
Abstract
The purpose of this study was to evaluate the use of three different superparamagnetic iron oxide (SPIO) particles for labeling of ovine and equine bone marrow (BM)-derived multipotent stromal cells (MSCs) in vitro. MSCs were obtained from five adult sheep and horses, respectively. After three passages (p3), cells were labeled with either 1) Molday ION Rhodamine B, 2) Endorem, 3) Resovist, or 4) remained unlabeled as control. Labeling efficiency, marker retention, and long-term detectability in MRI until p7 were evaluated. Further, proliferation capacity and trilineage differentiation as indicators for potential impact on stromal cell characteristics were assessed. MSCs of both species were successfully labeled with all three SPIO products. A high, exclusively intracellular, iron uptake was achieved by Molday ION Rhodamine B only. Labeling with Resovist led to prominent extracellular iron presence; labeling with Endorem was less efficient. During MRI, all labeled cells showed strong hypointense signals, contrary to unlabeled controls. Resovist induced the largest areas of hypointense signals, followed by Molday ION Rhodamine B and Endorem. MRI signal detectability decreased from p4 to p7. Proliferation, adipogenic, and osteogenic differentiation potential were not reduced by cell labeling compared to unlabeled cells. Chondrogenic differentiation capacity decreased with increasing amount of iron associated with the cells. Among the three products, Resovist and Molday were identified as promising labeling agents. While Resovist achieved superior results in most of the assessed parameters, Molday ION Rhodamine B ensured intracellular iron uptake without extracellular SPIO complexes and consistent hypointense signals on MRI.
Keywords
Introduction
Owing to their regenerative and immunoregulatory potential, multipotent stromal cells (MSCs) provide promising opportunities for the therapy of various diseases such as cartilage-related disorders (7). To translate the experimental application into clinical reality, labeling and tracking of MSCs is crucial to evaluate their distribution and homing (31). Magnetic resonance imaging (MRI) has emerged as one of the leading modalities for tracking of transplanted cells (1). To visualize specific cells using MRI, they must be labeled with a magnetic contrast agent. The ideal labeling agent remains exclusively intracellular, does not disturb cell function or behavior, and allows for long-term detection. Superparamagnetic iron oxide (SPIO) may provide most or all of these characteristics, as iron particles within SPIO-labeled cells induce signal-void areas in T2 and T2*-weighted MR images (17,19,23,30).
The aim of the study was to compare three commercially available and, at that time, relevant SPIO products for labeling of ovine and equine bone marrow (BM)-derived MSCs: a) Molday ION Rhodamine B (further referred to as “Molday”; Biopal, Worcester, MA, USA), b) Endorem (Guerbet, Sulzbach, Germany), and c) Resovist (Schering, Berlin, Germany). Endorem (agent name: AMI-25), a standard SPIO (SSPIO), is coated with dextran; the iron oxide crystal is 4.3-5.6 nm, and the hydrodynamic diameter is 80-150 nm (32,34). The agent is approved for clinical application and distributed in Europe and Brazil. It was further distributed in the US as Feridex I.V. by Bayer HealthCare Pharmaceuticals, Inc., but went off market in 2008 (26). Resovist (agent name: SHU 555A), also classified as a SSPIO, is coated with carbodextran; the crystal is 4 nm, and the hydrodynamic diameter is 62 nm (34). It was EU approved in 2001 but was withdrawn from the market in 2009. Molday is still a relatively new ultrasmall SPIO (USPIO) product; the iron magnetic core is 8 nm, and the hydrodynamic diameter is 35 nm (1,26). It is formulated for cell-labeling applications and not approved for clinical applications. The products were compared with regard to their labeling efficiency, longitudinal detectability via MRI, and effects on cell biology.
The treatment of equine orthopedic conditions with MSC-based regenerative therapies gains more attention, while sheep represent a widely accepted animal model in the field of orthopedic research. These aspects are the rationale to test labeling on both equine and ovine MSCs in the present study. Results will help to determine the most appropriate SPIO product for in vivo cell tracking of intra-articularly applied MSCs in a large animal model of osteoarthritis.
Materials and Methods
Bone Marrow Aspiration, MSC Isolation, and Culture
All procedures were approved by the University of Leipzig animal care committee. One female and four male, healthy Warmblood horses (one horse with 1.5 years of age, four horses with 13—15 years of age; University of Leipzig, Germany) as well as four female and one male sheep (3—4 years of age; University of Leipzig, Germany) were included in this study. Equine BM aspirates were obtained from the sternum as described previously (9), while ovine BM aspirates were obtained from the ileac crest. All BM samples were kept at room temperature and were processed within 6 h after sampling.
