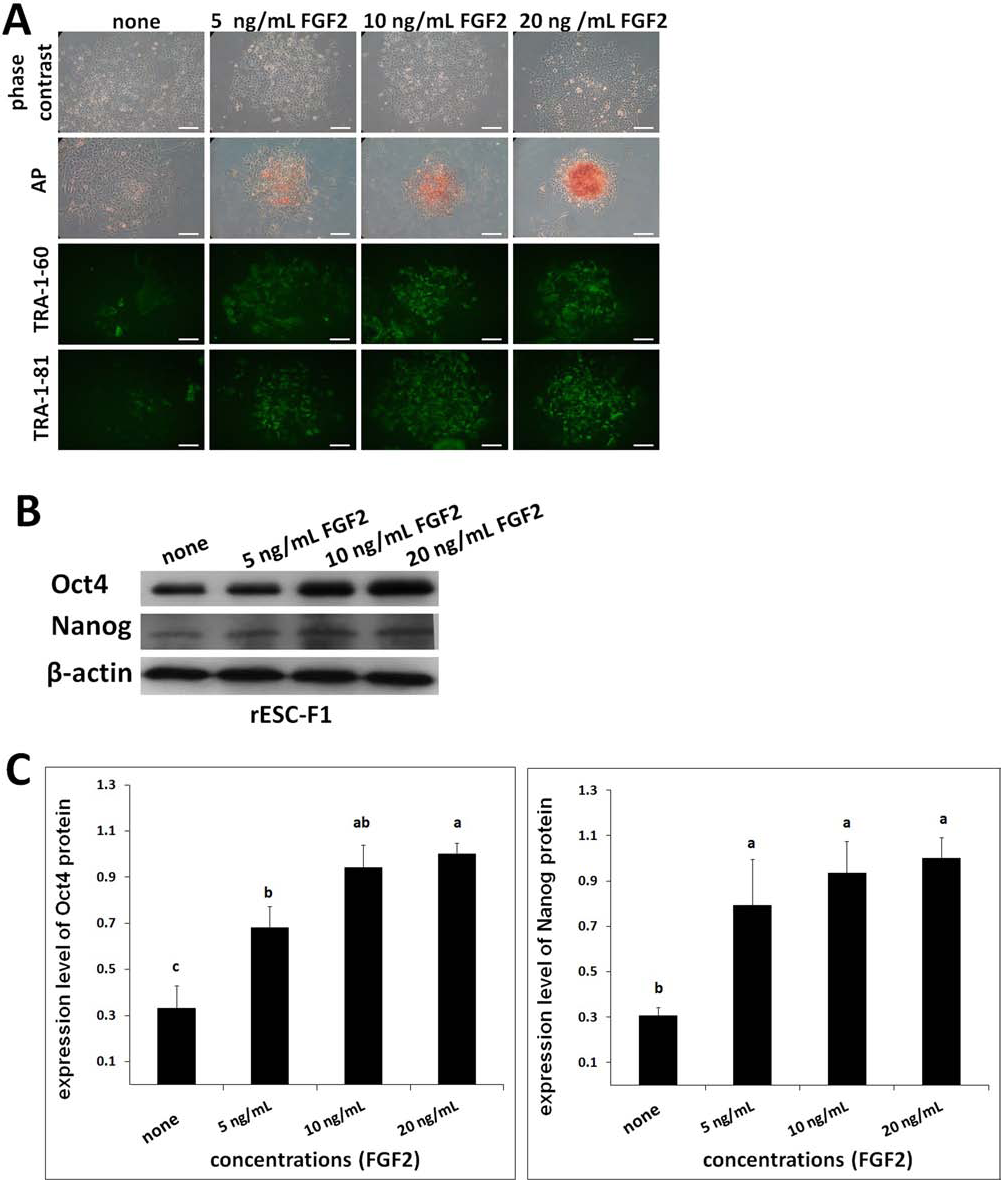
FGF2 maintained pluripotency of rESC lines. Representative images of the colony morphology of ESC line F1 (passage 17) cultured in the FBS-containing medium supplemented with (5, 10, or 20 ng/ml) or without FGF2 for 4 days under feeder-free conditions. Expressions of AP, TRA-1–60, and TRA-1–81 were all detected in the undifferentiated colonies (A) with increased intensity as the FGF2 concentration increased. (B) Based on Western blotting, there were increased levels of Oct4 and Nanog expressions of the ESCs in the medium supplemented with FGF2 for 4 days. β-Actin, the loading control. (C) Oct4 and Nanog expressions of ESC lines were largely increased in the FGF2-supplemented groups (5, 10, and 20 ng/ml) compared to the control. Three replicates. Scale bars: 100 μm. Bars without a common superscript differed (p < 0.05).
