
Abstract
Magnetic resonance (MR) imaging of superparamagnetic iron oxide (SPIO)-labeled stem cells offers a non-invasive evaluation of stem cell engraftment in host organs. Excessive cellular iron load from SPIO labeling, however, impairs stem cell differentiation. The purpose of this study was to magnetically label human embryonic stem cells (hESCs) via a reduced exposure protocol that maintains a significant MR signal and no significant impairment to cellular pluripotency or differentiation potential. hESCs were labeled by simple incubation with Food and Drug Administration-approved ferumoxides, using concentrations of 50–200 μg Fe/ml and incubation times of 3–24 h. The most reduced exposure labeling protocol that still provided a significant MR signal comparable to accepted labeling protocols was selected for subsequent studies. Labeled hESCs were compared to unlabeled controls for differences in pluripotency as studied by fluorescence staining for SSEA-1, SSEA-4, TRA-60, and TRA-81 and in differentiation capacity as studied by quantitative real-time PCR for hOCT4, hACTC1, hSOX1, and hAFP after differentiation into embryoid bodies (EBs). Subsequent MR and microscopy imaging were performed to evaluate for cellular iron distribution and long-term persistence of the label. An incubation concentration of 50 μg Fe/ml and incubation time of 3 h demonstrated a significantly reduced exposure protocol that yielded an intracellular iron uptake of 4.50 ± 0.27 pg, an iron content comparable to currently accepted SPIO labeling protocols. Labeled and unlabeled hESCs showed no difference in pluripotency or differentiation capacity. Ferumoxide-labeled hESCs demonstrated persistent MR contrast effects as embryoid bodies for 21 days. Electron microscopy confirmed persistent lysosomal storage of iron oxide particles in EBs up to 9 days, while additional microscopy confirmed the iron distribution within single and multiple EBs. Labeling hESCs with ferumoxides by this tailored protocol reduces exposure of cells to the labeling agent while allowing for long-term visualization with MR imaging and the retention of cellular pluripotency and differentiation potential.
Keywords
Introduction
The first human embryonic stem cells (hESCs) were isolated in 1998 (55). Just more than a decade later, in 2009, the Food and Drug Administration (FDA) approved the first-phase I clinical trial of spinal cord injury in humans based on treatment with oligodendrocyte precursor cells derived from human embryonic stem cells (17). Unlike adult stem cells, embryonic stem cells exhibit unique pluripotency, plasticity, unlimited capacity for self-renewal, and the ability to grow into any cell type of the human body. It is these characteristics that give embryonic stem cells their particular potential for applications in regenerative medicine (5, 44). Stem cell transplantations, however, have demonstrated low survival rates in vivo (7, 32, 36). A noninvasive method that could evaluate stem cell migration dynamics, homing and engraftment longitudinally in vivo, could offer critical insight into factors that may facilitate or impair successful stem cell treatment outcomes.
Magnetic resonance (MR) imaging is a noninvasive, radiation-free, and immediately clinically applicable imaging modality with high anatomical resolution and high soft tissue contrast. MR imaging cannot distinguish transplanted stem cells from host tissues; cells must be labeled with specific contrast agents or reporter genes to allow in vivo tracking with MR imaging techniques. Although a useful tool for preclinical studies, stem cell labeling via MR reporter genes requires genetic manipulation, that is, the integration of reporters into the target cells' genome, which could be a barrier for potential clinical translations (18, 35, 58, 61). Labeling stem cells with MR contrast agents, on the other hand, can provide quick, easy, and effective cell tracking, which would be, in principle, immediately clinically applicable. The FDA-approved iron oxide nanoparticle-based contrast agent ferumoxide has been extensively studied for this purpose (2, 4, 13, 14, 25). Monocytes, macrophages, and various stem cell populations spontaneously phagocytose ferumoxides, which alter local magnetic fields, thus leading to a strong signal effect from iron-labeled cells on MR images (22, 38, 53).
In addition to a high clinical translational potential and ease of use of iron oxide nanoparticles, these agents are appealing for cell labeling because iron is considered a “safe” cell marker because the core component of superparamagnetic iron oxides (SPIOs) is an innate and essential element in a number of human cellular functions including metabolic processes such as electron transport, deoxyribonucleotide synthesis, oxygen transport, and redox reactions involving hemoproteins (45, 46). On the other hand, overload of cellular iron poses risks such as oxidative stress and the formation of reactive oxygen species (24, 43). It is therefore essential to investigate effective and safe cellular iron-loading techniques.