The MSCs were isolated by density gradient centrifugation as described previously, with slight modifications of the protocol (11). Briefly, BM was diluted 1:1 with phosphate-buffered saline (PBS; PAA, Pasching, Austria), and 22.5 ml BM aspirate was placed on 15 ml Ficoll PREMIUM (GE Healthcare, Uppsala, Sweden) and centrifuged (1000 × g, 20 min, 20°C). Mononuclear cells were collected and washed twice with PBS. Cells were plated at a density of 1.5 × 105 cells/cm2 into T175 tissue culture flasks (Greiner Bio-One GmbH, Frickenhausen, Germany) in Dulbecco's modified Eagle's medium (DMEM; PAA) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich, Steinheim, Germany), 1% penicillin/streptomycin (PAA), and 8.9 μg/ml ascorbic acid (Sigma-Aldrich). Cultures were maintained at 37°C in humidified atmosphere at 5% CO2. After 24 h, cells were washed with PBS, and culture medium was added again. Subsequently, culture medium was changed twice weekly. MSCs were trypsinized [trypsin-ethylenediaminetetraacetic acid (EDTA); 0.05%/0.02% in PBS; PAA] at subconfluency and cultured to the third passage (p3).
Cell Labeling
MSCs of p3 were washed with PBS and labeled according to the manufacturer's instructions or previously published studies (3,4,19) and were refined in our own pilot studies. Molday was added to culture medium to a final iron concentration of 100 μg/ml (50 μl stock solution; 1 ml culture medium), and MSCs were labeled for 10 h under standard culture conditions. Endorem was added to culture medium to a final iron concentration of 25.2 μg/ml and supplemented with 6 μg/ml of the transfecting agent protamin sulfate (2.25 μl Endorem stock solution; 0.6 μl protamin sulfate, 1 ml culture medium; Sigma-Aldrich). The solution was incubated for 1 h on a rotating shaker and subsequently placed onto the cells for 12 h. Resovist was added to culture medium to a final iron concentration of 933 μg/ml (30 μl stock solution; 1 ml culture medium) for 24 h. After labeling, all culture media was removed, and cells were washed three times with PBS. Owing to different labeling times (10, 12, and 24 h) labeling was timed to finish at a common end point (referred to as “day 0”).
For further steps, labeled cells and unlabeled controls (p3) were subdivided and conveyed to the following parts of the study. For cell expansion, all samples were routinely passaged after 7 days until p7 without reaching confluency.
Labeling Efficiency and Long-Term SPIO Retention
On day 0, labeled and unlabeled cells (p3) were seeded as p4 into 24-well plates (Greiner Bio-One GmbH) at a density of 5,000 cells/cm2 and allowed to adhere under standard culture conditions. After 24 h (referred to as “day 1 after labeling”) cells were washed with PBS, fixed with 4% paraformaldehyde (PFA; Roth, Karlsruhe, Germany) for 1 h, and stained with Prussian blue stain (Biopal, Worcester, MA, USA) to dye iron particles blue. Efficiency was evaluated at 20× magnification (Nikon Eclipse Ti-S; Nikon Instruments, Melville, NY, USA) in three randomly selected fields of view per sample. The applied semiquantitative scoring system accounted for stain localization (extra- vs. intracellular), average intracellular stain content (averaged stained area per cell), and percentage of stained cells among all cells per field of view. For each of the three parameters, the examiners awarded a grade from 0 to 4 (Table 1). The three grades were summarized to a labeling efficiency score ranging from 0 (lowest labeling efficiency) to 12 (highest labeling efficiency).
Scoring Systems to Evaluate Suitability of Superparamagnetic Iron Oxide (SPIO) Products for Intra-articular Use in a Long-term In Vivo Experiment

MRI, magnetic resonance imaging; cPDs, cumulative population doublings.
To evaluate long-term SPIO retention within the cells, the above-mentioned steps were repeated at weekly intervals from p4 until p7. All samples were evaluated at equal time points: p4 = 1 day after labeling, p5 = 8 days after labeling, p6 = 15 days after labeling, p7 = 22 days after labeling.
In Vitro MRI Detectability
MRI detectability of labeled cells was evaluated at each passage from p3 to p6 using a custom-made agarose gel phantom: 50 ml of molten 2% agarose gel (Sigma-Aldrich) was poured into a Petri dish (10 cm diameter; Greiner Bio-One GmbH), while 20 microcentrifuge tubes (Eppendorf, Hamburg, Germany) served as space holders until the gel solidified. Then, 1 × 106 cells were suspended in 2 ml 1.5% agarose, transferred into the agarose wells, and covered with a final layer of 2% agarose. Each phantom was loaded with cells from all four experimental groups (in four columns) of all five individuals per species (in five rows). Phantoms were maintained at 8°C for a maximum of 24 h until MRI evaluation.
MR images were obtained with a 0.5T MRI system (Gyroscan NT Compact Plus; Philips HealthCare, Hamburg, Germany). A 3DT2*FFE sequence [echo time (TE) 41 ms, repetition time (TR) 27 ms, flip angle 25°, 1 mm slice width, no gap] and a C3 surface coil was used to obtain images in all three orientations.
The hypointense signal area on MR images produced by the SPIO particles (Fig. 1) was measured digitally: a grid was laid over a representative transverse MR image of the phantom, separating the individual sample wells. Subsequently, a gray threshold value was defined to differentiate the hypointense signal from the surrounding. Within each individual square, the area with a hypointense signal below the gray threshold was subsequently quantified in mm2. Again, all samples were evaluated on a weekly routine at equal time points: p3 = 0 days after labeling, p4 = 7 days after labeling, p5 = 14 days after labeling, and p6 = 21 days after labeling.

