
Abstract
The treatment of laryngotracheal stenosis remains a challenge as treatment often requires multistaged procedures, and successful decannulation sometimes fails after a series of operations. Induced pluripotent stem cells (iPSCs) were generated in 2006. These cells are capable of unlimited symmetrical self-renewal, thus providing an unlimited cell source for tissue-engineering applications. We have previously reported tracheal wall regeneration using a three-dimensional (3D) scaffold containing iPSCs. However, the efficiency of differentiation into cartilage was low. In addition, it could not be proven that the cartilage tissues were in fact derived from the implanted iPSCs. The purpose of this study was to evaluate and improve the use of iPSCs for the regeneration of tracheal cartilage. iPSCs were cultured in vitro in a 3D scaffold in chondrocyte differentiation medium. After cultivation, differentiation into chondrocytes was examined. The ratio of undifferentiated cells was analyzed by flow cytometry. The 3D scaffolds were implanted into tracheal defects, as an injury site, in 24 nude rats. Differentiation into chondrocytes in vitro was confirmed histologically, phenotypically, and genetically. Flow cytometric analysis demonstrated that the population of undifferentiated cells was decreased. Cartilage tissue was observed in the regenerated tracheal wall in 6 of 11 rats implanted with induced iPSCs, but in none of 13 rats implanted with the control and noninduced iPSCs. The expression of cartilage-specific protein was also demonstrated in vivo in 3D scaffolds containing iPSCs. The presence of the GFP gene derived from iPSCs was confirmed in samples of cartilage tissue by the combination of laser microdissection (LMD) and polymerase chain reaction (PCR) techniques. Our study demonstrated that iPSCs have the potential to differentiate into chondrogenic cells in vitro. Cartilage tissue was regenerated in vivo. Our results suggest that iPSCs could be a new cell source for the regeneration of tracheal cartilage.
Keywords
Introduction
Reconstruction of the upper airway after the resection of malignancies or stenotic inflammatory lesions is thought to be difficult. In conventional treatments, several types of flaps and grafts, made from cartilage, muscle, skin, etc., have been used in the repair of defects after the resection of tracheal lesions (6, 8). However, these treatments cause postoperative scarring, and granulation is sometimes observed, leading to airway stenosis. Our group has developed and clinically applied an artificial trachea made from a collagen sponge with a polypropylene scaffold frame for the regeneration of tracheal tissue (18, 25, 29). This technique was used in four patients (30), and all cases have shown good progress without restenosis. However, the premade artificial trachea cannot be utilized for pediatric airways because the tracheal frame needs to be expanded as the child develops. Various studies have focused on the differentiation of chondrogenic cells in vitro, using mesenchymal cell lines and embryonic stem (ES) cell lines as potential cell sources (2, 16, 21, 22, 33, 43). However, mesenchymal stem cell lines have limited ability to proliferate and differentiate (26), and ethical issues exist in relation to the clinical application ES cell lines.
In 2006, induced pluripotent stem cells (iPSCs) were generated from mouse skin fibroblasts by the introduction of four transcription factors (38). These cells are capable of unlimited symmetrical self-renewal, thus providing an unlimited cell source for tissue-engineering applications. In addition, the use of iPSCs obtained from patient-derived somatic cells can prevent transplant rejection.
We have previously reported tracheal wall regeneration using a three-dimensional (3D) scaffold containing iPSCs (15) to resolve difficulties associated with conventional treatments. Our previous experiments confirmed that iPSCs have the potential to differentiate into chondrocytes, as shown by the appearance of Alcian blue-stained colonies and the in vitro expression of a cartilage-specific gene of type II collagen. Cartilage-like tissue was observed in vivo in the 3D scaffold containing differentiated iPSCs (15). However, their differentiation into chondrocytes could not be confirmed, and phenotypic analysis is essential. In addition, we could not prove that the cartilage-like tissues observed in the scaffolds were in fact derived from the implanted cells, and no phenotypic analysis was performed. To resolve these problems, in our current study, we developed a technique to improve differentiation in vitro and examined the time course of chondrocyte differentiation medium (CDM)-induced morphological changes in iPSCs.
In addition, to determine whether the cartilage-like tissues observed in vivo were in fact derived from the implanted cells, the presence of the green fluorescent protein (GFP) gene derived from iPSCs was examined in microdissected tissue sections. The ratio of differentiated/ undifferentiated cells, which we considered to be associated with the problem of tumor formation, was also analyzed using flow cytometric analysis for future effective tracheal regeneration.
The purpose of this study was to assess the effectiveness of iPSCs for the regeneration of the tracheal cartilage through the evaluation of both in vitro and in vivo experiments.