Although some studies have demonstrated normal differentiation of ferumoxide-labeled cells, including neuronal stem cells into glia (31), embryonic stem cells into neuronal cells (52), and mesenchymal stem cells into chondrocytes (3), other studies have demonstrated disrupted differentiation following labeling with iron oxides, including hindrance of mesenchymal stem cell (MSC) differentiation into chondrocytes (6, 21, 41). Reported impairments in differentiation, however, have been dose dependent, with negative effects directly corresponding to increased iron load of labeled cells (6, 21, 30, 41).
Previous MR imaging studies of magnetically labeled ESCs have been performed with high iron concentrations, incubation times, and/or transfection agents and resulted in high cellular iron load, on average greater than 10 pg/cell, or failed to comprehensively investigate MR signal corresponding to cellular iron load (23, 25, 51, 53). Both our group and others have investigated the iron content–MR signal correlation and have found that stem cells labeled with ferumoxides with an iron load of 4.1 pg Fe/cell caused a significant signal decline on T2-weighted MR images (12, 22). It is further suggested that this iron load is a favorable cellular iron load for significant and sustained MR detectability (59). The purpose, therefore, of this study was to evaluate if a reduced iron exposure labeling protocol could be applied to hESCs, which would lead to a similar iron load (about 4 pg/cell) and at the same time provide preserved MR signal effects and unimpaired pluripotency and hESC differentiation. To the best of our knowledge, this is the first study that attempts to reduce the exposure protocol for labeling hESCs with SPIOs while studying the influence of iron oxide nanoparticle labeling on hESCs plasticity. If successful, this technique would, in principle, be readily clinically applicable.
Materials and Methods
Culture of Undifferentiated hESCs
The study was approved by the Committee on Human Research at our institution. HSF-6 cells (UC06, an NIH-approved cell line; University of California, San Francisco, CA, USA) were cultured as undifferentiated, pluripotent stem cells in 10-cm petri dishes (BD Falcon, Sparks, MD, USA) on irradiated mouse embryonic fibroblasts (~800,000 cells/plate). Cell culture dishes were coated with 0.1% gelatin (Sigma-Aldrich, St. Louis, MO, USA), and cells were grown in hESC medium at 37°C in a 5% CO2 atmosphere. hESC medium was prepared as follows: knockout Dulbecco's modified Eagle's medium (DMEM), 20% knockout serum replacement (KSR), 1% nonessential amino acids, 1 mM l-glutamine, 4 ng/ml human bFGF (basic fibroblast growth factor; all from Invitrogen, Carlsbad, CA, USA), and 0.1 mM β-mercaptoethanol (Sigma-Aldrich). Every 4–5 days, cells were split manually by dissection at a ratio of approximately 1:3.
Contrast Agent
Ferumoxides (Endorem, Guerbet, Aulnay-sous-Bois, France) are SPIOs composed of a nonstoichiometric magnetite core and a dextran coat (26). At 37°C and 0.47 T, ferumoxides have an r1 relaxivity of 40.0 mM−1 s−1 and an r2 relaxivity of 160 mM−1 s−1, with a hydrodynamic diameter of 80-150 nm (57).
Ferumoxides are FDA approved for MR imaging of the liver in patients. The iron oxide particles are phagocytosed by reticuloendothelial cells and stored in secondary lysosomes within the cytoplasm (38).
Cellular Labeling
Our group along with other investigators has found that ferumoxide-labeled human MSCs and mESCs with a cellular iron load of 4.1 pg Fe/cell caused a significant signal decline on T2-weighted MR images (12, 22). In order to achieve a similar iron load in hESCs, stepwise studies were performed to reduce SPIO labeling concentrations and incubation times as compared to those previously applied to hESCs (9, 20, 34, 53). All labeling experiments were performed in triplicates.
Ferumoxide concentration: Undifferentiated hESCs were labeled while the cells were in a monolayer and incubated with decreasing concentrations of 200, 100, and 50 μg Fe/ml in serum-free hESC medium for an incubation time of 12 h.
Incubation time: hESCs were incubated for decreasing durations of 24, 12, 6, and 3 h at 50 μg Fe/ml.
Cell samples were collected as follows: Plates with labeled and unlabeled colonies of undifferentiated hESCs were treated with 4 ml of collagenase IV (1 mg/ml; Sigma-Aldrich) for approximately 6–8 min until colonies partly detached. Colonies were transferred to a centrifuge tube and allowed to sit for 2 min for selective sedimentation of colonies, while single cells (detached feeders, dead cells) remained in suspension.