MR images of gel phantoms containing unlabeled and superparamagnetic iron oxide (SPIO)-labeled ovine (a—d) and equine (e—h) multipotent stromal cells (MSCs) (n = 5, in rows) at different time points. (a, e) 0 days after labeling (p3); (b, f) 7 days after labeling (p4); (c, g) 14 days after labeling (p5); and (d, h) 21 days after labeling (p6). Left columns present unlabeled control cells (U); remaining columns present MSCs labeled with Molday (M), Endorem (E), or Resovist (R). Philipps 0.5 Tesla, GRE 3DT2*.
Proliferation Capacity
On day 0, labeled and unlabeled cells (p3) were plated as p4 at a density of 500 cells/cm2 into T25 tissue culture flasks (Greiner Bio-One GmbH) in culture medium. After 7 days in culture, the total number of MSCs per sample was counted in a hemocytometer (Neubauer-improved; Marienfeld-Superior, Lauda-Koenigshofen, Germany). The process was repeated at weekly intervals, from p4 (evaluated 7 days after labeling) to p7 (evaluated 28 days after labeling). The population doublings (PDs) were calculated with the equation PDs = log10 (N/N0) × 3.33, where N is the number of harvested cells and N0 the number of plated cells (12). Cumulative population doublings (cPDs) for each time point were calculated by adding values of all previous PDs.
Trilineage Differentiation Capacity
For adipogenesis, cells were seeded as p4 in duplicates at a density of 5,000 cells/cm2 in 12-well plates (Greiner Bio-One GmbH). At confluence, differentiation was induced according to standard protocol (18,27), comprising four cycles of 72-h cultivation in an induction medium consisting of DMEM/Ham's F12, 10% FBS, 1% penicillin/streptomycin, 1 μM dexamethasone (Sigma-Aldrich), 100 μM indomethacin (Sigma-Aldrich), 500 μM 3-IBMX (3-isobutyl-1-methylxanthine; Sigma-Aldrich), and 1,700 nM insulin (Invitrogen, Karlsruhe, Germany) followed by 24-h incubation in maintenance medium consisting of DMEM/Ham's F12 (PAA), 10% FBS, 1% penicillin/streptomycin, and 1,700 nM insulin. After the fourth cycle, cells were cultured in maintenance medium. After 21 days of adipogenic induction, the production of intracellular lipid droplets was assessed qualitatively. For that, samples were fixed in 4% PFA for 60 min, washed twice with distilled water, and incubated with 60% isopropanol (Roth) for 5 min at room temperature. Cells were stained in 0.35% Oil Red O (0.35 g Oil Red O; Sigma-Aldrich; 100 ml 60% isopropanol) for 15 min at room temperature under light protection and then washed four times with distilled water. To counterstain cells, they were incubated for 5 min with Mayer hematoxylin (Sigma-Aldrich), washed with tap water, and air dried. Staining produces red lipid droplets, light orange cytoplasm, and blue nuclei. To assess adipogenic differentiation, samples were evaluated qualitatively under light microscope at 20× magnification for the presence of lipid droplets.
For osteogenesis, cells were seeded in duplicates at a density of 5,000 cells/cm2 in 12-well plates. At confluence, differentiation was induced in DMEM/Ham's F12, 10% FBS, 1% penicillin/streptomycin supplemented with 10 nM dexamethasone, 10 mM ß-glycerophosphate (Sigma-Aldrich), and 0.1 mM ascorbic acid. Medium was changed twice weekly. Osteogenesis, characterized by extracellular calcium deposition, was qualitatively assed after 21 days via Alizarin red staining. For that, cells were fixed in 4% PFA for 60 min at room temperature, washed twice with distilled water, stained in 2% Alizarin red (100 mg Alizarin red; Sigma-Aldrich; 5 ml distilled water) for 5 min at room temperature protected from light. Finally, cells were carefully washed twice with distilled water and air dried. To assess osteogenic differentiation, samples were evaluated qualitatively under light microscope (Leica DMIL; Leica Microsystems, Wetzlar, Germany) at 20× magnification for the presence of extracellular calcium depositions that appeared intensively red.