Effects of Combined LIF and FGF2 Treatments on rESC Self-Renewal
We further investigated whether the combination of LIF and FGF2 also affected rESC pluripotency marker expression. The two rESC lines (F1 and 10) were cultured under one of the four feeder-free treatments: (a) control, culture medium without LIF and FGF2; (b) medium supplemented with 1,000 U/ml LIF; (c) medium with 10 ng/ml FGF2; and (d) medium with both LIF (1,000 U/ml) and FGF2 (10 ng/ml). Cells treated with LIF or FGF2 alone maintained colony morphology for 2–3 days in both the F1 and F10 lines. However, when cells (F1) were cultured in the medium supplemented with both LIF and FGF2, their morphology was defined and compacted, far better than that of the cells treated with LIF or FGF2 alone (Fig. 5A). In addition, rESCs treated with both factors had marked expressions of AP, TRA-1–60, and TRA-1–81 (p < 0.05). In contrast, AP, TRA-1–60, and/or TRA-1–81 could barely be detected among cells maintained in the medium simultaneously lacking LIF and FGF2 (Fig. 5A). Similarly, low expression patterns were also noted for Oct4 and Nanog in the control group (p < 0.05) by Western blot analyses (Fig. 5B, C).
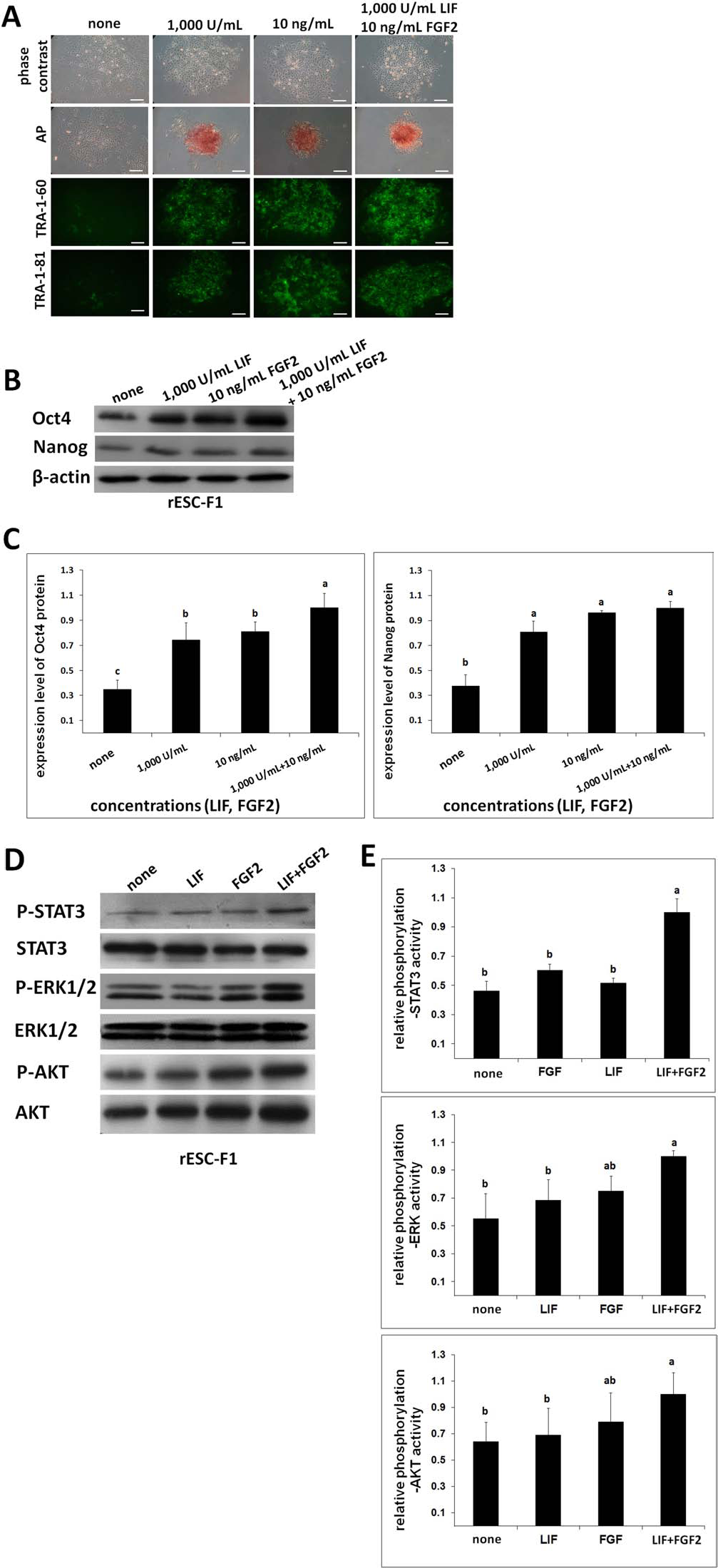
Combined effects of LIF and FGF2 treatments on pluripotency marker expressions of the rESCs cultured in a feeder-free system. The ESCs were maintained in the FBS-containing medium supplemented with or without LIF (FGF2), or