Materials and Methods
Cell Culture
iPSCs were cultured as described previously (15). Briefly, mouse iPSCs from the iPS-MEF-Ng-20D-17 line (20D-17 was purchased from Riken BioResource Center, Tsukuba, Japan) were routinely cultured on a feeder layer of mitomycin C-inactivated mouse embryonic fibroblasts, SNL 76/7 (DS Pharma Biomedical Co., Ltd., Osaka, Japan), in cultivation medium consisting of Dulbecco's modified Eagle's medium (DMEM; Gibco, Invitrogen, Grand Island, NY) supplemented with 15% fetal bovine serum (FBS; SAFC Biosciences, Lenexa, KS), 2 mM l-glutamine (Gibco, Invitrogen), 0.4 ml of β-mercaptoethanol (Sigma-Aldrich, St. Louis, MO), and nonessential amino acids (Gibco, Invitrogen). iPSCs were passaged every second day using 0.25% trypsin–EDTA (Gibco, Invitrogen). To maintain iPSC pluripotency, we prepared the puromycin-resistant SNL 76/7 cells. Briefly, the Streptomyces alboniger puromycin N-acetyl-transferase gene was subcloned into a retrovirus shuttle vector, pCX4-bsr (3), and introduced into SNL 76/7 cells by infecting the recombinant retrovirus vector as described previously (14). The puromycin-resistant SNL cells were selected by culturing in DMEM in the presence of 2 μM puromycin. iPSCs were cultured in the above-mentioned medium with the puromycin-resistant SNL 76/7 cells containing 1.5 μg/ml puromycin (Sigma-Aldrich) during selection of undifferentiated cells, to select GFP-expressing undifferentiated cells, on a gelatin-coated dish (6 cm in diameter). GFP expression became negative when differentiation was induced, whereas GFP expression remained in undifferentiated cells (27).
Fabrication of Bioengineered Scaffold and Differentiation of iPSCs (Fig. 1)

Fabrication of bioengineered scaffold and implantation into tracheal defects in rats. Induced pluripotent stem cells (iPSCs) were cultured in the 3D scaffold containing collagen gel with DMEM and chondrocyte differentiation medium (CDM; bioengineered scaffold model). After confirming the chondrogenic differentiation, these scaffolds were implanted into a rat tracheal defect model. At 1 and 4 weeks postimplantation, the rats were humanely killed, and several analyses were performed. See Materials and Methods for more details. Three-dimensional scaffold (3D scaffold) consisting of collagenous sponge and polypropylene mesh. Control model: 3D scaffold without iPSCs. Noninduced model (DMEM culture): 3D scaffold with noninduced iPSCs. Induced model (CDM culture): 3D scaffold with induced iPSCs. Noninduced and induced scaffold: bioengineered 3D scaffold.
To induce efficient differentiation of iPSCs into chondrocytes in vitro, we prepared a 3D scaffold. The procedure for the production of the 3D scaffold was described by Nakamura et al. (25). The sponge, with a pore size range of 100–500 μm, was made from porcine dermal atelocollagen consisting of type 1 (70–80%) and type 3 collagen. The atelocollagen was dissolved in a hydrochloric acid solution (pH 3.0) at a concentration of 1.2%, stirred at 8,000 rpm for 15 min, freeze-dried, and heated at 140°C in a vacuum for 24 h. In our current study, a 3D scaffold (diameter, 10 mm; thickness, 3 mm) made from a piece of artificial trachea was utilized. Collagen gel containing iPS cells was prepared as follows. Type I collagen solution (Nitta Gelatin, Osaka, Japan), fivefold concentrated DMEM/F-12, and reconstituted buffer (25 mM Hepes, 0.15 M sodium hydroxide) were mixed at a ratio of 7:2:1. iPSCs were suspended in the reconstituted collagen solution at a density of 5.0 × 106 cells/ml. To cultivate the iPSCs, a 3D scaffold made from the above-mentioned collagen sponge scaffold with polypropylene mesh was utilized. Four hundred microliters of 0.3% collagenenous solution containing the iPSCs was introduced into the 3D scaffold (2.0 × 106 cells per scaffold) in Millicell® Culture Plate inserts (Millipore Co., Billerica, MA) with a membrane filter (0.4 μm in pore size), and the collagenenous solution was allowed to infiltrate the scaffold. The bioengineered scaffolds were then incubated at 37°C for at least 30 min, and the collagenous solution formed a gel. These bioengineered scaffolds were placed in six-well polystyrene plates (Becton Dickinson, Franklin Lakes, NJ) containing 2 ml of chondrocyte differentiation medium (CDM; Cell Applications, Inc., San Diego, CA) for the induced model or Dulbecco's modified Eagle's medium (DMEM; Invitrogen, USA) supplemented with 10% fetal bovine serum (FBS; SAFC Biosciences) for the noninduced model. As a control, collagen gel without iPSCs was used for the control model. At 2, 4, and 6 weeks of cultivation, noninduced and induced models were selected for histological and genetic examination. These models were also selected at 5 weeks of cultivation for immunohistochemical analysis to confirm the decrease in GFP-expressing undifferentiated iPSCs and flow cytometric analysis to confirm the decrease in undifferentiated cells.