Supernatant was discarded, and the cells were washed three times with prewarmed phosphate-buffered saline (PBS). Following washing, cells were resuspended in warm hESC medium. For pilot MR studies, for analysis by inductively coupled plasma atomic emission spectroscopy (ICP-AES), and for trypan blue staining, cells were further digested into single cells by 0.1% trypsin (Invitrogen) for 5 min. Labeled cells and unlabeled controls underwent quantification of iron content by ICP-AES in order to demonstrate iron deposition. Parallel samples underwent MR imaging to assess for contrast-agent effects as described below.
MR Imaging of Labeled hESCs
The aim of these experiments was to ensure a significant MR signal effect for iron oxide-labeled cells via a protocol that reduced the iron oxide load of cells. Samples of ferumoxide-labeled hESCs and unlabeled controls underwent MR imaging, and signal effects were compared for significant differences. Triplicate samples were scanned as cell pellets in Eppendorf tubes. Cell pellets were imaged because pellets provide maximal MR signal effects and the supernatant above the pellet serves as a marker for free iron oxides in suspension when compared to the signal of the surrounding water bath. Dispersed cells in solution do not allow differentiation of MR signal effects from labeled cells and potential residual free iron oxides in suspension. In addition, evaluation of cell pellets ensures consistent cell concentrations between different experiments. In a first experiment, 8 × 106 cells per pellet were investigated after incubation with decreasing concentrations of ferumoxides.
In a second experiment, 2 × 106 cells per pellet were examined after incubation with ferumoxides at decreasing incubation times. The cell number per sample in the second study was reduced compared to the first study in order to investigate more time points with the limited quantity of available cells. Test tubes were immersed in a water bath to avoid susceptibility artifacts from surrounding air and then scanned at room temperature (20°C). MR imaging was performed on a 3T clinical MR scanner (Signa Excite HD 3T, GE Medical Systems, Milwaukee, WI, USA) using a standard circularly polarized quadrature knee coil (Clinical MR Solutions, Brookfield, WI, USA). MR images were obtained using T1- and T2-weighted spin echo (SE) sequences with respective TR values of 500 ms/4,000 ms and TE values of 15 ms/15 ms. Acquisition times were 2:16 and 17:52 min, respectively. T2*-weighted axial and coronal gradient echo images were obtained with a flip angle of 30°, a TR of 500 ms, a TE of 14.4 ms, and an acquisition time of 2:12 min. All sequences were acquired with a field of view (FOV) of 120 × 120 mm, a matrix of 256 × 256 pixels, a slice thickness of 2 mm, and one acquisition.
MR images were analyzed using a DICOM imaging software (OsiriX, UCLA, Los Angeles, CA, USA). Signal intensities of cell pellets were determined via user-defined regions of interests and divided by the background noise to obtain the signal-to-noise ratio (SNR). SNRs were tested for significant differences between labeled and unlabeled cells. Based on the results of initial optimization experiments, an incubation concentration of 50 μg Fe/ml and an incubation time of 3 h were used for all further experiments.
Evaluation of Pluripotency
To examine if hESCs labeled under the experimental conditions retained expression patterns consistent with undifferentiated/pluripotent hESCs and to account for potential delayed effects, cells were examined during the first three passages after labeling.
Colonies of unlabeled and labeled cells were plated in gelatin-coated 24-well plates on feeder cells and allowed to grow for 2–3 days. Cells were then stained according to the manufacturer's instructions (ES Cell Characterization Kit, Chemicon, Billerica, MA, USA) with the primary antibodies [stage specific embryonic antigen-1 (SSEA-1), SSEA-4, TRA-1-60, and TRA-1-81; 1:50], and goat serum (4%) was added as a blocking solution. Goat anti-mouse IgG (Invitrogen; 1:200) was used as secondary antibody. Cells were then counterstained with Vectashield® mounting medium containing DAPI (Vector Laboratories, Inc., Burlingame, CA, USA; 1:15) and imaged on a Nikon TE 2,000 E (Nikon, Melville, NY, USA).
Induction of Embryoid Bodies
The formation of embryoid bodies was induced using previously established protocols that were adapted for our purposes (10, 16). In brief, colonies of undifferentiated hESCs were resuspended in high-glucose Dulbecco's modified Eagle's medium (Invitrogen), supplemented with penicillin/streptomycin and 20% fetal calf serum. The cell suspension was then added to ultralow attachment dishes (Corning, Inc., Corning, NY, USA). At different time points as specified below, embryoid bodies (EBs) were harvested and used for follow-up MR studies, microscopy visualization, or quantitative real-time PCR (qPCR).