with both LIF and FGF2 for 4 days. Expressions of TRA-1–60/81 and AP activity were detectable with a relatively stronger level in the combined treatment group (A). Pluripotency markers Oct4 and Nanog were similarly expressed (B, C). The expression level was maximal when both LIF and FGF2 (1,000 U/ml + 10 ng/ml) were present in the culture medium. (D, E) The total and phosphorylated protein expressions of STAT3, ERK1/2, and AKT of the rESCs were examined. β-Actin was used as a loading control. Three replicates. Scale bars: 100 μm. Bars without a common superscript differed (p < 0.05).
Thereafter, we examined the phosphorylation statuses of LIF- and FGF-related downstream molecules including STAT3, MEK/ERK, and AKT under the influence of their ligand binding in the feeder-free culture system. rESC line F1 was FBS starved overnight and subjected to priming with LIF and/or FGF ligands for phosphorylation. In the presence of LIF, rESCs had normal levels of STAT3, AKT, and ERK1/2 phosphorylation under a feeder-free condition, as opposed to the relatively weak expression of AKT and ERK1/2 in untreated cells (Fig. 5D, E). Similarly, rESCs cultured in the medium containing only FGF2 or combined LIF and FGF2 had higher levels of ERK1/2 and AKT phosphorylation than untreated ones (p < 0.05). Furthermore, the combined effects of LIF with FGF2 on ERK and AKT phosphorylations were not as pronounced as on STAT3 phosphorylation (p < 0.05).