Chondrogenesis was induced in micromass pellet cultures as previously described (11). Briefly, pellets were prepared in duplicates of 5 × 105 cells, placed in 15-ml tubes, and centrifuged (280 × g, 5 min, 4°C). Pellets were then incubated in chondrogenic medium consisting of high-glucose (4.5 g/L) DMEM (PAA) supplemented with 1% ITS+ Premix (insulin, transferrin, selenous acid, bovine serum albumin, and linoleic acid; BD Biosciences, Erembodegem, Belgium), 0.1 mM ascorbic acid, 0.4 mM proline (Roth), 100 nM dexamethasone, and 10 ng/ml human recombinant transforming growth factor (TGF)-β1 (BD Biosciences). Medium was replaced twice weekly. After 21 days in chondrogenic culture, pellets were fixed for 24 h in 4% PFA, dehydrated, embedded in paraffin, and cut into 4-μm-thick sections. Histologic sections were stained with Safranin O-fast green (Sigma-Aldrich), as described by Grogan and coworkers (14), and Alcian blue (Sigma-Aldrich), to detect extracellular glycosaminoglycan depositions. For Alcian blue stain, deparaffinized and hydrated slides were rinsed in 3% acetic acid (3 ml glacial acetic acid; Sigma-Aldrich; 97 ml distilled water) for 3 min and immersed in Alcian blue solution (0.1 g Alcian Blue 8GX; Sigma-Aldrich; 100 ml 3% acetic acid) for 1.5 h followed by washing in tap water. Slides were further stained in Nuclear Fast Red Solution (Sigma-Aldrich) for 10 min, washed in distilled water, and dried between filter paper. Subsequently, slides were rinsed in absolute alcohol for 3 min, washed in xylene (Roth) for 10 min, and air dried. A cover slip with Roti-Histokitt (Roth) was placed over the stained tissue. Masson's trichrome staining was performed to evaluate collagen synthesis. For that, deparaffinized and hydrated slides were fixed in Bouin's fluid (Sigma-Aldrich) overnight and afterwards washed in running tap water for 10 min. Slides were stained in Weigert's iron hematoxylin (Sigma-Aldrich) for 10 min and washed in tap water and 1% acetic acid (1 ml glacial acetic acid; Sigma-Aldrich; 99 ml distilled water) for each 10 min. For staining with Ponceau-fuchsin (stock solution: 0.75 g xylidine ponceau, 0.25 g acid fuchsin; Sigma-Aldrich; 100 ml 1% acetic acid; working solution: 10 ml stock solution; 90 ml 1% acetic acid), slices were immersed in ponceau—fuchsin working solution for 30 min, rinsed in 1% acetic acid, placed in 2% tungstophoric acid (2 g tungstophoric acid hydrate; Sigma-Aldrich; 100 ml distilled water) for 2 min and rinsed again in 1% acetic acid. Slides were finally stained in 0.2% fast green (0.2 g fast green FCF; Sigma-Aldrich; 100 ml 1% acetic acid) for 5 min and again rinsed in 1% acetic acid. Subsequently, slides were rinsed in absolute alcohol for 3 min, washed in xylene for 10 min, and air dried. A cover slip with Roti-Histokitt was placed over the stained tissue.
A visual scoring system (Bern Score) based on Safranin O-fast green staining was used to assess chondrogenesis semiquantitatively as described by Grogan and others (11,14).
Histologic Scoring and Statistical Analysis
All histological samples were scored independently by two examiners (H.J., C.V.) who were blinded toward the experimental treatment of the examined samples. Where results from the two investigators differed, the mean of the two values was used as the final result.
Statistical analysis was performed with IBM® SPSS® Statistics (version 20; Armonk, New York, NY, USA). Data was tested for normal distribution using the Shapiro—Wilk test, and significance was set at p < 0.05. To compare the parameters of the four labeling conditions, the nonparametric Kruskal—Wallis and Mann—Whitney U-tests were applied. Bonferroni correction was used to adjust the p value. Thus, significance was set at p < 0.0083. Data were expressed as median ± median absolute deviation.
Results
Labeling Efficiency and Long-Term SPIO Retention
Prussian blue staining revealed varying amounts of iron particles in the cytoplasm of ovine and equine MSCs on a histological level (Fig. 2a-1). Evaluation of labeling efficiency and SPIO retention showed significant differences between labeled and unlabeled cells and between SPIO products (Table 2, Fig. 3). In both species, Molday-labeled cells received the highest score for marker retention 1 day after labeling, followed by Endorem and Resovist. Importantly, labeling with Resovist led to marked extracellular accumulations (Fig. 2c, f). For ovine cells 8 days after labeling, Resovist-labeled cells showed the highest scores, followed by Molday labeling, while Endorem-labeled cells showed the lowest blue iron stain. Interestingly, the score for Resovist-labeled cells was higher at day 8 compared to day 1. This improved score is due to less extracellular iron attached to the cells resulting in higher scores in the responsible variable “localization.” Fifteen days after labeling of ovine cells, only the Resovist-labeled samples consistently showed strong blue iron stain (Fig. 2g-i), and 22 days after labeling, none of the SPIO products exhibited retention in ovine MSCs. In equine samples, SPIO retention was very similar in all three SPIO products on days 1 and 8 after labeling. However, 15 days after labeling, only Resovist-labeled cells followed by Molday-labeled cells revealed detectable intracellular iron (Fig. 2j—l). Twenty-two days after labeling, only equine MSCs labeled with Resovist exhibited intracellular iron.

Ovine (a—c, g—i) and equine (d—f, j—l) MSCs labeled with three different SPIO products. Images show cells 1 day after labeling with high SPIO content (a—f) and 15 days after labeling when SPIO content decreased (g—l). Only Resovist-labeled cells show remnants of intracellular SPIO particles. Cells were stained with Prussian blue to visualize iron particles (arrows indicate individual cells containing intracellular iron particles stained in blue). Scale bar: 100 μm. MSCs, multipotent stromal cells; SPIO, superparamagnetic iron oxide.

Labeling efficiency of SPIO products and unlabeled control in ovine (a) and equine (b) MSCs. The semiquantitative scoring system was based on Prussian blue-stained samples 1, 8, 15, and 22 days after labeling. Elements show Tukey box plots of the determined values and correspond to numbers shown in Table 2. *p < 0.0083. MSC, multipotent stromal cells; SPIO, superparamagnetic iron oxide.
