
Abstract
Cell therapy is a promising treatment modality to improve heart function in acute myocardial infarction. However, the mechanisms of action and the most suitable cell type have not been finally determined. We performed a study to compare the effects of mesenchymal stem cells (MSCs) harvested from different tissues on LV function and explore their effects on tissue structure by morphometry and histological staining for species and lineage relationship. MSCs from skeletal muscle (SM-MSCs) and adipose tissue (ADSCs) were injected in the myocardium of nude rats 1 week after myocardial infarction. After 4 weeks of observation, LVEF was significantly improved in the SM-MSCs group (39.1%) and in the ADSC group (39.6%), compared to the placebo group (31.0%, p < 0.001 for difference in change between groups). Infarct size was smaller after cell therapy (16.3% for SM-MSCs, 15.8% for ADSCs vs. 26.0% for placebo, p < 0.001), and the amount of highly vascularized granulation tissue in the border zone was significantly increased in both groups receiving MSCs (18.3% for SM-MSCs, 22.6% for ADSCs vs. 13.1% for placebo, p = 0.001). By in situ hybridization, moderate engraftment of transplanted cells was found, but no transdifferentiation to cardiomyocytes, endothelial cells, or smooth muscle cells was observed. We conclude that MSC injections lead to improved LVEF after AMI in rats predominantly by reduction of infarct size. After 4 weeks, we observed modulation of scar formation with significant increase in granulation tissue. Transdifferentiation of MSCs to cardiomyocytes or vascular cells did not contribute significantly in this process. MSCs from skeletal muscle and adipose tissue had similar effects.
Keywords
Introduction
Despite advances in revascularization and medical therapy in the last decade, acute myocardial infarction (AMI) and heart failure are still important causes of morbidity and mortality in industrialized countries (18). Although recent evidence suggest the presence of cardiac stem cells and a slow turnover of cells in the heart throughout life (5,6), AMI leads to a permanent loss of contractile elements and the subsequent formation of a fibrous scar.
Regeneration of contractile myocardium has been a target for cell therapy research for more than a decade (24). In the clinical setting, skeletal myoblasts and bone marrow cells have been used in different applications, but clinical trials have so far not provided the evidence needed for routine implementation of this approach. Mesenchymal stem cells (MSCs) are present in many tissues, can be expanded rapidly when cultured, and have been shown to differentiate also to muscular tissues, including smooth muscle cells and cardiomyocytes (2,4,28,38). MSCs tolerate hypoxia, secrete angiogenic factors, and have been shown to improve vascularization and perfusion (3,30). MSCs also lack constitutive surface expression of major histocompatibility complex (MHC) class II, making the cells less immunogenic in allotransplantation. Thus, MSCs have properties that suggest that they may be beneficial in AMI, chronic heart failure, and angina pectoris, perhaps also as allogenic off-the-shelf therapy in acute settings (1,9,37).
Previous studies have investigated the therapeutic effects of MSCs isolated predominantly from bone marrow and adipose tissue and have shown that both may stimulate myocardial regeneration and recovery of left ventricular (LV) function (20,21,25,27,31,35). However, the mechanisms responsible for the beneficial effects remain incompletely understood. Also, MSCs obtained from different tissues may have different regenerative capacity (16), but this has not been broadly investigated in the setting of intramyocardial injection after AMI. Consequently, we designed the present study to compare the effect of MSCs derived from human adipose tissue (ADSCs) with MSCs isolated from human skeletal muscle (SM-MSCs) following injection into the border zone and infarct area of immunodeficient rats 1 week after the induction of large myocardial infarctions. We used echocardiography to measure myocardial functionality and histological analyses combined with tissue morphometry to determine the size of the postinfarct scar, the density of blood vessels in the scar, the border zone, and the healthy myocardial tissues, and, finally, the fraction of the border zone identifiable as granulation tissue. In addition, we used fluorescence in situ hybridization (FISH) and immunohistochemistry to identify human cells in the rat heart and to determine if they had transdifferentiated into cells normally resident in the heart.
