
Abstract
Induced pluripotent stem cells (iPSCs) were originally generated by forced ectopic expression of four transcription factors genes—OCT4, KLF4, SOX2, and c-MYC—in fibroblasts. However, the efficiency of iPSCs obtention is extremely low, and reprogramming takes about 20 days. We reasoned that adult cells showing basal expression of core embryonic stem (ES) cell regulator genes could be a better cell source for reprogramming. Menstrual blood-derived mesenchymal cells (MBMCs) are multipotent cells that show detectable levels of some of the core ES cells regulators. The aim of this study was to determine whether reprogramming efficiency could be increased by using MBMCs as a cell source to generate iPSCs. MBMCs were transduced with recombinant retroviruses expressing the coding regions of OCT4, SOX2, and KLF4 genes. Cells with high nucleus/cytoplasm ratio can be detected about 5 days of posttransduction, and colonies of typical ES-like cells begun to appear after 7 days. At day 15, colonies were picked up and expanded for characterization. Most of the clones were morphologically identical to ES cells and positive at the mRNA and protein levels for all pluripotency markers tested. The clones are capable of forming embryoid bodies and to differentiate in vitro into cells of the three germ cell layers. Our results show that the reprogramming was faster and with efficiency around 2–5%, even in the absence of ectopic expression of c-MYC. To date, this is the first study showing MBMCs as a cell source for nuclear reprogramming.
Keywords
Introduction
Reprogramming of human adult somatic cells to induced pluripotent stem cells (iPSCs) has the potential to be used in regenerative medicine to repair tissues damaged by disease or injury. Although human iPSCs have been generated from various types of somatic cells, such as skin fibroblasts (17), keratinocytes (1), neural stem cells (NSCs) (9), blood cells (13,19), and cord blood-derived endothelial cells (CBECs) (7), there is still a large debate about the best sources from which to derive iPSCs. Subsequent studies have shown that age, origin, and cell type used profoundly affect reprogramming efficiency. A desirable protocol should aim not only to optimize all these parameters but also to reduce the number of factors used, the time required for reprogramming and to reduce the difficulty to obtain the cells. For example, mouse neural and adipose tissue-derived stem cells could be more efficiently reprogrammed than fibroblasts, and the reprogramming occurs by the superexpression of only two [Octamer-binding transcription factor 4 (OCT4) and Krüppel-like factor 4 (KLF4)] or even one (OCT4) of the Yamanaka's factor genes, most likely due to their consistent endogenous expression of sex-determining region Y-box 2 (SOX2) and c-MYC genes (10,11,18). In these reports, human embryonic stem (ES) cell-like colonies appeared ~10–15 days after transduction of the factors, which is a substantially shorter time than the ~16–30 days required for the human fibroblasts. However, NSCs are rare cells available in small quantities and not easily accessible without highly invasive procedures, making them poor candidates for reprogramming in a clinical setting.
Mesenchymal stromal cells (MSCs) are nonhematopoietic cells characterized by plastic adherence in culture, differentiation potential under specific conditions, and a cell surface marker gene expression pattern [presence of cluster of differentiation 105 (CD105), CD73 and CD90, and absence of CD45, CD34, CD14 or CD11b, CD79a or CD19, and human leukocyte antigen (HLA) class II] (4,5). MSCs have been used as an adult somatic cell source for nuclear reprogramming and, in general, were shown to be reprogrammed faster and/or with higher efficiency. The faster and more efficient reprogramming of MSCs derived from distinct sources, such as third molar teeth (15), umbilical cord matrix, amniotic membrane and amniocytes (2,3), and bone marrow (12,13), are usually attributed to the endogenous expression of some of the core pluripotency genes and their overall epigenetic landscape (20).
In the present work, we have generated iPS cell lines from recently characterized human menstrual blood-derived MSCs (hMBMCs) (16) using retroviral vectors containing human OCT4, SOX2, and KLF4 genes. Our results show that the resulting iPSCs are obtained in a very short period of 15–17 days and at a high efficiency (2–5%) when compared to that of reprogrammed fibroblasts (0.01–0.1%) (17). The shorter and more efficient reprogramming of these cells, together with the ease of obtention, since they can be obtained from discarded material by noninvasive procedures, make hMBMCs an attractive cell source to produce high-quality human iPS cells.