Results of Quantitative and Semiquantitative Evaluation of SPIO-Labeled and Unlabeled Ovine and Equine Multipotent Stromal Cells (MSCs)
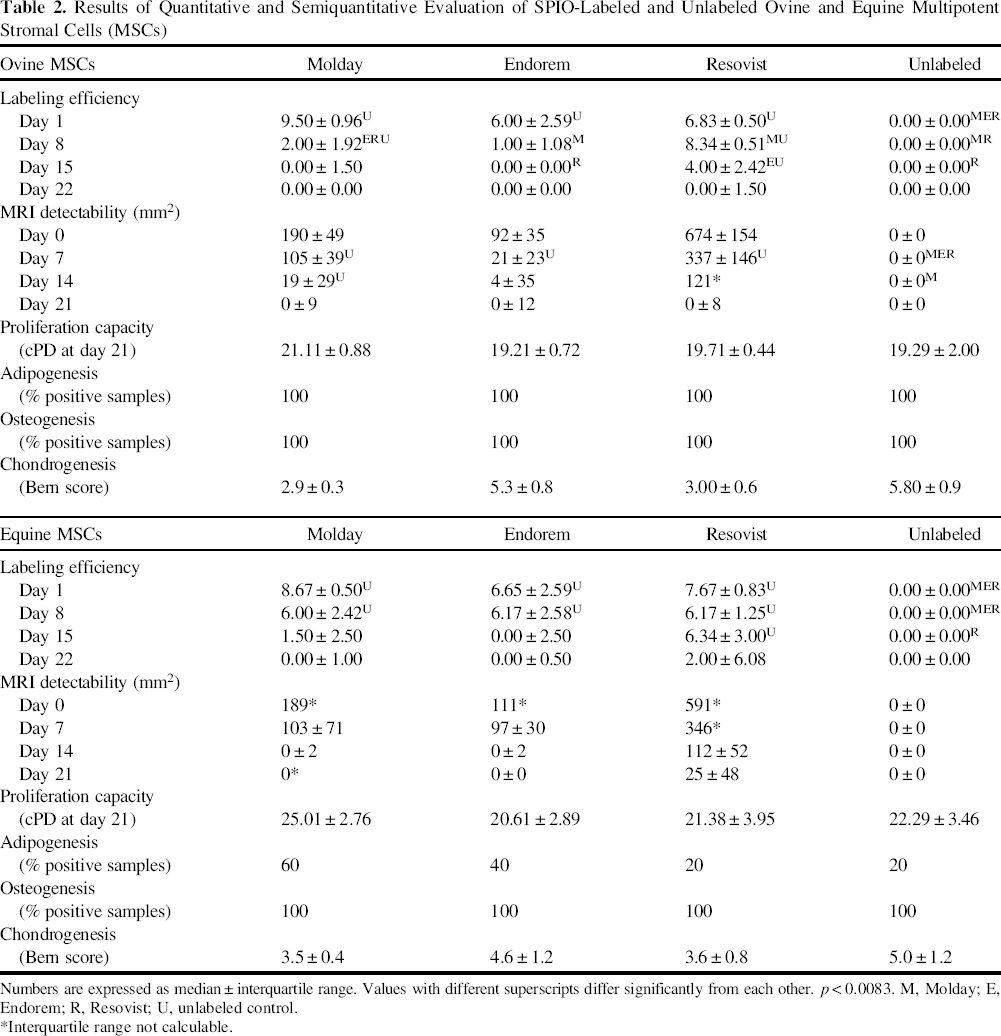
Numbers are expressed as median ± interquartile range. Values with different superscripts differ significantly from each other. p < 0.0083. M, Molday; E, Endorem; R, Resovist; U, unlabeled control.
Interquartile range not calculable.
In Vitro MRI Detectability
MR images of samples 0 days after labeling showed well-defined areas of hypointense signal for SPIO-labeled cells in contrast to unlabeled controls. The signal-void area related to the labeling condition and changed time dependently (Table 2, Figs. 1 and 4). In both species, labeling with Resovist produced distinctly larger signal areas 0 days after labeling, followed by Molday- and Endorem-labeled cells. This ranking of the products remained constant for both species during the observation period. Interestingly, while 14 days after labeling all three SPIO products caused MRI signals in the ovine samples, only Resovist-labeled MSCs were detectable via MRI in the equine samples. After 21 days, only Resovist-labeled equine MSCs produced a hypointense signal area.

MRI detectability due to SPIO-labeled and unlabeled ovine and equine MSCs over time. MRI detectability measured in size of hypointense signal area (in mm2) due to SPIO-labeled and unlabeled ovine (a) and equine (b) MSCs in a gel phantom 0, 7, 14, and 21 days after cell labeling *p < 0.0083. Elements show Tukey box plots of the determined values. MRI, magnetic resonance imaging; SPIO, superparamagnetic iron oxide; MSCs, multipotent stromal cells.
Proliferation Capacity
In both species, the progression of the four curves showing the cell proliferation was very similar. Cumulative population doublings did not significantly differentiate between labeled and unlabeled cells or between the SPIO products (Table 2).