Long-Term MR Signal Effects of EBs
Longitudinal MR imaging of EBs was performed to assess the long-term signal effects of labeled hESCs. EBs derived from labeled and unlabeled hESCs were collected after 1, 4, 7, 14, and 21 days of differentiation and digested into a single-cell suspension. Cell pellets of 2 × 106 cells were scanned in Eppendorf tubes using the MR imaging protocol described above.
Intracellular Iron Oxide Uptake and Retention
To localize the iron oxide nanoparticles in the cells, Prussian blue staining was performed at days 1, 4, 7, 14, and 21. Ferumoxide-labeled EBs were fixed with 10% formalin. The samples were dehydrated, embedded in paraffin, and cut into 5-μm sections, which were then mounted on glass slides and stained with the Prussian blue kit (Sigma-Aldrich).
In addition, EBs derived from unlabeled and labeled cells at day 9 of differentiation were fixed with 2% glutaraldehyde in 0.1 M sodium cacodylate buffer, then postfixed with 1% osmium tetroxide followed by 2% aqueous uranyl acetate. The samples were dehydrated with ethanol and embedded in epoxy resin. Ultrathin sections were stained with 2% uranyl acetate and Reynolds lead citrate. They were examined and photographed at 80 kV by transmission electron microscopy (TEM) (JEOL 100CX II). Additional microscopy (Zeiss Axio Observer Z1, Carl Zeiss IMT, Maple Grove, MN, USA) was performed to examine iron contrast-agent distribution within single and multiple EBs.
Evaluation of the Differentiation Potential of Labeled EBs
Retained pluripotency of labeled cells was determined by somatic differentiation into the three germ layers. EBs were harvested after 16 days of differentiation and were then subjected to qPCR expression analysis for the following markers: octamer-binding transcription factor 4 (OCT4) as embryonic marker; sex-determining region Y box 1 (SOX1) as ectodermal marker; α-fetoprotein (AFP) as endodermal marker; and α-actin, cardiac muscle 1 (ACTC1) as mesodermal marker. Control markers were glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and mediator complex subunit 1 (MED1) (primers sequences are described in Table 1). Total cellular RNA was prepared from each sample with the Pure Link™ Micro-to-Midi™ Total RNA Purification System (Invitrogen). cDNA was prepared from 5 μg of total RNA in a 100-μl reaction using the ProtoScript cDNA preparation kit (NEB, Beverly, MA, USA). All quantitative real-time PCRs (qPCRs) were carried out and analyzed on an Applied Biosystems 7300 Real-Time PCR System. Briefly, each PCR reaction was carried out with TaqDNA Polymerase (Invitrogen, Carlsbad, CA) and consisted of the following reaction mixture: 1.1 μM primers, 1/210th of the cDNA reaction (corresponds to cDNA derived from 10 ng of total RNA), 125 μM dNTPs, 1.5 mM MgCl2, and 1x reaction buffer [20 mM Tris (pH 8.4), 50 mM KCl]. Taq DNA polymerase [0.15 μl (0.75 units)] was used for each reaction. Formation of the double-stranded DNA product was monitored using SYBR Green (Molecular Probes, Eugene, OR, USA); ROX served as reference dye. Expression data were collected as single-sample Ct values, where Ct is the number of PCR cycles that elapse before a threshold concentration of PCR product is reached, thus providing a direct measure of the input concentration of nucleic acid. mRNA from each experiment was normalized to two different control genes, MED1 and GAPDH.
List of Primers Used for Quantitative Real-Time PCRs (qPCRs) to Evaluate the Differentiation Potential of Labeled EBs

Statistical Analyses
Statistical analysis was performed to compare the varying labeling protocols. Quantitative viability, intracellular iron content, and MR data from triplicate experiments were compared for significant differences between labeling protocols and unlabeled controls, using a t test and a value of p < 0.05. Values of p for comparisons of multiple experimental groups were adjusted with the Bonferroni correction. Calculations were performed using the statistical software SPSS (SPSS, Inc., Chicago, IL, USA).
Results
Cellular Labeling
Spectrometry revealed a significant (p < 0.05) iron uptake for all labeling protocols (Fig. 1), while trypan blue staining showed no significant (p > 0.05) decrease in cellular viability (Fig. 1). Labeling hESCs with decreasing concentrations of ferumoxides (200, 100, and 50 μg Fe/ml) resulted in decreased iron uptake (Fig. 1A). In addition, labeling of hESCs with decreasing incubation time (24, 12, 6, and 3 h) resulted in decreased cellular iron uptake (Fig. 1B). Labeling with 50 μg Fe/ml for 3 h yielded a cellular iron uptake of 4.50 ± 0.27 pg Fe/cell, which was in the order of previously reported iron quantities that led to significant MR signal effects in other stem cell types (12, 22).