Materials and Methods
Derivation of hMBMCs
Menstrual blood samples were collected after personal hygiene from healthy young donors, 24 h after the beginning of menses. The samples were collected in 5 ml of 1x calcium magnesium-free PBS (CMF), 100 U/ml penicillin, 100 mg/ml streptomycin, and 5 mM EDTA. Mononuclear cells were fractionated in Ficoll (Histopaque 1077) and maintained in DMEM high-glucose medium (1 g/L) supplemented with 20% FBS, 2 mM l-glutamin, 100 U/ml penicillin, and 100 mg/ml streptomycin. The medium was changed twice a week and the cells were passaged after reaching 70–80% confluence. All reprogramming experiments were carried out using third or fourth passage cells. The mesenchymal nature of the cells was confirmed by the following characteristics: adherence to plastic in culture, ability to differentiate into adipocytes, chondrocytes and osteocytes, and a typical set of cell surface markers. All experiments were approved by our local institutional review board (Hospital Universitário Clementino Fraga Filho, Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ, Brazil) protocol No. 056/09. All donors signed an informed consent form.
hMBMCs Reprogramming and Maintenance of Undifferentiated iPS-hMBMCs
Reprogramming experiments, summarized in Figure 1A, were carried out by pseudo-typed retroviral transduction of three out of the four Yamanaka's transcription factor genes: OCT4, SOX2, and KLF4 using standard protocols (17). Briefly, Phoenix™ Ampho Cells (Orbigene) were transfected separately with each of the recombinant retrovirus vector pMX-Oct4, pMX-SOX2, and pMX-KLF4 (Addgene, Cambridge, MA, USA) using fugene-6 transfection reagent (Roche, Indianapolis, IN, USA) following manufacturer's instructions. The combined retroviral-containing supernatants were filtered in 0.22-μm low protein binding filters (Millipore) and concentrated by ultracentrifugation (45,000 x g, 4°C, 90 min) and used to transduce hMBMCs. Twenty-four hours after we repeated the transduction using a second supernatant harvest, hMBMCs were trypsinized and replated onto a mitomicin-C inactivated mouse embryonic fibroblast (MEF) feeder layer, and the medium was changed to the human ES (hES) cell medium containing basic fibroblast growth factor (bFGF) [DMEM/F12, 20% knockout serum replacement (KSR), 2 mM glutamine, 0.1 mM nonessential amino acids, 0.1 mM β-mercaptoethanol and 8 ng/ml bFGF]. Fifteen to 17 days after transduction, colonies with typical hES cell morphology (iPS-MBMC colonies) were picked up for individual analysis, mechanically dissociated into small clumps, and plated on MEF feeder cells. Derived iPS-MBMCs and H9 ES cells (WiCell, used as positive controls) were maintained either on MEF feeder layer or on Matrigel-coated tissue culture dishes (ES qualified; BD Biosciences) with mTESR-1 hES growth medium (Stemcell Technology).
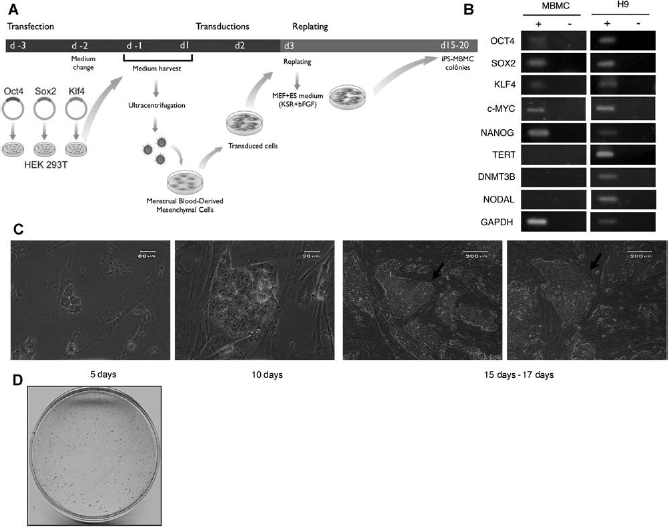