Materials and Methods
Isolation and Culture of Human Skeletal Muscle-Derived MSCs
Skeletal muscle tissue was obtained from the gracilis and semitendinosus muscles removed from patients undergoing surgery of the anterior cruciate ligament. The donors provided written informed consent, and the collection and storage of muscle tissue and SM-MSCs was approved by the regional committee for ethics in medical research (approval no. 1.2006.740). Ten to 20 g of skeletal muscle was washed three times in PBS containing 100 IU/ml penicillin, 100 IU/ml streptomycin, and 2.5 μg/ml amphotericin B (Sigma Aldrich, St. Louis, MO), minced with scissors, rewashed, and centrifuged at 100 x g for 5 min. The muscle fragments were digested for 60 min at 37°C in 1.5 mg/ml collagenase IA (Sigma) in 20–40 ml DMEM/F12 (Gibco, Paisley, UK). The digested fragments were centrifuged at 300 x g for 10 min and resuspended in 5–10 ml trypsin-EDTA (Sigma) in a new culture flask for 20 min at 37°C. The enzymatic reaction was stopped by adding 1 ml fetal bovine serum (FBS, Cambrex, Verviers, Belgium), and the cell solution was centrifuged at 300 x g for 10 min. The isolated cells were washed once and subsequently cultured in DMEM/F12 containing 20% FBS, amphotericin B, and antibiotics. On day 5, the medium containing nonadherent cells was discarded, and fresh culture medium was added. When the cells reached 50% confluence, plastic adherence was interrupted with trypsin-EDTA, and the cells were plated into new flasks at 5,000 cells/cm2. After the first passage (P1), amphotericin B was removed and 10% FBS was used instead of 20% for the duration of the cultures. Muscle cells were expanded up to P5 for two donors and P7 for one donor. At P2 and immediately before transplantation, CD56+ cells were removed using magnetic beads directly coupled to mouse anti-human CD56 monoclonal antibody (MAb) (Miltenyi Biotech, Bergisch Gladbach, Germany) and LS columns as described by the manufacturer (Miltenyi Biotech), leaving a CD56- population, which has previously been described as MSCs (8). Cell viability was always >90% (data not shown). Flow cytometry showed that no more than 3% of CD56+ cells were left in the suspension at P2 and last passage.
Isolation and Culture of Human Adipose Tissue-Derived MSCs (ADSCs)
Adipose tissue (AT) was obtained as part of routine liposuction procedures from healthy donors. The donors provided written informed consent, and the collection and storage of AT and ADSCs was approved by the regional committee for ethics in medical research (approval No. 2.2007.132). The stromal vascular fraction (SVF) was separated from AT as described previously (7). Briefly, lipoaspirate (300–1,000 ml) was washed repeatedly with Hanks' balanced salt solution (HBSS) (Life Technologies-BRL, Paisley, UK) containing 100 IU/ml penicillin, 100 IU/ml streptomycin, and 2.5 μg/ml amphotericin B and digested for 45 min on a shaker at 37°C using 0.1% collagenase A type 1 (Sigma). After centrifugation at 400 x g for 10 min, floating adipocytes were removed. The remaining SVF cells were resuspended in HBSS containing 2% FBS. Tissue clumps were allowed to settle for 1 min. Suspended cells were filtered through 100-μm and then 40-μm cell sieves (Becton Dickinson, San Jose, CA). Cell suspensions (15 ml) were layered onto 15 ml Lymphoprep gradient separation medium (Axis Shield, Oslo, Norway) in 50-ml tubes. After centrifugation (400 x g, 30 min), cells at the gradient interface were collected, washed, and resuspended in culture medium containing 10% FBS and antibiotics. Immediately after separation, ADSCs were separated from the remaining cells using magnetic cell sorting. Endothelial cells (CD31+) and leukocytes (CD45+) were removed using magnetic beads directly coupled to mouse anti-human CD31 and CD45 Mab (Miltenyi Biotech) and LS columns as described by the manufacturer. Flow cytometry showed that no more than 5% of CD31+ and CD45+ cells were left in the suspension. Cells were washed, resuspended and seeded in DMEM/F12 containing 20% FBS and antibiotics. On day 7, culture medium containing nonadherent cells was discarded, and fresh culture medium was added. When the cells reached 50% confluence, plastic adherence was interrupted with trypsin-EDTA, and the cells were plated into new flasks at 5,000 cells/cm2. After P1, amphotericin B was removed and 10% FBS was used instead of 20% for the duration of the cultures. ADSCs from three donors were expanded to P5. Prior to injection, cell viability was always >90% (data not shown).