Cellular iron uptake and cell viability after labeling. Iron concentrations used for labeling (A) (incubation time, 12 h) and incubation time (B) (50 μg Fe/ml) are proportional to the intracellular iron deposition. Iron uptake was significant for all labeling protocols. There was a minor dose-dependent but nonsignificant decrease of cell viability in labeled cells immediately after labeling (A, B). *Significant difference in relation to unlabeled control (p < 0.05).
MR Imaging of Labeled hESCs
MR images of labeled cell pellets showed a strong negative (dark) contrast effect when compared to unlabeled controls for all protocols and for all pulse sequences used (T1, T2, and T2*) (Fig. 2A, B). This was confirmed by the corresponding SNR values that were significantly lower than unlabeled controls (p < 0.05) (Fig. 2C, D). The SNR of labeled cell pellets showed no significant difference between samples (p > 0.05) (Fig. 2C). Cells incubated for 12 h with varying concentrations of the contrast agent demonstrated a chemical shift artifact on T1 and T2 sequences and a blooming effect on T2* sequences (Fig. 2A). SNR quantification of cell pellets labeled for 12 h with varying concentrations showed no significant difference between labeled samples (p > 0.05) (Fig. 2C).

MR imaging for evaluation of different labeling protocols. (A, B) All incubation protocols caused a strong visible contrast-agent effect on MR images. (C, D) Corresponding quantitative analysis of MR images: Signal-to-noise ratios (SNRs) showed significant contrast-agent effects on T1, T2, and T2* sequences for all labeling protocols. *Significant difference in relation to unlabeled control (p < 0.05).
The lower cell count (2 × 106 vs. 8 × 106 cells) used for the investigation of the effects of varying incubation times led to reduced artifacts and increased sensitivity (Fig. 2B). T1 and T2* images showed only a minor decrease in contrast-agent effect as incubation time decreased. Differences in SNR measurements were not significant (p > 0.05) (Fig. 2D).
Evaluation of Pluripotency
Immunohistochemistry analyses revealed that iron oxide-labeled and unlabeled HSF-6 cells expressed similar patterns in surface antigens SSEA-4, TRA-1-60, and TRA-1-81, and neither group expressed SSEA-1 for up to three passages after labeling (Fig. 3). In passage 3, minor inhomogeneities in the distribution of pluripotency markers were demonstrated in both control samples and labeled samples. There were no observed morphological abnormalities of labeled hESCs (colonies had clearly defined borders and the cells within each colony were homogenously sized) compared to unlabeled controls for a period of three passages after labeling. Overall, expression patterns were consistent with undifferentiated hESCs or “pluripotent” stem cells.
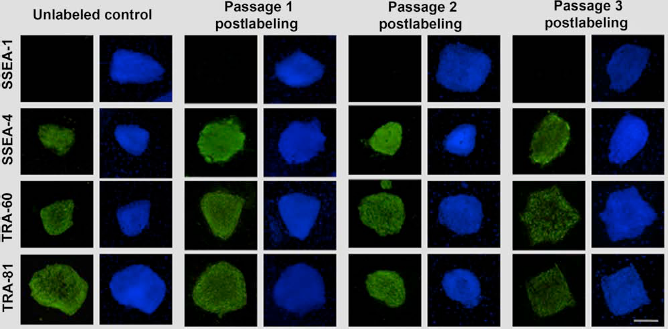
Retained pluripotency of labeled human embryonic stem cells (hESCs). Fluorescent staining of labeled hESCs and unlabeled control showed negative staining for stage-specific embryonic antigen-1 (SSEA-1) and positive staining for SSEA-4, TRA-60, and TRA-81, consistent with pluripotent cells. This was shown for three passages after labeling. Scale bar: 100 μm.
Long-Term MR Imaging Signal Effects of EBs
Longitudinal MR imaging studies of EBs from cells labeled with the optimized protocol showed a marked MR signal effect on all sequences at all time points of observation, up to 21 days after differentiation (Fig. 4). A T2 effect was seen on both T2- and T1-weighted sequences. The labeled cells did not show any T1 effect on T1 sequences, likely due to the relatively high iron load, which led to confounding T2 effects. On T2*-weighted images, a blooming artifact was seen at all time points. The contrast-agent effect showed only a minor decrease after 21 days. By contrast, unlabeled controls showed no contrast-agent effect on any pulse sequence.