Chondrogenic Differentiation of iPSCs and Confirmation of the Decrease in GFP-Expressing Undifferentiated iPSCs In Vitro
In Vitro Morphological and Immunohistochemical Analyses
Culture cells in the collagen gel complex were fixed with 4% paraformaldehyde and embedded in paraffin. Sections were stained with hematoxylin and eosin (H&E) and Alcian blue to detect acidic proteoglycans produced by chondrocytes (35). In addition, immunohistochemical analysis was performed using antibodies to S-100 protein (DAKO, Kyoto, Japan), a chondrocyte-associated protein (36), and an antibody against GFP as a marker of undifferentiated iPSCs (27) (MBL, Nagoya, Japan). Immunohistochemical staining was performed using an indirect streptavidin-biotin immunoperoxidase method [SAB-PO(M) kit, Nichirei Corp., Tokyo, Japan]. After blocking endogenous peroxidase activity in 0.3% hydrogen peroxide in methanol for 30 min, slides were incubated with primary antibodies overnight at 4°C, washed with PBS, and then incubated with biotin-labeled secondary antibodies for 30 min at room temperature. Antibody localization was visualized with peroxidase-conjugated streptavidin for 30 min at room temperature, followed by the diaminobenzidene reaction. The slides were then counterstained with hematoxylin.
Detection of Gene Expression by Quantitative Reverse Transcription-Polymerase Chain Reaction (RT-PCR) Analysis
PCR analysis was performed as described previously (15), using the following premade primers purchased from TaKaRa Bio (Shiga, Japan): aggrecan (Acan; MA073560), type II collagen (Col2a1; MA067811), type I collagen (Col1a1; MA093358), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; MA050371). The primer sequences for genes encoding the matrix protein aggrecan were 5′-CGCCACTTTCATGACCGAGA-3′ and 5′-TCATTCAGACCGATCCACTGGTAG-3′, those for type II collagen were 5′-AGGGCAACAGCAGGTTCA CATAC-3′ and 5′-TGTCCACACCAAATTCCTGTTCA-3′, and those for bone matrix protein collagen I were 5′-ATGCCGCGACCTCAAGATG-3′ and 5′-TGAGGCA CAGACGGCTGAGTA-3′. The primer sequences for the housekeeping gene GAPDH, which was used as an internal standard, were 5′-TGTGTCCGTCGTGGATCTGA-3′ and 5′-TTGCTGTTGAAGTCGCAGGAG-3′.
Flow Cytometric Analysis
About 5 × 104 cells were suspended in PBS/1% FBS. Fluorescence-activated cell sorting (FACS) analysis of GFP-expressing cells was performed on a FACSCalibur system (Becton-Dickenson), and their green fluorescent emission (FL1 height) was used to distinguish between populations of undifferentiated and differentiated iPSCs. GFP fluorescence was excited at 488 nm and emission was measured with a 530/30 nm bandpass filter. Differences in fluorescence intensity between noninduced model and induced model derivatives were then analyzed. The data were processed using a FACSCalibur system with CellQuest software (BD Biosciences, San Jose, CA).
Implantation Into Tracheal Defects in Rats (Fig. 1)
This study was carried out under the control of the Animal Care and Use Committee in accordance with the Guidelines for Animal Experiments of Fukushima Medical University, the Japanese Government Law Concerning the Protection and Control of Animals, and the Japanese Government Notification on Feeding and Safekeeping of Animals.