(A) Experimental outline of MBMCs nuclear reprogramming by the ectopic expression of OCT4, SOX2, and KLF4 genes. In day -3, Phoenix™ Ampho Cells (Orbigene) were transfected separately with pMXs vectors containing Oct4, Sox2, and Klf4 cDNAs. At 48 and 96 h after (d-1 and d1), the retrovirus-containing supernatant were mixed and concentrated to transduce the MBMCs. In day 3 (d3), the transduced MBMCs are trypsinized and split to mitomicin-C inactivated MEF cells plate. One day after (d4), the medium is changed to the human embryonic stem cell medium containing bFGF (HuES + bFGF). Next, the medium is changed everyday with HuES + bFGF, until the colonies reach the correct size for manual passage, which occurs at days 15–20. (B) Pluripotency-associated gene expression in MBMCs. MBMCs were used for total RNA extraction and RT-PCR assays, H9 cell line were used as positive control. Amplified products were analyzed in 2% agarose gel electrophoresis and visualized by ethidium bromide staining. The RT (+) and (-) samples refer to the presence or absence of reverse transcriptase in transcriptase reaction, respectively. GAPDH was used as internal amplification control. (C) Morphology of iPS-MBMCs colonies during reprogramming. Phase-contrast micrographs of different representative iPS-MBMCs colonies show that they have morphology comparable to hES and distinct from parental MBMCs. The kinetics of colony formation shows that the colonies start to appear as early as 5 days (scale bar: 60 μm) after the first retroviral transduction of MBMCs; at 10 days (scale bar: 30 μm). In 15–17 days typical iPS-MBMCs colonies were chosen for the first manual passage (arrows) (scale bar: 200 μm). (D) Alkaline phosphatase activity staining demonstrates the ES-like feature of iPS-MBMCs colonies. The estimated efficiency of reprogramming with our methodology is approximately 2–5%.
RT-PCR and qRT-PCR Analysis
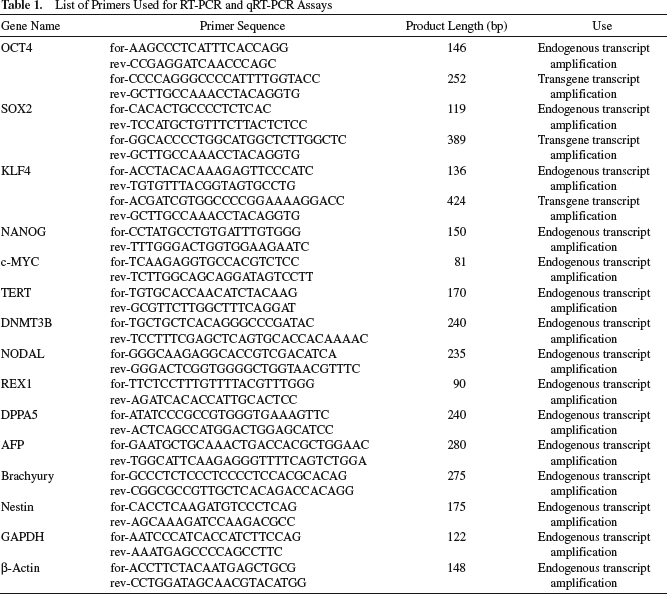
Total RNA was extracted from cell lines using RNeasy Plus Kit (Qiagen). cDNA synthesis was performed using the High-Capacity Reverse Transcriptase Kit (Applied Biosystems) according to the manufacturer's instructions using 0.5 μg of total RNA. Specific primer pairs were designed to span introns and avoid eventual DNA contamination detection (Table 1). RT-PCR reactions were conducted in a 50-μl reaction mixture containing 2 μl of each 10x diluted cDNA reaction volume, 1x PCR buffer (Invitrogen), 1.5 mM MgCl2, 0.2 mM of each dNTP, 0.2 μM of each forward and reverse primer, and 1 U of Platinum Taq Polymerase (Invitrogen). The amplification conditions were denaturing at 95°C for 5 min, 30 cycles of denaturing at 95°C for 1 min, annealing at 56°C for 1 min, and extension at 72°C for 1 min, followed by a final extension at 72°C for 10 min. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as internal amplification control. For quantitative RT-PCR (qRT-PCR), real-time PCR amplifications were carried out using the 7500 Real-Time PCR System (Applied Biosystems). The amplification reactions were performed in triplicate in 96-well plates in 25 μl final volume that contained 1 μl of 10x diluted cDNA reaction volume, 12.5 μl of 2x Power SYBR Master Mix (Applied Biosystems), and 150 nM of each primer, forward and reverse (Table 1). The amplification conditions were 55°C for 2 min, 95°C for 10 min, followed by 40 cycles of 95°C for 30 s and 58°C for 1 min. qRT-PCR efficiency and specificity were evaluated using serial dilutions of the template cDNA and melting curve data. Transcript levels of the target genes were normalized using β-actin as an internal control. Relative gene-specific transcript levels were calculated by the 2-(ΔΔCt) method determined by the Applied Biosystems SDS 7500 software v1.4.