Trilineage Differentiation Capacity
Adipogenesis was observed in all labeled and unlabeled ovine cells without any differences between SPIO products (Table 2, Fig. 5). In contrast, adipogenesis was not observed in all equine MSC samples, but there were no significant differences between the labeled and unlabeled cells.

Adipogenic differentiation capacities of SPIO-labeled and unlabeled ovine (a—d) and equine (e—h) MSCs stained with Oil red O. Note the red-stained intracellular lipid droplets. Scale bar: 100 μm. SPIO, superparamagnetic iron oxide; MSCs, multipotent stromal cells.
Osteogenesis was observed in all ovine and equine samples without any significant differences between labeled and unlabeled cells or between SPIO products (Table 2, Fig. 6).
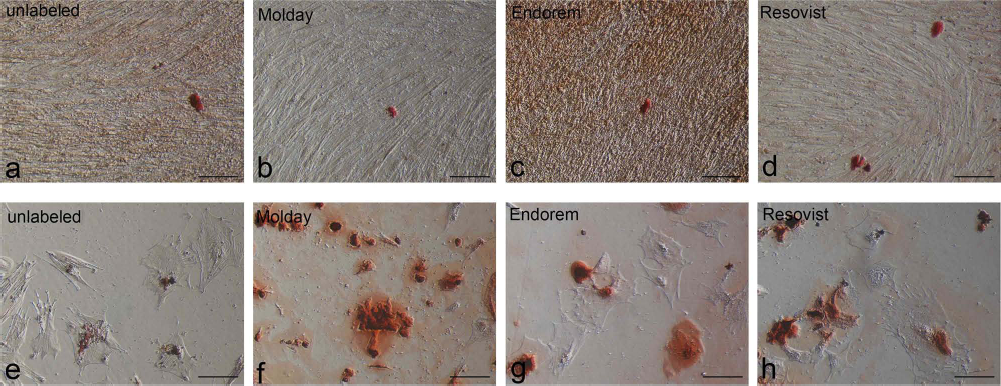
Osteogenic differentiation capacities of SPIO-labeled and unlabeled ovine (a—d) and equine (e—h) MSCs stained with Alizarin red. Note the red-stained extracellular calcium depositions. Scale bar: 100 μm. SPIO, superparamagnetic iron oxide; MSCs, multipotent stromal cells.
Micromass pellet cultures for evaluation of chondrogenic capacities showed a remarkable, grossly detectable discoloration due to iron in SPIO-labeled samples (Fig. 7a). Histologically, production of extracellular glycosaminoglycan was detected in labeled and unlabeled pellets (Fig. 7b—i). SPIO particles were visible within labeled pellets, most obviously in Resovist-labeled samples (Fig. 7e, i). The Bern score revealed differences in chondrogenic capacities between labeled and unlabeled cells (Table 2). Among ovine MSCs, unlabeled cells were most capable of producing neocartilagineous tissue, while chondrogenesis was slightly reduced when cells were labeled with Endorem and markedly, albeit not significantly, reduced in MSCs labeled with Resovist and Molday (Fig. 8A). Equine MSCs showed similar patterns: Unlabeled cells achieved the highest score followed by Endorem-, Resovist-, and Molday-labeled cells (Fig. 8B).

Chrondrogenic differentiation of labeled and unlabeled ovine and equine MSCs. (a) Pellets of chondrogenically differentiated ovine and equine MSCs (U: unlabeled control, M: Molday, E: Endorem, R: Resovist; scale bar: 5 mm). Note the brown discoloration of pellets due to iron within the pellets. (b—e) Chondrogenically differentiated ovine MSCs stained with Safranin O-fast green (b) unlabeled, or labeled with (c) Molday, (d) Endorem, and (e) Resovist. (f—i) Chondrogenically differentiated equine MSCs stained with Safranin O-fast green (f) unlabeled, or labeled with (g) Molday, (h) Endorem, and (i) Resovist. Note the decrease in proteoglycan deposition (orange) upon SPIO labeling as well as large amounts of iron particles (brown) within the pellet after Resovist labeling (e, i). Scale bar: 200 μm. MSCs, multipotent stromal cells; SPIO, superparamagnetic iron oxide.