A total of twenty-four 8- to 10-week-old male F344/ NJcl-rnu/rnu nude rats were used as implant recipients in this study. The 21 rats each received one of the three types of 3D scaffold (noninduced-6 weeks, induced-6 weeks, and control-6 weeks) after 6 weeks of cultivation in vitro. The three rats received the 3D scaffold (induced-3 weeks) after 3 weeks of cultivation in vitro as an insufficient differentiation model to confirm the relationship between insufficient cell differentiation and tumor formation. Implantation into tracheal defects in rats was performed as follows. Tracheostomy and implantation of the four types of scaffold were performed under general anesthesia induced by inhalation of isoflurane. The cervical tracheas were exposed through a vertical skin incision, and the sternohyoid and sternothyroid muscles were split along the median line. Tracheal defects, approximately 1.5 mm in width by 2.5 mm in length, were then formed. The scaffolds were laid onto the tracheal defects. To prevent shifting of the graft, the sternohyoid and sternothyroid muscles were then replaced over the graft and sutured. Finally, the incised skin was sutured. At 7 and 28 days postimplantation, the rats were humanely killed, and the tracheas and sternohyoid and sternothyroid muscles were extirpated en bloc. The samples were fixed in 4% paraformaldehyde, embedded in paraffin, and sliced for H&E staining and immunohistochemical analysis. Paraffin-embedded tissues containing the 3D scaffold were cut into 500 sections at a thickness of 5 μm, which were then mounted on hydrophilic silanized glass slides (DAKO, Tokyo, Japan). To confirm cartilage differentiation in the tissue components, immunohistochemical analysis was performed using an antibody against type II collagen (Abcam, Cambridge, MA).
Confirmation of iPSCs-Derived Tissue
Sample Preparation for Laser Microdissection (LMD)
To determine whether the cartilage tissues observed in the scaffolds were in fact derived from the implanted cells, we confirmed the presence of the GFP gene in the microdissected tissue sections. To remove the paraffin, sections were immersed in 100% xylene (three times) and 100% ethanol for 10 min each and then air-dried completely at room temperature. Using ArcturusXT Laser Microdissection System LCC1704 (Molecular Devices, Ontario, Canada), cartilage tissue, teratoma tissue, and rat sternohyoid muscle tissue samples were captured by an infrared laser onto CapSure HS Caps (Molecular Devices) and were subsequently cut using a UV laser (Fig. 2).

Sample preparation for laser microdissection (LMD). A sample (C) was collected from the targeted tissue area (A and B) by LMD procedure for PCR analysis of the bioengineered scaffold tissue sections. The boxed region in (A) is magnified and shown in (B and C).
PCR Analysis
Genomic DNAs for these tissues and iPSCs, as a positive control, were extracted in 50 μl of solution (0.5% SDS, 10 mM EDTA, 150 μg/ml proteinase K) overnight at 65°C. One microliter of each DNA solution was subjected to PCR to detect the GFP gene. The premade primer set, GFP primer-1 in the Transgene Detection Primer Set for Real Time (Mouse) (TaKaRa Code 3788), was used to detect the GFP gene. A primer set for the GAPDH gene was used as a positive control. The PCR products were electrophoresed on 10% polyacrylamide gel, followed by ethidium bromide staining.
Results
Chondrogenic Differentiation of iPSCs and Confirmation of the Decrease in GFP-Expressing Undifferentiated iPSCs In Vitro
In Vitro Morphological and Immunohistochemical Analyses
The time course of CDM-induced morphological changes in iPSCs was as follows. In the noninduced model, the tissue showed no histological evidence of cartilage formation throughout the experimental period (Fig. 3A–F). In the induced model, at 2 weeks after induction, Alcian blue-stained areas appeared with no chondrocytic changes (Fig. 3G, H). At 4 and 6 weeks, the cells revealed cartilaginous characteristics with condensed hyaline matrices that were positive for Alcian blue staining and round chondrocytic cells in the lacunae (Fig. 3I–L).

Morphological examination of chondrocyte differentiation in vitro. The time course of CDM-induced morphological changes in iPSCs was evaluated. Sections were stained with hematoxylin and eosin (H&E) and Alcian blue. The tissue in the noninduced model showed no histological evidence of cartilage formation throughout the experimental period (A–F). At 2 weeks after induction, Alcian blue-stained areas appeared (H) in the induced model, with no cellular changes (G). At 4 and 6 weeks, the cells revealed cartilaginous characteristics, with avascular tissue showing condensed hyaline matrices that were positive for Alcian blue staining and round chondrocytic cells in the lacunae (I–L). (A, C, E, G, I, and K were stained with H&E, and B, D, F, H, J, and L with Alcian blue.) Scale bars: 100 μm.
In the noninduced model, the tissue showed no histological evidence of S-100 protein expression throughout the experimental period (Fig. 4A–C). In the induced model, no expression of the chondrocyte-associated S-100 protein was detected at 2 weeks after induction (Fig. 4D). The expression of S-100 protein was, however, detected in a few cells at 4 weeks after induction (Fig. 4E), whereas at 6 weeks, most of the chondrocytic cells expressed S-100 protein (Fig. 4F).