List of Primers Used for RT-PCR and qRT-PCR Assays
Alkaline Phosphatase (AP) Staining and Immunocytochemistry Fluorescence Analysis
iPS-MBMC colonies were AP stained with Alkaline Phosphatase Detection Kit (Millipore) according to the manufacturer's instructions. For immunocytochemistry, cells grown on Matrigel-coated coverslips were fixed with 4% paraformaldehyde at room temperature. After three washes in 10 mM phosphate-buffered saline (PBS) pH 7.4, cells were incubated in PBS with 5% normal goat serum (Invitrogen) and 0.3% Triton-X100 for 30 min at room temperature. Cells were then incubated overnight at 4°C with anti-OCT4, anti-NANOG, anti-acid sphingomyelinase (ASM), anti-α-fetoprotein (AFP), anti-TRA1, anti-stage-specific embryonic antigen (SSEA), or mouse anti-nestin (1:200; Millipore) antibodies in PBS with 0.3% Triton. After washing, secondary antibody cyanine 3 (Cy3)-conjugated goat anti-mouse IgG (1:1,000; Jackson, West Grove, PA, USA) in PBS 0.3% Triton-X 100 was added, and the cells were incubated for 2 h at room temperature. Following three washes with PBS, nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole; Sigma) for 3 min and the coverslips were mounted on slides using PPD (2,5-diphenyl-1,3,4-oxadiazole; Sigma). Fluorescence images were recorded using a digital camera attached to an inverted microscope (Carl Zeiss Apotome; Zeiss, GmbH, Germany).
EB Formation and In Vitro Differentiation
For embryoid body (EB) formation, iPS-MBMC colonies were harvested by digestion with collagenase IV (Invitrogen). The cell clumps were transferred to poly (2-hydroxyrthyl methacrylate)-coated dishes in DMEM/F12 containing 20% KSR, 2 mM l-glutamine, 0.1 mM nonessential amino acids, 0.1 mM β2-mercaptoethanol, and 0.5% penicillin and 0.5% streptomycin (all from Invitrogen). The medium was changed every 2 days. After 8 days as a floating culture, EBs were transferred to gelatin-coated plates and cultured in the same medium for another 8 days.
Results
iPSCs Can Be Generated From hMBMCs Efficiently and Rapidly with Three Factors
To reprogram these cells, a hMBMC culture was first established as described in methods. After three to four passages in vitro, the cells displayed a homogeneous morphology and all the characteristics of mesenchymal cells (not shown). We also investigated the gene expression profile of hMBMCs using RT-PCR to detect transcripts of several core pluripotency genes, including those used in the reprogramming experiments. As can be seen in Figure 1B, transcripts from OCT4, SOX2, KLF4, c-MYC, and NANOG, but not from telomerase reverse transcriptase (TERT), DNA (cytosine-5-)-methyl-transferase-3-β (DNMT3B), or NODAL, were detected in MBMCs. Although the profile is different from that of the ES cell line H9 (Fig. 1B), as expected, the presence of transcripts from some pluripotency marker genes could lead to a high reprogramming efficiency of MBMCs.