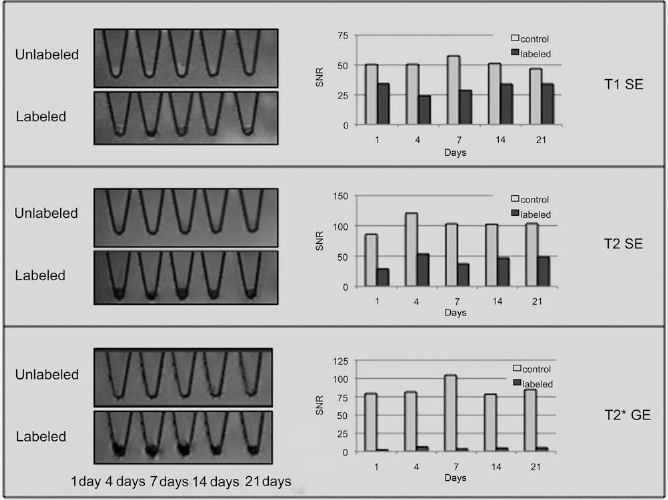
Long-term MR contrast effect of labeled hESCs. Representative MR images and corresponding signal-to-noise (SNR) values of embryoid bodies acquired with different MR pulse sequences. Samples were collected at different intervals of embryoid body formation. A marked contrast-agent effect was found for all sequences and all time points for up to 21 days.
Intracellular Iron Oxide Uptake and Retention
Prussian blue staining showed the presence of the iron oxide particles in EBs (Fig. 5). To confirm that the iron oxide particles are localized in the cytoplasm and not on the surface of the cells, TEM studies were performed at day 9 of EB differentiation (Fig. 6). TEM confirmed the intracellular deposition of iron oxide particles within lysosomes of labeled cells (Fig. 6B, C). By contrast, there were no SPIO particles in unlabeled controls (Fig. 6A). No morphological difference was noted between unlabeled and labeled cells, and the cellular ultrastructure appeared normal with typically large nuclei. Additional microscopy confirmed no iron deposition within unlabeled EBs (Fig. 6D), significant distribution of iron within multiple EBs (Fig. 6E), and uneven distribution of iron within a single EB (Fig. 6F).
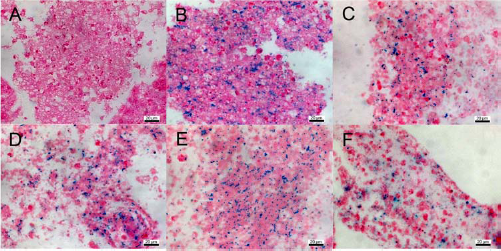
Cellular iron storage. Prussian blue staining of unlabeled (A) and labeled hESCs in embryoid bodies at different time points: day 1 (B), day 4 (C), day 7 (D), day 14 (E), and day 21 (F). Scale bars: 20 μm.

Intracellular iron deposition. Transmission electron microscopy of unlabeled (A) and labeled (B, C) hESCs in EBs at day 9 of differentiation. Arrows indicate intracellular compartments: Nucleus (open black arrow), nucleolus (black arrrow), mitochondria (dotted arrow), and lysosomes containing iron oxide particles (white arrowhead). Scale bars: 5 μm (A, B) and 1 μm (C). (D–F) Microscopy of (D) unlabeled control EBs (original magnification: 5x) and (E, F) ferumoxide-labeled EBs derived from hESCs clearly displaying iron distribution within EBs (white arrows) [original magnification: 5x (E) and 20x (F)].
Evaluation of the Differentiation Potential of the Labeled EBs
qPCR of EBs after 16 days of somatic differentiation in EBs showed a decreased expression of the embryonic marker OCT4 (–6.7-fold) and increased expression of the endodermal marker AFP (2-fold), the ectodermal marker SOX1 (20-fold), and the mesodermal marker ACTC1 (189-fold) (Fig. 7). This pattern was similar for both unlabeled controls and labeled hESCs and is consistent with a successful somatic differentiation of pluripotent hESCs.

Differentiation capacity of labeled hESCs. Cell marker expressions in 16-day-old embryoid bodies were assessed for embryonic (hOCT4) and somatic markers (hACTC1, hSOX1, and hAFP) by qPCR. Expression was normalized to GAPDH, and change is indicated relative to undifferentiated hESCs. Marker expression is consistent with somatic differentiation into all three germ layers and remains unchanged for labeled cells.