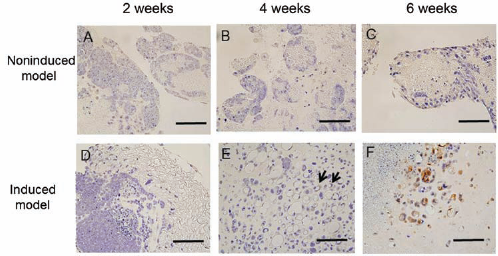
Immunohistochemical analysis of chondrocyte differentiation in vitro. No S-100 protein was detected in the noninduced model throughout the experimental period (A–C). In the induced model, no expression of the chondrocyte-associated protein, S-100 protein, was detected at 2 weeks after cultivation (D). The expression of the S-100 protein was detected in a few cells at 4 weeks (E). After 6 weeks, most cells expressed S-100 protein (F). Arrows show S-100 protein-expressing cells. Scale bars: 100 μm.
At 5 weeks after induction, expression of GFP as a marker of undifferentiated iPSCs was detected in most colonies in the noninduced model (Fig. 5A). In the induced model, a few colonies showed histological evidence of GFP expression (Fig. 5B).

Immunohistochemical analysis of green fluorescent protein (GFP)-expressing undifferentiated cells in vitro. Many GFP-expressing iPSCs colonies were detected in the tissue in the noninduced model (A). In the induced model, a few GFP-expressing iPSCs colonies were detected (B). Arrows show GFP-expressing colonies. Scale bars: 100 μm.
Detection of Gene Expression by Quantitative RT-PCR Analysis
In the induced model, mRNA expression of type II collagen and aggrecan was increased at 4 weeks after cultivation. In contrast, in the noninduced model, mRNA expression of type II collagen and aggrecan was not increased at any time point. In both models, no increase in the expression of type I collagen, an osteogenic differentiation or chondrogenic dedifferentiation marker, was observed throughout the experimental period (Fig. 6A–C).

PCR-based analysis of chondrocyte differentiation in vitro. In the induced model, mRNA expression of type II collagen (A) and aggrecan (B) was increased at 4 weeks after cultivation. There was no increase in the expression of type I collagen (C), an osteogenic differentiation marker, throughout the experimental period. N, noninduced model; I, induced model.
Confirmation of a Decrease in GFP-Expressing Undifferentiated iPSCs Using Flow Cytometric Analysis
To distinguish between populations of undifferentiated and differentiated cells, we analyzed iPSCs after 5 weeks of cultivation by FACS. The cell samples of the induced model and noninduced model were characterized according to their fluorescent emission. GFP expression remained observable in undifferentiated cells (27). The changes in CDM induction in the population of GFP-expressing iPSCs were as follows. After 5 weeks of cultivation, there was a marked difference in the population of GFP-expressing iPSCs between the noninduced model (59.06%) and induced model (27.19%) (Fig. 7). These data reveal that the decrease in undifferentiated cells in the induced model was greater than that in the noninduced model at 5 weeks. This shift in population represents a transition, from undifferentiated to differentiated, in the state of the cells.
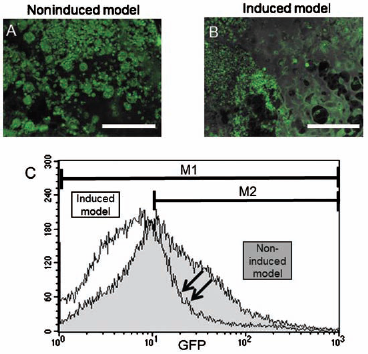
Flow cytometric analysis of GFP-expressing undifferentiated cells in vitro. To distinguish between populations of undifferentiated and differentiated cells, iPSCs were analyzed by fluorescence activated cell sorting (FACS) after cultivation for 5 weeks. The cell samples from noninduced (A) and induced (B) models were characterized according to their GFP expression. We considered the cells in the M2 area as GFP-positive undifferentiated cells. As expected, a clear difference in fluorescent intensity exists between the noninduced (shaded histogram) and induced (open histogram) models (C). GFP expression was observed in 59.06% (M2/ M1) of iPSCs in the noninduced model and in 27.19% (M2/M1) in the induced model. This shift in population represents a transition, from undifferentiated to differentiated, in the state of the cells. Arrows show the decrease in the GFP-expressing iPSCs population and fluorescence intensity. Scale bars: 100 μm.