An outline of the reprogramming approach is shown in Figure 1A. MBMC cultures containing 104 cells were subjected to two rounds of retroviral transduction, at an interval of 24 h between rounds, with equal proportions of each of the OCT4-, SOX2-, and KLF4-containing recombinant retrovirus. One day after the second transduction, hMBMCs were replated onto inactivated MEF, and the medium was changed to ES cell culture medium. The ES cell medium was then replenished everyday. Interestingly, small cell clusters with a high nucleus-to-cytoplasm ratio appeared 5 days after the first transduction and typical ES cell-like colonies could be seen at day 10 after first transduction (Fig. 1C). These ES cell-like colonies reached the optimum size to be picked up around days 15–17 after the first transduction (Fig. 1C), which is a substantially shorter period when compared to that required to reprogram fibroblasts (17). The reprogrammed MBMCs were then referred to as iPS-MBMCs. In order to quantify the reprogramming efficiency, we performed AP staining of iPS-MBMCs at day 15. Figure 1D shows a typical 100-mm dish containing reprogrammed MBMC colonies after AP staining. After four independent reprogramming experiments, the reprogramming efficiency was then estimated by dividing the number of AP-positive colonies (which possessed cells with the high nucleus-to-cytoplasm ratio and the ES cell-like morphology) by the number of transduced MBMCs. The calculated reprogramming efficiency was approximately 2–5%, which is also higher than the efficiency of 0.01–0.1% achieved with fibroblasts (17). Thus, hMBMCs can be a good cell source for reprogramming since they can be obtained by noninvasive procedures.
Phenotypic and Molecular Characterization of iPS-MBMCs
Fifteen single iPS-MBMCs colonies were mechanically selected, and five were successfully expanded up to 10 passages. Each of the five iPS-MBMC lines exhibited and maintained a morphology distinct from the original MBMCs and resembling hES cells in morphology as well as in AP staining capacity (Fig. 2A, B). We chose three cell lines for further characterization as described in methods (clones 1, 3, and 7). The three iPS-MBMC lines propagated at a very similar rate but varied in the percentage of spontaneous differentiation during culture procedures, suggesting particular differences between them. Immunocytochemistry assays showed that they expressed the transcription factor genes OCT4 and NANOG as well as the surface protein genes SSEA-4 and TRA-1-60 (Fig. 2C). To assess the gene expression pattern in the iPS-MBMC lines, we isolated RNA from iPSCs cultured under feeder-free conditions. Figure 3 shows the detection and quantification by RT-PCR and qRT-PCR of transcripts from the endogenous pluripotency-associated genes OCT4, SOX2, and KLF4 using specific primers designed to detect the endogenous mRNAs of these genes. Transcripts from NANOG, DNMT3B, and TERT genes were detected in iPS-MBMCs (Fig. 3A) while undetectable in MBMCs (Fig. 1A). Moreover, similar to what has been previously described (17), the three exogenous genes OCT4, SOX2, and KLF4 (referred to as OCT4 tg, SOX2 tg, and KLF4 tg) were all silenced in iPS-MBMCs lines after 10 passages, as shown by RT-PCR assays using primer sets specifically designed to amplify the transgenes' mRNAs (Fig. 3B). GAPDH was used as internal amplification control. The transcript levels of the three iPS-MBMCs lines were evaluated by qRT-PCR. Robust transcript levels of the pluripotency-associated genes OCT4, NANOG, DNMT3B, SOX2, zinc finger protein 42(zfp42 or REX1) and developmental pluripotency associated 5 (DPPA5), normalized to that of β-actin (Fig. 3C), were observed, comparable to that of the H9 hES cell line and distinct from that of MBMCs. To exclude transforming events during MBMCs nuclear reprogramming, karyotype analysis of iPS-MBMCs was performed. The analysis showed that 85–90% of the cells have a normal karyotype (data not shown).
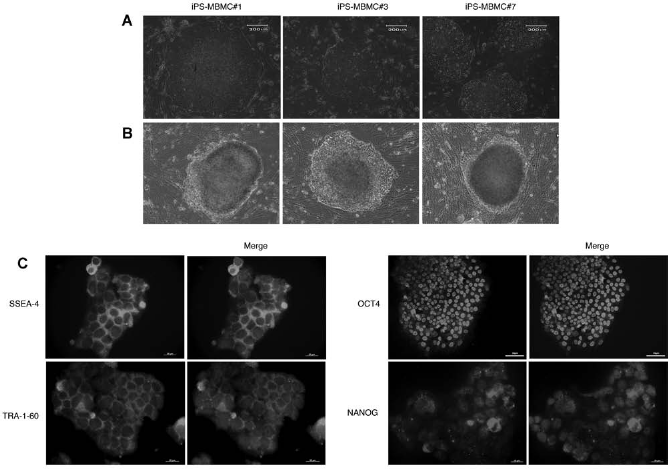
Phenotypic characterization of iPS-MBMC clones. (A) Phase-contrast micrographs of clones 1, 3, and 7 show that they have typical hES morphology. Scale bar: 200 μm. (B) Alkaline phosphatase staining showing the ES-like feature of these three iPS-MBMCs clones. Scale bar: 200 μm. (C) Immunocytochemistry of iPS-MBMCs colonies, counterstained by DAPI (blue), confirmed the presence of the ES cell markers TRA-1-60, SSEA-4, NANOG (scale bar: 20 μm), and OCT4 throughout the colonies.