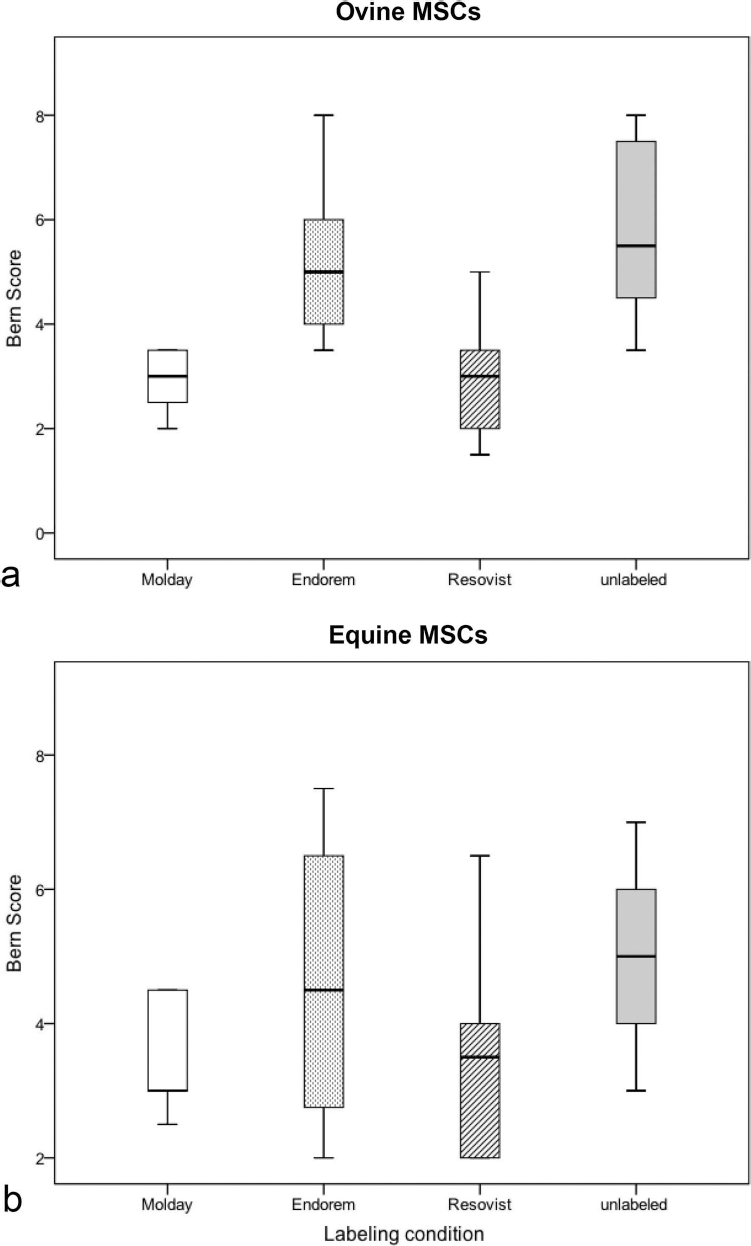
Chondrogenic differentiation capacities of SPIO-labeled and unlabeled ovine (a) and equine (b) MSCs evaluated via Bern score. *p < 0.0083. Elements show Tukey box plots of the determined values. SPIO, superparamagnetic iron oxide; MSCs, multipotent stromal cells.
Using the Alcian blue and Masson's trichrome stain as a measure of immature proteoglycan and collagen deposition, respectively, all samples showed positive staining.
Discussion
In the present study, we used three commercially available SPIOs to label ovine and equine MSCs and compared these with regard to labeling efficiency, longitudinal detectability in labeled cells, and biologic effects on labeled MSCs.
Based on Prussian blue stain, all three SPIO products labeled ovine and equine MSCs successfully. However, there was a distinct difference in labeling efficiency between the three products. The labeling protocols for the SPIO products had been refined in pilot studies. Serial dilutions (Molday: 1, 10, 25, 50, 100 μg Fe/ml; Resovist: 0.9, 9, 93, 933 μg Fe/ml) identified optimal iron concentrations in culture medium. Increase in incubation times led to more intense SPIO uptake (Endorem: 4, 12 h). This led to different final iron concentrations in the incubation media (Endorem 25.2 μg Fe/ml, Molday 100 μg Fe/ml, Resovist 933 μg Fe/ml) and incubation times (Endorem 12 h, Molday 10 h, Resovist 24 h). Therefore, differences in SPIO uptake may be caused by varying iron concentrations and incubation times, rather than by the contrast agents itself. Age and gender of the animals used might have another possible influence on cell-staining characteristics. Whereas all sheep were young adult, the horses' ages varied considerably between immature (one animal) and middle aged (four animals). This might impede comparison between sheep and horses. Accordingly, gender of the animals may have also had an impact on cell behavior.
Our study further revealed large variations regarding SPIO particle localization. While Molday and Endorem showed a selective intracellular presence of SPIO, labeling with Resovist led to marked extracellular accumulations. Those may have caused the large signal void areas during MRI detection in Resovist samples. Repeated washing steps after labeling and addition of 10 IU/ml heparin to PBS as suggested by Arbab et al. for Endorem (2) did not dissolve extracellular accumulations in our study. A suggested interaction of iron and proteins within the culture medium (containing FBS) is considered as a negligible factor since the medium conditions remained constant between the three labeling products. Stromal cells are considered nonphagocytic; however, intracellular SPIO uptake into MSCs is thought to occur via endocytosis (2,16), as observed in macrophages (28). To increase SPIO internalization, the use of transfecting agents such as the cationic compound protamine sulfate has been described (3). In our study, protamine sulfate was part of the labeling protocol for Endorem only. An increased uptake of Resovist by adding protamine sulfate has been described recently (13,15) and may have improved intracellular labeling and decreased extracellular accumulation of Resovist in our study.