Implantation Into Tracheal Defects in Rats
Cartilage was observed in the regenerated tracheal wall tissue (Fig. 8A, B), and the expression of the cartilage-specific protein type II collagen was detected by immunohistochemical analysis (Fig. 8C) in 6 of 11 rats implanted with induced-6 weeks scaffolds. At 1 week after implantation, three of three rats showed cartilage formation, whereas at 4 weeks after implantation, cartilage formation was observed in three of eight rats. Cartilage tissue was found within the edges of the scaffolds. Tumor formation was confirmed in the bioengineered scaffold in 14 of 18 rats (Table 1). On the other hand, no cartilage tissue was observed in any of the six rats implanted with the control scaffolds at either 1 or 4 weeks after implantation (Fig. 8D), in any of the four rats implanted with noninduced-6 weeks scaffolds (Fig. 8E), or in any of the three rats implanted with induced-3 weeks scaffolds as an insufficient differentiation model to confirm the relationship between insufficient cell differentiation and tumor formation. Three of four noninduced-6 weeks scaffold-implanted rats and two of three induced-3 weeks scaffolds-implanted rats were suffocated due to tumor formation (Fig. 8E). Therefore, the regeneration of the tracheal wall could not be evaluated entirely in these rats. One of four noninduced-6 weeks scaffold-implanted rats survived for 4 weeks without tracheal stenosis. Tumor formation was not confirmed in that rat, suggesting the absence of iPSCs in the scaffold.
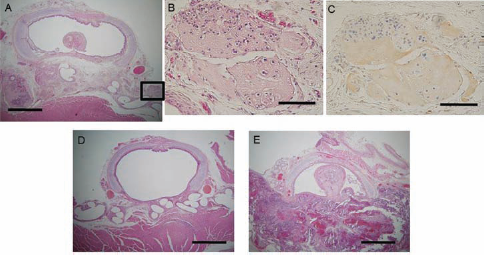
Histological findings of the implanted scaffolds at 4 weeks postimplantation in vivo. Tissues including the control, noninduced, and induced model scaffolds were excised and fixed for histological analyses. Paraffin sections were stained with H&E. The boxed region in (A) is magnified and shown in (B) and (C). In induced model-implanted rats (A), the bioengineered scaffold contained cartilage tissue (B). The expression of cartilage-specific protein, type II collagen, was detected by immunohistochemistry (C). No cartilage tissue was observed in control model-implanted rats (D). Noninduced model-implanted rats were suffocated due to tumor formation (E). Scale bars: 1 mm (A–C) and 100 μm (D, E).
Summary of Results (Cartilage Tissue, Rat Death, and Tumor Formation)
Confirmation of iPSCs-Derived Tissue
To confirm that these cartilage tissues were derived from the implanted iPSCs, the presence of the GFP gene sequence in the excised sections was examined. As shown in Figure 9, the GFP gene was detected in iPSC samples and in sections of teratoma and cartilage tissue, but not in rat tissue section.

PCR analysis of laser microdissected tissue samples. Portions of the cartilage tissues, teratomas, and rat sternohyoid muscle tissues were excised from paraffin sections by laser microdissection as described in Materials and Methods. Genomic DNA from these tissues and iPSCs was extracted, and the presence of the GFP sequence was examined by conventional PCR. The glyceraldehyde 3-phosphate dehydrogenase (GAPDH) gene was used as a positive control. The GFP gene was detected in samples from iPSCs and in sections of teratoma and cartilage tissue, but not in the rat tissue sections.
Discussion
The trachea is indispensable to the maintenance of respiratory function. Resection of part of the trachea due to a malignant tumor or inflammatory lesion produces various functional disorders involving speech and deglutition, as well as respiration. Even now, the treatment of pediatric laryngotracheal stenosis remains a challenge because treatment often requires multistaged procedures, and successful decannulation sometimes fails after a series of operations (12, 13).
This study demonstrated the regeneration of the tracheal cartilage through the use of iPSCs. iPSCs have the potential to differentiate into chondrogenic cells in our cell culture system, as shown by the appearance of Alcian blue-stained areas, the expression of chondrocyte-associated protein, and the expression of cartilage-specific genes of collagen type II and aggrecan in vitro. Strong stainability with Alcian blue suggests that proteoglycan is highly expressed in differentiated iPSCs. mRNA expression of type II collagen and aggrecan in cultivated iPSCs after 6 weeks was lower than that after 4 weeks, suggesting that the expression level of proteins reached a peak at 4 weeks, before decreasing thereafter. No expression of type I collagen, an osteogenic differentiation or chondrogenic dedifferentiation marker (4, 32, 41), was detected. This result showed that no chondrogenic dedifferentiation occurred under these culture conditions. This study further suggests the potential for iPSCs to be used as a new cell source for tracheal regeneration therapy in vivo because cartilage tissue could be confirmed in implanted 3D scaffolds.