Induced Dephosphorylation of STAT3, MEK/ERK, and AKT Blocked Self-Renewal of rESCs
In this study, inhibitors against LIF and FGF2 stimulations were used to suppress LIF and FGF2 signaling pathways. The rESCs (F1) grown in the feeder-free system with LIF (1,000 U/ml) and FGF2 (10 ng/ml) supplementation were further treated with one of the inhibitors for JAK kinase (JAKi), MEK/ERK1/2 (U0126), or PI3K/AKT (LY294002). Treatment with JAKi (10 μM/ml), LY294002 (10 μM/ml), or U0126 (10 μM/ml) dramatically reduced phosphorylation of STAT3, AKT, or ERK1/2, respectively (Fig. 6A). The ESC colonies failed to maintain normal morphology and the cell numbers being stained positively with AP, TRA-1–60, and/or TRA-1–81were reduced (Fig. 6B) when either inhibitor was present in the medium. There were also indications that coexistence of multiple inhibitors exacerbated differentiation of ESCs. Protein levels of the pluripotency markers Oct4 and Nanog were also drastically reduced by the inhibitors (Fig. 6C, D).

Specific kinase inhibitors suppressed expression and phosphorylation of the downstream signaling molecules of the LIF and FGF2 pathways in rESCs. The ESC lines were treated with DMSO as a negative control (vehicle only), or specific inhibitors JAKi (10 μM, for Jak kinase 3), LY294002 (10 μM for AKT), and/or U0126 (10 μM for ERK1/2) for 4 days. (A, B) When each inhibitor was present in the culture medium, phosphorylation of the targeted signaling molecules (p-STAT3, p-AKT, or p-ERK1/2) was reduced specifically, but the expressions of AP and TRA-1–60/81 were not affected. (C, D) Oct4 and Nanog expression levels exhibited a similar pattern of reduction as in AP and TRA-1–60/81 expressions (B). Three replicates. β-Actin was used as a loading control. All treatments were performed in triplicate. Scale bars: 100 μm. Bars without a common superscript differed (p < 0.05).
Discussion
Recently, several rESC lines, including those derived from parthenotes in our laboratory, have been established (24). However, there were some interesting points that needed to be addressed in the present study. First, p-rESCs lack of biparental imprinting gene (no paternal genome). Fertilized rESCs, on the contrary, have a more complete genetic background (containing both maternal and paternal genomes) (24). Second, in vitro derivation of rESCs from fertilized embryos was more efficient than that of parthenote-derived ESCs (17.2% vs. 4.6%), although both conventionally derived ESCs and parthenote-derived ESCs were maintained in an undifferentiated state with different genetic compositions and gene expressions respectively (24). Third, f-rESCs appeared to have a higher teratoma formation rate than parthenote-derived ESC lines. Higher cell numbers (20 × 106) of p-rESCs were required for a nude mouse to form teratoma (24), compared to that for the f-rESCs (5.0 × 106). Fourth, efficient germline transmission by an ESC line is a basic protocol to produce continuous generations of transgenic animals. Fertilized rESCs rather than p-ESCs can produce germline transmissible progenies of both sexes (49). Mammalian pathenogenetic embryos have apparently not yet been reported to achieve full-term development after embryo transfer due to the lack of a paternal genome. Therefore, fertilized rESCs are still the primary choice for production of transgenic animals. Last but not least, on the basis of proteomics profiling, we recently reported that many proteins in parthenote and fertilized embryoderived ESCs were differentially expressed by either up-or downregulatory mechanisms (26). Heat shock protein 60 isoform 4 protein and tubulin β-5 chain protein are upregulated in f-rESCs, whereas six other proteins [tubulin, β 2A class IIa (TUBB2A) protein, keratin 8 (KRT8) protein, α-enolase protein, 14–3-3 protein sigma, heat shock protein 60 (HSP60), and myosin-9] were highly expressed in p-rESCs (26). These observations strongly indicated that f-rES and p-rESCs are in essence two different cell types.