Molecular characterization of iPS-MBMCs. (A) RT-PCR analysis of endogenous transcript profiles shows that an iPS-MBMC clone exhibits a similar profile to that of the hES cell line H9. GAPDH amplification was used as internal control. (B) Retroviral transgene silencing in iPS-MBMCs. Transgene-specific primers were used to detect the exogenous Oct4, Sox2, and Klf4 transcripts. iPS-MBMC 7d, a pool of colonies collected 7 days after transduction; iPS-MBMC #3 and #7, established clones 3 and 7, respectively; MBMC, original MBMCs; H9, H9 ESC line used as positive control. (C) Quantification of pluripotency markers transcript levels in iPS-MBMCs. qPCR was used to evaluate the expression of OCT4, NANOG, DNMT3B, SOX2, REX1, and DPPA5 in the three iPS-MBMC lines compared to their parental MBMC. H9 ES cells were used as calibrator sample, and reactions were normalized to that of β-actin transcript.
In Vitro Differentiation
In order to verify the developmental capabilities of iPS-MBMCs, we generated embryoid bodies (EBs) from all three iPS-MBMCs lines. In Figure 4A and B, it can be seen typical round-shaped EBs formed after 5 days in suspension culture. After 14 days, the expression of differentiation-specific genes was analyzed by RT-PCR. As shown in Figure 4C, transcripts from germ layer-specific marker genes were detected in the EBs, indicating differentiation to cell types from all three embryonic germ layers. In addition, immunostaining assays (Fig. 4D) show clearly the presence of nestin (ectoderm), smooth muscle actin (mesoderm), and α-fetoprotein (endoderm) in the EBs. These results indicate that iPS-MBMCs possess the ability to differentiate into cell types from all three germ layers in vitro.

Functional characterization of iPS-MBMCs. (A) Phase-microscopy of EBs from iPS-MBMC#7 after 5 days in suspension culture. Scale bar: 200 μm. (B) Typical cystic EBs from the same cell line. Scale bar: 200 μm. (C) RT-PCR analysis to detect the transcripts from endoderm, mesoderm, and ectoderm germ layers: Afp, brachyury, and nestin, respectively. GAPDH was used as internal control. (D) Immunostaining showing expression of the lineage markers nestin (ectodermal) (scale bar: 50 μm), smooth muscle actin (mesodermal) (scale bar: 20 μm), and α-fetoprotein (endodermal) (scale bar: 20 μm) in iPS-MBMCs clones subjected to differentiation.
iPS-MBMCs Already Possess a Pluripotent Character 10 Days of Posttransduction
Due to the fact that typical hES-like colonies appeared very early in our reprogramming experiments (Fig. 1C), we investigated whether these early iPS-MBMCs colonies (10 days of posttransduction) already possessed some of the pluripotency-associated characteristics. If confirmed this would represent an even more reduced time in iPSC generation. We therefore carried out immunocytochemistry staining for OCT4, NANOG, and TRA-1-60 in 10 days of posttransduction iPS-MBMC colonies. As can be seen in Figure 5A, these three pluripotency-associated proteins could be detected at the correct subcellular localization. Moreover, to verify whether the transcript levels of the two major pluripotency-associated genes OCT4 and NANOG in the 10 days of posttransduction iPS-MBMCs are similar to that of the 17 days of posttransduction iPS-MBMCs (time at which we usually picked up colonies from the plates), we measured by qRT-PCR the transcript level kinetics of these genes during the reprogramming procedure. Figure 5B shows NANOG and OCT4 transcript levels normalized to that of β-actin, in total RNA extracted from iPS-MBMCs 0, 5, 10, and 17 days of posttransduction and from hES cell line H9. Transcript levels at 0 day correspond to that of MBMCs. As can be seen in Figure 5B, there is a sharper increase in both NANOG and OCT4 transcript levels between 5 and 10 days of the reprogramming procedure. Also, transcript levels at 10 days of posttransduction are not much lower than those at 17 days of posttransduction or than those showed by the hES cell line H9. These results suggest that reprogramming to pluripotency may occur before 17 days of posttransduction.