Long-term detectability of labeled cells is highly desirable after their in vivo application. Our in vitro study demonstrated positive Prussian blue stain and MRI detection of Resovist in equine MSCs for up to 21 days, which would not allow for tracking of labeled cells in vivo over longer time periods. This raises the important question of the marker's fate. Decrease in intracellular SPIO concentration may occur due to intracellular biodegradation, cell death, exocytosis, or cell division (2,5,8,24,33). A recent in vivo study showed that SPIO-labeled Kupffer cells lost their staining after 8 to 16 days while iron was recycled into the blood pool (35). However, SPIO-labeled MSCs were detectable for up to 12 weeks after intra-articular application (19). Our MSCs were examined in vitro, where rapid cell proliferation took place. It is possible that higher intracellular SPIO levels are sustained when cells are administered in vivo, but further studies are necessary to confirm this assumption.
In addition to successful SPIO uptake and long-term detectability via MRI, unaffected cell biology is a mandatory prerequisite for MSC-tracking studies. Therefore, we assessed proliferation capacities and trilineage differentiation. Our study did indeed not reveal any adverse effects of SPIO labeling on proliferation rate, adipogenic, and osteogenic differentiation of equine or ovine MSCs. This is consistent with reports of human MSCs labeled with Resovist (15,21) or Endorem (3,6,22,25), and Molday-labeled monkey or mouse MSCs (1,26). Other studies found inhibition of osteogenic differentiation of Resovist-labeled human MSCs in a dose-dependent manner (8). Further, Ren and coworkers showed decreased proliferation capacity of Molday-labeled monkey MSCs via bromodeoxyuridine incorporation assay (29). Direct comparison of the varying results of the studies might be difficult due to different labeling conditions and read out parameters. In our study, the adipogenic differentiation was successfully induced in all ovine MSCs. However, this protocol was not consistently effective in equine samples irrespective of labeling status. We therefore accept the poor adipogenic potential of labeled and unlabeled equine cells as a random effect.
We observed markedly, albeit not significantly, reduced chondrogenic differentiation potential of Molday-and Resovist-labeled ovine MSCs based on histologic evaluations. Even though several authors observed undisturbed chondrogenesis with SPIO labeling (3,10,19,29), our data support previously published studies on human MSCs labeled with Endorem (22,25) and Resovist (15) that describe inhibited chondrogenic potential. Interestingly, decreased chondrogenesis has been described to occur in a dose-dependent manner (6,15,22), which is further confirmed by our study; samples of both species show an inverse relationship with iron presence (assessed by the parameter “labeling efficiency”) and the rate of chondrogenic differentiation. Although it has not been fully explored how chondrogenesis is being inhibited by SPIO, different mechanisms are currently discussed. Kostura et al. suspect iron interferes with intracellular signaling processes (22). However, Henning et al. refer to the fact that chondrogenic differentiation highly depends on surface-linked cellular interactions and suggest that extracellular, surface-bound iron may interfere with mechanisms or structures essential for these interactions (15). Further studies investigating the exact mechanism are warranted, especially for in vivo studies with SPIO-labeled cells focusing on cartilage repair.
Recently, Khurana and coworkers examined the new FDA-approved USPIO Ferumoxytol for cell labeling of adipose-derived stem cells (20). According to the labeling protocol, Ferumoxytol is used with a final iron concentration of 500 μg Fe/ml supplemented with 10 μg/ml protamine sulfate. Labeling leads to significant cellular iron uptake, and cells can be detected in MRI in vitro and in vivo. An inclusion of Ferumoxytol into our study would have led to more comprehensive information; unfortunately, experiments were started before the product came onto market.
The aim of the study was to compare the three labeling products Molday, Endorem, and Resovist to determine the most appropriate candidate for in vivo cell tracking of intra-articularly applied MSCs in a large animal model of osteoarthritis. In the majority of assessed parameters, labeling with Resovist led to superior values: labeling efficiency, long-term SPIO retention, and longitudinal detectability via MRI were superior, while cell proliferation, as well as adipogenic and osteogenic differentiation, were undisturbed. However, high amounts of SPIO particles were shown to decrease the chondrogenic capacities of MSCs. Thus, Resovist appears to be a recommendable candidate for in vivo applications. Nevertheless, high amounts of extracellular iron accumulations in Resovist-labeled samples may lead to questionable results when used in preclinical in vivo studies, and further studies are needed to improve the labeling protocol. Molday-labeled MSCs could be an acceptable alternative due to selective intracellular iron uptake and convincing results in labeling efficiency, long-term SPIO retention, longitudinal detectability via MRI, cell proliferation as well as adipogenic and osteogenic differentiation.
Footnotes
Acknowledgments
The authors would like to thank Dr. Habil. Johannes Seeger and Gabriele Lindner for their support during histological processing, Dipl.-Inform. Patrick Scheibe for his assistance during the digital quantification of MR images, and Dipl.-Inform. Karsten Winter for his support during statistical analysis. We also would like to thank Dr. Katja Duesterdieck-Zellmer, Ph.D., for critical reading of the manuscript. The work presented in this article was made possible by funding from the German Federal Ministry of Education and Research (BMBF, PtJ-Bio, 0313909/0315883). Results of this project have been published previously as a poster at the Ninth World Congress of the International Cartilage Repair Society (ICRS), Barcelona, September 2010, and it was part of the requirements to obtain a Dr. med. vet. degree by Christin Veit (2011) through the University of Leipzig. The authors declare no conflict of interest.