Intrinsic self-renewal capability and maintenance of stemness define the uniqueness of each ESC line. As reported, LIF signaling pathway maintained symmetrical self-renewal in mESC lines (50) but not in hESC lines (54). Our previous study also demonstrated that LIF signaling was sufficient to maintain self-renewal of rESCs; furthermore, the major players within the pathway, including LIFR/gp130, JAK kinases, and STAT3, were all expressed in rESCs. Such a LIF-dependent characteristic of rESCs is similar to that of mESCs (25). However, rESCs derived from fertilized embryos can be effectively maintained under a comparable culture condition to hESCs with low FGF2 concentration (4 ng/ml), as previously reported (54). In this study, although a wide range of FGF2 concentrations (0.1 to100 ng/ml) was used, optimal efficiency was in the range of 5–20 ng/ml (Table 3). The ESC lines derived from the low (0.1 and 1.0 ng/ml) and high (50 and 100 ng/ml) concentrations of FGF2 lost typical ESC morphology and became differentiated fibroblast-like cells.
In the present study, we examined FGF2 signaling associated with the mitogen-activated protein kinases ERK1/2 and phosphatidylinositol-3 kinase (PI3K)-AKT pathways. According to findings in hESCs (based on the FGF2 binding assay), FGF2 signaling was predominantly executed via binding with FGFR2 to activate MAPKs and AKT kinases (13). In the present study, MEK/ERK and PI3K/AKT signalings were essential in maintaining pluripotency and self-renewal of rESCs, similar to hESCs on MEF feeders (4,13,28,32). It has been demonstrated that LIF or FGF2 alone can maintain self-renewal of rESCs (12,16,19,25). Both LIF and FGF2 were also used to maintain rESCs by several groups (9,22–24,72), although optimized dosages and their combinatory effects were not determined. In mESCs and hESCs, LIF or FGF2 combined with BMP were reported to sustain undifferentiated proliferation (67,69). Similarly, FGF2 together with Activin/Nadal or insulin-like growth factors were both added to maintain self-renewal of hESCs (5,55).
To eliminate other confounding paracrine molecules in culture, we used a feeder-free system to test the roles of LIF and FGF2 signaling in rESCs. In the presence of both LIF and FGF2, rESCs maintained normal colonial morphology and expressed greater TRA-1–60, yet similar levels of AP and TRA-1–81, compared to cells supplemented solely with LIF or FGF2 in culture (Fig. 5). Furthermore, phosphorylation status of LIF and FGF2 downstream molecules, including STAT3, ERK1/2, and AKT, was drastically influenced by the presence of LIF and/or FGF2. Expression levels of pluripotency markers and signaling proteins were also affected by the presence of various inhibitors in the feeder-free condition (Fig. 6). Therefore, we inferred that LIF and FGF2 both acted to maintain rESC pluripotency and self-renewal, even though other complicated cascades involving additional signaling molecules could not be completely excluded. It was noteworthy that all the rESC pluripotency characteristics were maintained by either LIF or FGF2 alone at a rather basal level, although they were improved to some extent by coexistence of both factors, or by a single factor at higher concentrations.
In mESCs, LIF signaling was integrated into the core regulatory circuitry of pluripotency mainly via two parallel pathways. The JAK-STAT3 pathway activated transcription of Kruppel-like factor 4 (Klf4) gene and then Sox2, but not Nanog. Second, both PI3K/AKT and MAPK-growth factor receptor-bound protein 2 (GRB2) pathways regulated expression of the transcription factor-encoding gene T-box 3 (Tbx3), which is upstream of Nanog (40). In this study, treatments of rESCs with LIF and FGF2 in culture medium increased their Nanog expression and AKT phosphorylation (Fig. 6). Supplementary inhibitor LY294002 to suppress the PI3K/AKT pathway had apparent effects on AKT phosphorylation (Fig. 6A) and decreased Nanog expression (Fig. 6C), suggesting that the PI3K/AKT pathway was of importance to the downstream pathways of LIF and FGF2 in the rESC lines examined in the present study. This observation was similar to that reported in mESCs and hESCs that Nanog expression was regulated via the PI3K/AKT pathway (52) and that the PI3K/AKT signaling prevented cell death (60,65). In the present study, rESCs were more likely to die after being treated with an AKT inhibitor compared to those of the untreated group. It was therefore evident that the PI3K/AKT signaling was crucial for rESCs in both maintaining self-renewal and preventing cell death. In brief, inhibition of PI3K/AKT signaling augmented LIF-induced phosphorylation of ERKs and induced differentiation of mESCs, whereas simultaneous inhibition of PI3K/AKT and MEK/ ERK1/2 reversed the negative effect of PI3K inhibition on mouse ESC self-renewal, suggesting that the elevated ERK1/2 activity observed upon PI3K inhibition also caused differentiation (41).