Differentiation into cartilage has not been well examined or established in iPSCs research. Only a few studies have addressed the possibility of cartilage differentiation from iPSCs (23, 40). Kramer et al. (21) showed that differentiation of ES cells in vitro into chondrocytes can be modulated by members of the transforming growth factor-β family [TGF-β1, bone morphogenetic protein (BMP-2), and BMP-4]. A combination of various factors might be needed to induce effective differentiation into chondrocytes. Bigdeli et al. (5) showed that the coculture of ES cells and chondrocytes improved chondrogenic differentiation, with no evidence of teratoma formation. Further studies to induce the efficient differentiation of iPSCs into chondrocytes are expected.
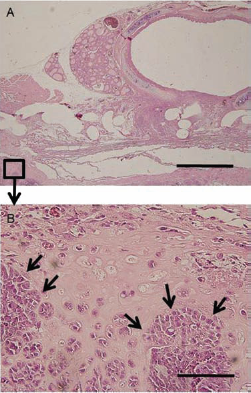
Previously, similar studies using mesenchymal and ES cells failed to demonstrate cartilage tissue formation in vivo, although histological and PCR analyses had demonstrated chondrogenic differentiation in vitro prior to cell implantations (9, 16). The reason suggested for this was that predifferentiation in vitro was insufficient to maintain a stable chondrogenic phenotype in vivo. In these studies, grafts were implanted ectopically in the subcutaneous tissue of the back of nude mice. Thus, the lack of chondrogenic signals in the ectopic implantation site may cause the dedifferentiation or cell death of the implanted cartilaginous tissue. In our study, differentiated iPSCs were implanted in the tracheal cartilage defect. The conditions in this case may induce the release of various appropriate factors, such as keratinocyte growth factor (KGF) (1), from the surrounding tissue to maintain a chondrogenic phenotype. To confirm this possibility, it is thought to be essential to implant iPSCs ectopically in the subcutaneous tissue in nude rats as a comparison. In this study, tracheal glandular epithelium and cartilage tissue were observed in the area of the tracheal defect (Fig. 10). This result confirms the above-mentioned hypothesis: tracheal defect as an injury site induced iPSCs into these various differentiated tissues. In addition, we compared the models at 1 and 4 weeks postimplantation and found that, at 1 week, cartilage tissue was confirmed in all rats implanted with Induced scaffolds, whereas cartilage tissue was confirmed in only three of eight rats at 4 weeks. These results show that differentiated iPSCs may dedifferentiate or die as time passes in vivo.

Histological findings of the area of tracheal defect in vivo. Tracheal glandular epithelium and cartilage tissue were observed in the area of the tracheal defect. Arrows show cartilage (CA) and glandular epithelium (GE). Scale bar: 500 μm.
Unfortunately, the regenerated tissue in this study did not always fit the area of the tracheal cartilage defects. In future, we intend to further investigate scaffold size and thickness and gel volume to better match the tracheal defect area. Various kinds of scaffolds have been reported for tracheal regeneration, including poly(lactic-co-glycolic acid) (PLGA)–collagen hybrid scaffolds (39), biodegradable scaffolds (20), and collagen vitrigel scaffolds (37). Further research is required to identify the scaffold that offers the best results in terms of tracheal wall regeneration.
Apart from our previous report, there have been no reports on the use of iPSCs for tracheal cartilage regeneration. In our previous study, the survival of iPSCs was confirmed by teratoma formation. It has also been reported that there is a significant correlation between teratoma size and undifferentiated iPSCs content (24). In addition, to confirm how iPSCs form tumors in vivo, we implanted an insufficient differentiation model to investigate the relationship between undifferentiated cells and tumor formation. As expected, most rats in the insufficient differentiation model were suffocated due to the formation of tumors derived from iPSCs. As a result of insufficient induction, a large number of undifferentiated cells were implanted into the tracheal defect, and the rapid proliferation of these cells might have caused death due to tumor formation. Information regarding the ratio of differentiated/undifferentiated cells would also be useful in dealing with the problem of teratoma formation. To prevent teratoma formation, undifferentiated cells can be isolated by FACS using the expression of GFP. Flow cytometric analysis confirmed the decrease in undifferentiated cells in the induced model. Additional research on the time course of CDM-induced morphological changes in GFP-expressing cell populations is proposed. In the noninduced model, an undifferentiated cell model, the population of GFP-expressing iPSCs was 59.06%, not 100%, at 5 weeks of cultivation. This may show that iPSCs spontaneously differentiated into other differentiated cells. For further study of flow cytometric analysis, it would be useful to distinguish iPSCs from differentiated cells using antibodies, such as the TRA-1-81 cell surface marker, an undifferentiated cell marker (42), and live cells from dead cells using fluorescent dyes such as calcein-AM and propidium iodide (10, 31).