Discussion
The results of this study indicate that hESCs labeled by a reduced iron exposure, simple incubation protocol with the FDA-approved contrast agent ferumoxides effectively label cells for detection with MR imaging. The labeled cells showed marked MR signal up to 21 days after labeling and preserved cell pluripotency and differentiation capacity. Iron oxide nanoparticles are an attractive agent for cell labeling because they not only provide sensitive cell detection on MR images, but their contrast effect depends upon a naturally occurring physiological substrate; thus, these agents do not introduce substantial foreign components into cells during labeling. Iron, the primary component of SPIOs, is an essential component of human cells and plays a key role in many cellular metabolic processes such as electron transport, deoxyribonucleotide synthesis, oxygen transport, and essential redox reactions involving hemoproteins (45, 46). Depletion of iron inhibits cellular proliferation and induces apoptosis (49). Iron overload, on the other hand, causes the formation of toxic reactive oxygen species. Furthermore, recent studies have found that chronic cellular iron overload is associated with progressive damage to mitochondrial DNA, which in turn leads to loss of normal cellular respiration (15).
Overloading stem cells with iron oxide-based contrast agents might pose similar adverse effects. While some authors have described unimpaired neurogenic differentiation of iron oxide nanoparticle-labeled embryonic stem cells (31, 52), other authors have described an inhibited chondrogenic differentiation of iron oxide-labeled MSCs (21, 28). These inhibitory effects, however, have been demonstrated as concentration dependent, with increasing cellular iron oxide concentrations resulting in increased inhibitory effects of cellular iron oxides (21). Therefore, we sought to reduce cellular exposure to iron oxide nanoparticles compared to previously accepted magnetic labeling protocols in order to reduce the potential risk of adverse effects while ensuring a significant MR effect and unimpaired viability, pluripotency, and differentiation capacity of labeled cells. Our data showed that a limited iron uptake of 4.5 pg/cell was sufficient to provide significant MR signal effects while not impairing the cells' pluripotency and differentiation capacity. Considering that this protocol carries potential translational applications, it is prudent to consider the in vivo iron dose this protocol would deliver and the potential implications of this iron load. The dose delivered to a patient by a stem cell transplant of 1 × 107 stem cells and our labeling protocol of 4.5 pg of iron per cell would result in delivery of 45 μg of iron or 1% of the total body's iron load. This is a significantly minimal amount, considering that ferumoxides are an established intravenous contrast agent for liver imaging that is administered at a dose of 0.8 mg of iron per kilogram of body weight, a dose that corresponds to 64 mg for 80-kg patients. Numerous clinical studies have demonstrated that the iron delivered by ferumoxides is incorporated into the body's iron metabolism and is safe in patients (14, 47, 56, 60).
Several investigators have reported that iron oxide nanoparticles provide long-term labeling of stem cells for up to several weeks or even months (29, 34, 48). As labeled cells proliferate, intracellular iron divides among daughter cells. This causes dilution of the initial contrast-agent concentration and potentially decreases the signal effects of viable, dividing, and metabolically active stem cells (37). We found that the contrast-agent effect of our labeled hESCs showed only a minor MR signal decline over 21 days during differentiation into embryoid bodies. This minor decrease can be explained by the characteristic slow proliferation and expansion rate of embryoid bodies (19, 27).
In order to investigate any delayed effects on pluripotency following labeling, we analyzed the expression of characteristic markers with immunofluorescence for three passages after labeling. This technique is routinely performed as proof of pluripotency (40). Unlabeled controls and labeled cells in the third passage after labeling showed inhomogeneities in the expression of pluripotency markers. Such inhomogeneities are common in colonies of embryonic stem cells and have been reported repeatedly (33, 40).
In our data, inhomogeneities are present in control samples and also correlate with the cell densities as shown on corresponding DAPI stains, thus suggesting that inhomogeneities are not correlated with reduced pluripotency. We conclude that iron oxide-labeled hESCs can be maintained as a culture of pluripotent and undifferentiated hESCs. Following undirected somatic differentiation into embryoid bodies, qPCR of differentiated hESCs showed increased cell marker expression of all three germ layers, thereby confirming retained differentiation potential of labeled cells. Moreover, qPCR analysis showed almost identical levels of marker expression between labeled and unlabeled cells. Retention of pluripotency following SPIO labeling is significant because labeling hESCs after differentiation via simple incubation with an SPIO has been shown as not possible (8); thus for clinical therapeutic purposes, cells would require labeling prior to differentiation. Our proposed simple incubation labeling protocol with an FDA-approved contrast agent offers a potential protocol that could be applied for investigation in a clinical setting.