Regardless of the presence of feeder cells, activation (p < 0.05) of MEK/ERK1/2 and PI3K/AKT was observed (Fig. 3E, F vs. 6D–F), both of which are downstream of the FGF2 pathway. Clearly, ERK1/2 and AKT signaling participated in maintaining rESCs in an undifferentiated state. Multiple levels of cross talks between the MEK/ERK1/2 and PI3K/AKT signaling pathways have been reported. Although some asserted that PI3K activity was essential for induction of Raf/MEK/ERK activity (29,62), others concluded that PI3K/AKT had an inhibitory role in MEK/ERK1/2 signaling. However, based on the current results, we concluded that U0126 and LY294002 had obvious inhibitory effects on phosphorylation of ERK1/2 and AKT, respectively, suggesting that both pathways were intricately managing a dialogue to balance these two pathways of the rESC lines.
To substantiate the joint effects of LIF and FGF2 signaling in maintaining rESCs in culture, a working model is proposed (Fig. 7). Based on our current study, we inferred that after binding of the LIF to the heterodimer formed by the LIF receptor and gp130, JAK/STAT3 became activated and phosphorylated, which was essential to sustain undifferentiation of rESCs, as well as mESCs. Conversely, we also demonstrated that FGF2-activated MEK/ERK1/2 and PI3K/AKT signaling pathways, which was as substantial to hESCs in maintaining self-renewal. When LIF and FGF2 were both present in culture, the levels of MEK/ERK1/2 and PI3K/AKT phosphorylation were maximized, since LIF alone could also increase phosphorylation in ERK1/2 via Grb2 (8). This activation was dependent on phosphorylation of the cytoplasmic tyrosine phosphatase Src homology region 2 domain-containing phosphatase-2 [SHP2 or tyrosineprotein phosphatase non-receptor type 11 (PTPN11)], which acted as an adaptor protein associated with growth factor receptor-bound protein 2 [GRB2 to activate the son of sevenless homolog (SOS)]/MEK/ERK1/2 pathway (3,8). We inferred that phosphorylation of Tyr757 at gp130 to recruit SHP2 was a JAK1-dependent process.
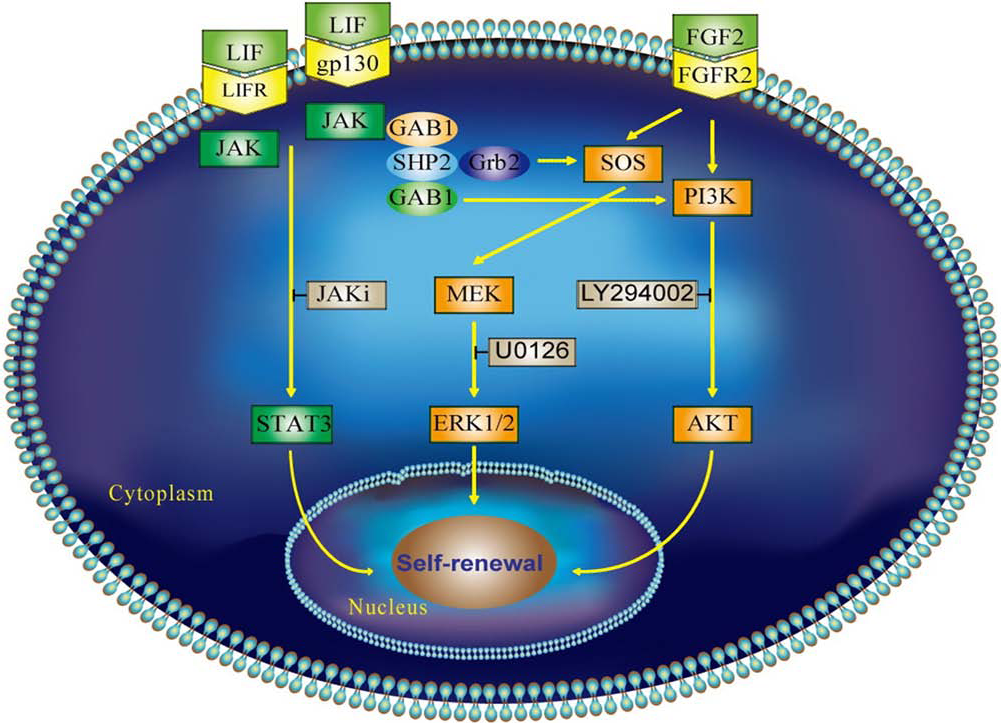
The proposed model illustrates the collaborative integration of LIF and FGF2 signaling pathways in sustaining stemness of rESCs. The arrow-ended lines indicate the potential route to transfer positive stimulations from upstream molecules, and the hammer-ended lines are positions at which inhibitors are effective. Both the LIF and FGF2 signaling pathways are integrated to maintain self-renewal of rESCs in an undifferentiated state. When LIF and FGF2 are both available in culture, their downstream kinases including