At 1 week postimplantation, no tumor formation could be confirmed macroscopically (Fig. 11A); however, a few colonies, derived from implanted undifferentiated cells, were confirmed microscopically (Fig. 11B). As it is well documented, implanting undifferentiated cells from any pluripotent stem cell sources (regardless of iPSCs or ESCs) results in uncontrolled differentiation and thus leads to formation of teratoma consisting of three germ layers in vivo. A few undifferentiated cell colonies were demonstrated in the induced model using an immunohistological technique in vitro. These few undifferentiated cells could form tumors after 4 weeks of cultivation in vivo. The first iPSCs were generated using single viruses to deliver each gene, resulting in multiple copies of individual viruses present in established iPSCs lines (38). Any vector integrated into the genome has the potential to disrupt the function of a gene at its site of entry, which can lead to a number of undesirable effects, such as tumorigenesis. In this study, iPSCs generated from mouse skin fibroblasts by the retroviral introduction of four transcription factors, Krüppel-like factor 4 (KLF4), c-MYC, octamer-binding transcription factor 4 (OCT4), and sex-determining region Y-box 2 (SOX2) (27), were utilized. Viral integration into the genome initially presented a formidable obstacle to the therapeutic use of iPSCs. To induce pluripotency without incurring genetic change, iPSCs have been derived via excisable lentiviral and transposon vectors or through repeated application of transient plasmid, episomal, and adenovirus vectors (7, 17, 28, 34, 45, 46). iPSCs have been derived using DNA-free methods: serial protein transduction with recombinant proteins incorporating cell-penetrating peptide moieties (19, 47). iPSCs have also been derived by RNA-based technology (44), which completely eliminates the risk of genomic integration and insertional mutagenesis inherent to all DNA-based methodologies.
Histological findings of the implanted scaffolds at 1 week postimplantation in vivo. At 1 week postimplantation condition, tumor formation could not be confirmed macroscopically; however, several colonies, formed by implanted undifferentiated cells, were confirmed microscopically. The boxed region in (A) is magnified and shown in (B). Arrows show undifferentiated cell colonies. Scale bars: 1 mm (A) and 100 μm (B).
A recent study has reported that, even in a syngenetic or autologous situation, pluripotent stem cells might become targets for cytotoxic natural killer (NK) cells (11). This could result in rejection after transplantation, as suggested by reduced teratoma growth after NK cell activation in vivo. It might also offer a strategy to deplete contaminating undifferentiated pluripotent cells before the grafting of differentiated cells. Generally, iPSCs could be used to generate autologous cells for therapeutic purposes that are expected to be tolerated by the recipient. Although we tried to implant mouse iPSCs into mouse tracheal defects as autologous implantations, we could only make very small defects, less than 1 mm, even when operating under a microscope. Any larger defects in mice would result in death. In this study, mouse iPSCs were implanted into tracheal defects in nude rats, an immunodeficient animal model, as xenograft implantations because we wanted to investigate how iPSCs differentiate and proliferate at an injury site, even if this required a xenograft implantation. In future studies, autologous iPSC implantation should be investigated to confirm whether tumor formation is decreased or not.
This study demonstrated the regeneration of the tracheal cartilage through the use of iPSCs. From the results of our current study, iPSCs could be a new cell source for the regeneration of tracheal cartilage.
Conclusions
Our study demonstrated that iPSCs have the potential to differentiate into chondrogenic cells in vitro. Cartilage tissue was regenerated in vivo in 3D scaffolds containing iPSCs. Our results suggest that our current study was an important step to develop a new approach that could in the future be used in cell therapy.
Footnotes
Acknowledgments
This study was supported in part by a Grant-in-Aid for Scientific Research (B) from Japan Society for the Promotion of Science and in part by a Grant-in-Aid for Research Activity start-up from Japan Society for the Promotion of Science. We thank Etsuko Sato for her technical assistance. M.I. and K.O. planned and directed the research. M.I. wrote the first draft of the paper together with K.O., T.S., Y.S., and I.W. M.I. and Y.N. designed the method and implemented this investigation. M.I. and Y.S. performed PCR analysis and flow cytometric analysis. M.I. and T.S. performed immunohistochemistry. M.I. and M.M. performed histological examination. M.I. and I.W. performed laser microdissection. I.W. provided puromycin-resistant SNL 76/7 cells. T.N. constructed 3D scaffolds. M.I., Y.N., Y.S., T.S., M.M., I.W., T.N., and K.O. contributed meaningfully to the final manuscript. The authors declare no conflict of interest.