Phenotypic and molecular characterization of 10-day iPS-MBMC colonies. Colonies formed 10 days after transduction were analyzed for the presence of typical ES cell markers. (A) Immunostaining assay of iPS-MBMC colonies, counterstained by DAPI, shows the expression of OCT4 and NANOG transcription factor genes throughout the colony nucleus, as well as surface marker TRA-1-60. (B) qRT-PCR analysis of endogenous OCT4 and NANOG transcript level kinetics during nuclear reprogramming. Total RNA was extracted from pools of colonies at each time point. The levels found in day 0 were used as calibrator samples, and reactions were normalized to that of β-actin transcript. H9 ESC line was used as positive control. Scale bars: 10 μm.
Discussion
In this study, we report the rapid and efficient generation of iPSCs from human menstrual blood-derived mesenchymal cells (hMBMCs) by the ectopic expression of OCT4, SOX2, and KLF4 genes. iPSC lines successfully characterized as pluripotent cells from MBMCs can be obtained and established as fast as 15–17 days after the first transduction. This makes hMBMCs, together with human keratinocytes, also reported to give rise to ES-like colonies in 10 days (1), the fastest cell types to reprogram. In routine reprogramming experiments, it is desirable to shorten the time needed for isolation of the individual iPSC colonies due to the limited time in which MEF cells are adequate as feeder cells, usually 1 week after inactivation. Moreover, the reprogramming efficiency of about 2–5% of hMBMCs, is 200–500 fold higher than that of human fibroblasts (17). Even though c-MYC is thought to accelerate nuclear reprogramming in fibroblasts, it is not necessary for efficient reprogramming of hMBMCs. Other cell types have also been reported to be reprogrammed faster and more efficiently than fibroblasts. For example, amniotic fluid-derived cells (AF) were reported to be reprogrammed twice as fast as skin fibroblasts and with an efficiency of 0.2% (6). AF cells express the reprogramming genes OCT4, SOX2, KLF4, and c-MYC but do not express NANOG, TERT, FGF4, growth differentiation factor 3 (GDF3), or DPPA5. The molecular mechanism underlying the speed and efficiency of reprogramming of these cells were not investigated, but the authors speculate that AF cells may have an embryonic-like epigenetic background, which may facilitate and accelerate pluripotency (6). A high reprogramming efficiency and speed was also reported for foreskin-derived keratinocytes (1). The authors showed that keratinocytes are reprogrammed with 100-fold higher efficiency and twofold faster compared to fibroblasts. They have also shown that keratinocytes contain higher levels of endogenous KLF4 and c-MYC transcripts when compared with fibroblasts and suggest it may be associated with transcriptional and epigenetic states favorable to keratinocyte reprogramming.
The molecular basis for the speed and efficiency of hMBMCs nuclear reprogramming is unclear. The possibility that the endogenous expression of some of the core ES marker genes, particularly those used for the transduction experiments, could make these cells more prone to nuclear reprogramming is a question that we are currently addressing. Moreover, it has also been described that a more relaxed chromatin structure is associated with regulatory elements of hES core genes in mesenchymal stromal cells (8,14). In any case, understanding the underlying mechanisms for the fast and efficient reprogramming of hMBMCs may shed some light on the molecular events governing nuclear reprogramming.
Conclusion
In this work, our findings provide evidence that MBMCs are an attractive cell source for nuclear reprogramming. By using three of the four Yamanaka's transcription factors (OCT4, SOX2, and KLF4), we were able to reprogram these cells with higher efficiency and in a shorter time when compared to other cell types. These cells can be used as a model to study the molecular events during nuclear reprogramming and to produce iPSCs with reduced time and cost.
Footnotes
Acknowledgments
We thank César Félix Schmidt and Cláudio Nunes Pereira for technical assistance. The present work was supported by grants from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Fundação de Apoio à Pesquisa do Estado do Rio de Janeiro (FAPERJ), and DECIT Ministério da Saúde, Brazil. The authors declare no conflicts of interest.