Janus kinase (JAK)/STAT3, ERK1/2, and PI3K/AKT may be activated to maintain an intricate homeostasis for self-renewal and proliferation of rESCs. The upstream molecules growth factor receptor-bound protein 2 (GRB2)-associated binding protein 1 (GAB1) and Src homology region 2 domain-containing phosphatase-2 (SHP2) may recruit PI3K to these cascades. It is likely that an alternative redundancy of these pathways was conserved during evolution. Once activated, the corresponding downstream molecules in each cascade are recruited and translocated to the nucleus to initiate the transcription associated with self-renewal and sustaining stemness. Conversely, when LIF and/or FGF2 are absent or blocked, only one or partial downstream pathways is functioning. The possibility of the coexistence or redundancy of these signaling pathways in rESCs requires more intensive study. Gp130, glycoprotein 130; SOS, son of sevenless homolog; MEK, mitogen-activated protein kinase kinase.
The LIF signaling also interacts with FGF2 signaling via the PI3K/AKT pathway, which has been associated with proliferation, survival, and self-renewal of mESCs (41,61). In mESCs, blocking the PI3K signaling substantially suppressed LIF-stimulated phosphorylation of AKT, glycogen synthase kinase 3α/β (GSK3α/β), and S6 proteins, yet it increases ERK1/2 phosphorylation (8). Therefore, perhaps various signaling molecules, for example, bone morphogenetic protein, Wnt, Activin/Nodal, and/or other transforming growth factor family ligands secreted by the feeder cells (23,58), may also contribute to maintaining rESC pluripotency, self-renewal, and proliferation. Nevertheless, we conclude that LIF or FGF2 per se are sufficient for isolation and proliferation of rESC lines derived from fertilized embryos, while keeping them in an undifferentiated state. When both factors are present in culture, there are additive effects on sustaining the stemness of rESC lines.
Footnotes
Acknowledgments
This study was supported in part by grants from the National Science Council (NSC# 96–2313-B-005–013, NSC# 98–2628-B005–019-MY3, NSC# 101–2313-B005 -013 -MY3 and NSC# 102–2313-B-059–002-MY2), Executive Yuan and the Ministry of Education, Taiwan, Republic of China, under the ATU plan. We are also grateful to Dr. John Kastelic, Professor, University of Calgary, Canada, for his critical reading and suggestions to improve this manuscript. The authors have no affiliations with or involvement in any organization or entity with financial interest or non-financial interest in the subject matter or materials discussed in this manuscript.