While variations in iron oxide labeling protocols have been previously investigated and established for somatic cells and adult stem cells (2, 4, 14, 23, 25, 38), few data are available on tailored techniques for magnetic labeling of hESCs (9, 34, 53). Previous investigators applied labeling protocols for adult stem cells to ESCs for the purpose of MR depiction but did not investigate the impact on the cell's pluripotency or differentiation capacity. Tallheden et al. successfully labeled hESCs with ferumoxides at 400 and 800 μg/ml and depicted these cells on MR images after engraftment into an explanted mouse heart (53).
The investigators reported no impairment to EB morphology (size and shape), viability, and proliferative activity, but they did not investigate how the proposed labeling technique might have affected pluripotency or the cell's differentiation capacity on a molecular level. In another study, Li et al. successfully labeled hESCs with ferumoxides and the transfection agent protamine sulfate to depict the labeled cells up to 4 weeks following implantation in murine hind limbs (34). These studies demonstrated the feasibility of long-term MR depiction of ferumoxide-labeled hESCs in vivo but relied on high concentrations of SPIOs and a transfection agent for cell labeling. Both studies did not address the pluripotency or differentiation capacity of the labeled cells. Our data showed no impairment to cellular pluripotency and EB formation and a persistent MR signal up to 21 days. Previous investigations showed no evidence of significant exocytosis of intracellular iron from labeled cells (38), thus suggesting that longitudinal MR signal effects can be attributed primarily to intracellular iron.
hESC labeling with ferumoxides for in vivo tracking with MR imaging is advantageous over other cell-tracking techniques because this technique permits longitudinal investigation and near microscopic anatomical resolution and is clinically translatable.
Additional imaging modalities for cellular visualization are available, including optical imaging (OI) and positron emission tomography (PET). However, while OI offers a high sensitivity and is inexpensive and fast, it is limited by low resolution, limited light penetration, and limited quantification and clinical availability. PET also offers a high sensitivity in addition to 3D imaging capabilities and wide clinical availability; however, it is associated with radioactive exposure to both labeled cells and the recipient of the labeled cells, is costly, and is limited in long-term monitoring applications due to limited half-lives of radiotracers (11, 50).
While MR imaging permits long-term tracking of iron-labeled cells, a challenge of longitudinal tracking is associated with death of some of the transplanted cells, resultant release of the iron oxide label, and secondary phagocytosis by resident macrophages. The detected MR signal at the transplantation site on long-term follow-up studies, as a result, may come from a combination of iron in originally labeled stem cells and iron in resident macrophages (1, 54). Pawelczyk et al. demonstrated that 5–15% of iron oxide label from transplanted stem cells might be incorporated by host tissue macrophages (42).
Conversely, other studies have demonstrated histological evidence of iron oxide retention in originally labeled stem cells up to 16 weeks posttransplantation (29, 48). Furthermore, our group is investigating distinct long-term signal characteristics of transplanted iron oxide-labeled viable stem cells versus apoptotic cells. Where viable cells slowly metabolize iron oxide labels thereby leading to persistent or slowly declining MR signal effects over time, released iron oxides from dead cells cause a transiently increasing signal effect, which subsequently vanishes in cases of phagocytosis and metabolization of iron by professional macrophages (39).
Of final interest, the ultrasmall superparamagnetic iron oxide (USPIO) ferumoxide is, to date, the only FDA-approved iron oxide-based MR contrast agent. Pharmaceutical companies, however, have recently discontinued production of ferumoxides due to limited marketing opportunities. While this contrast agent could potentially be specifically synthesized for cell-tracking applications, these agents are not easily accessible for translational studies. The SPIO Resovist is still available for clinical applications in Japan and would be expected to provide similar results as in this study. Furthermore, USPIOs such as ferumoxytol (FDA-approved iron supplement) and P904 (entering clinical trials in Europe) offer similar contrast-agent alternatives. Although USPIOs typically require longer incubation times and/or higher concentrations of the labeling solution in order to achieve similar labeling efficiencies as SPIOs, the iron core and coating of these iron oxides are similar and thus would suggest similar physiological effects. Further studies should be conducted to investigate if results from this study directly translate to these second-generation USPIOs.
Conclusion
In conclusion, our data demonstrate that hESCs can be labeled by a reduced exposure, simple incubation protocol with clinically applicable iron oxide nanoparticles to produce significant MR effects while preserving cellular viability, pluripotency, and differentiation capacity. This approach is, in principle, readily applicable for clinical investigations via “off-label” use of the FDA-approved iron oxide nanoparticle ferumoxide.
Footnotes
Acknowledgments
This study was supported by a research grant from the California Institute for Regenerative Medicine (CIRM) grant No. RS1-00381-1. We thank Jennifer Vancil for her excellent assistance with manuscript preparation and submission. The authors declare no conflict of interest.