Verification of the Cells as MSCs by Differentiation and Flow Cytometry Studies
To verify that the cultured cells were actually MSCs, SM-MSCs and ADSCs were differentiated along adipogenic and osteogenic lineages and analyzed by flow cytometry for MSC markers. For flow cytometric analysis, cells were incubated with directly conjugated antigen-specific or -irrelevant monoclonal antibodies (Mabs) at room temperature for 20 min, washed with PBS, and fixed in 1% paraformaldehyde. Mabs used were CD56/PE (phycoerythrin), CD105/APC (allophycocyanin), CD44/PE, human leukocyte antigen (HLA) ABC/Cy-Chrome, CD34/FITC (fluorescein isothiocyanate), CD45/FITC (all BD Biosciences, CA), CD105/APC and HLA DR/APC (Diatec, Oslo, Norway), 144/PE (eBioscience, http://www.ebioscience.com/), and CD146/FITC (AbD Serotec, Kidlington, UK). Cells were analyzed using a FACSCalibur flow cytometer (Becton Dickinson). For adipogenic differentiation, confluent cultures were incubated in DMEM/F12 containing 10% FBS, 0.5 μM 1-methyl-3 isobutylxanthine, 1 μM dexamethasone, 10 μg/ml insulin (Novo Nordisk, Copenhagen, Denmark), and 100 μMindomethacin (Dumex-Alpharma, Copenhagen, Denmark). For osteogenic differentiation, cells were incubated at 3,000 cells/cm2 in DMEM/F12 containing 10% AS or FBS, 100 nM dexamethasone, 10 mM β-glycerophosphate, and 0.05 mM l-ascorbic acid-2-phosphate. Fresh differentiation medium was replaced every third day. After 4 weeks, differentiated cells were examined by real-time (RT)-PCR and staining procedures. For real-time RT-PCR, total RNA was extracted from cells using Trizol reagent (Invitrogen, Carlsbad, CA). Following DNase treatment (Ambion, Austin, TX), RNA was quantified by spectrophotometry (Nanodrop, Wilmington, DE). Reverse transcription (RT) was performed using the High-Capacity cDNA Archive Kit (Applied Biosystems, Abingdon, UK) with 200 ng total RNA per 20 μl RT reaction. Relative quantification (RQ) was performed using the 7300 Real-Time RT PCR system (Applied Biosystems) and Taqman® Gene Expression assays following protocols from the manufacturer (Applied Biosystems). Taqman assays used were for osteomodulin (OMD; Hs00192325_m1) and peroxisome proliferator-activated receptor gamma (PPARG; Hs01115513_m1). All samples were run in triplicates (each reaction: 1.0 μl cDNA, total volume: 25 μl). The thermo cycling parameters were 95°C for 10 min followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. All samples were scaled relative to the expression level of glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Taqman assay no Hs99999905_m1), which was used as endogenous control. For staining procedures, cells were fixed with 4% formalin and subsequently incubated for 10 min with Oil-Red O and Alizarin Red S to visualize lipid droplets and calcium deposition, respectively.
Animal Model and Surgical Procedures
NiH rnu/rnu (Charles River, France) nude, athymic rats were bred at the Oslo University Hospital, Rikshospitalet animal facility. Animals were maintained in a specific pathogen-free environment in positive pressure rooms with a standard 12-h day/12-h night cycle. Eighty-two animals were included in the study, as 18 surviving animals in each group were required to achieve sufficient power.
Rats aged 8–11 weeks were anesthetized with 1% isoflurane, intubated, and ventilated by a rodent ventilator with 1% isoflurane in a mixture of 1/3 O2 and 2/3 N2O. A left thoracotomy was made in the fifth intercostal space, and the proximal portion of the left anterior descending (LAD) coronary artery was rapidly ligated by an intramural suture to induce anterior wall myocardial infarction (26,29). The chest was closed, 0.05 mg/kg buprenorphine was administered subcutaneously for analgesia, and animals were monitored for 1 h. On day 6 post-MI, rats were sedated with 1.2 % isoflurane inhalation, and transthoracic echocardiography was performed. Animals with LV fractional shortening (FS) < 25% were selected for further studies. The following day (day 7), animals were randomized to receive ADSCs, SM-MSCs, or placebo (cell growth medium without cells). Sedation, intubation, and anesthesia were performed as described. The chest was reopened by sternotomy, the infarct zone localized, and a total of 3 × 106 ADSCs or 3 × 106 SM-MSCs suspended in 150 μl medium or placebo (150 μl medium only) was injected with a 30-G needle as three equal aliquots in the border zone and one in the infarct zone. The chest was then closed and desufflated through an 18-G drain. Postoperative analgesia and monitoring were performed as described above. Four weeks after cell injection (day 35), repeated echocardiography was performed. Animals were then euthanized, and the hearts were removed for histological analysis.
Echocardiography
Transthoracic echocardiography was performed on sedated animals in supine position with a Vivid 7 scanner and a 14-MHz Linear Array Probe (GE Vingmed Ultrasound, Horten, Norway). Parasternal long-axis and two short-axis cine loops (midpapillary and basal) were acquired. M-mode registration including LV wall thickness, endocavitary diameter, and fractional shortening (FS) was obtained at the midventricular level. LV ejection fraction (LVEF) was calculated by the area–length method from the long-axis cine loop. We also calculated a wall motion score index (WMSI) in a 13-segment model using the long-axis and two short-axis cine loops (22). All analyses were performed blinded to treatment allocation. Measurements were performed on three consecutive heart cycles and averaged.
Histological Analysis
Hearts were fixed with 4% phosphate-buffered formalin for 24 h, dehydrated, and embedded in paraffin. Infarct size was assessed in 12 random hearts from each group, while assessment of vascular density and border zone quantification were performed in 10 random hearts from each group. The sample sizes were based on power calculations using the results from a preliminary analysis of four animals in each group.
Hearts were cut on a Leica microtome to obtain a stack of 3-μm short-axis sections covering the entire LV with 1-mm gap between sections for estimation of both infarct size and vascular density. For estimation of infarct size, we used Masson–Goldner staining to differentiate scar tissue (green-colored collagen) from cardiomyoctes (red/pink). Morphometry was performed by point counting images collected at 12.5x magnification on a randomly superimposed 250 × 250- μm grid, and intersections were recorded as hits on infarct zone or not (Fig. 1). The percentage infarct area on each section was determined by the number of infarct zone intersections on the grid divided by the total number of intersections. The sum of scores for all slides covering the entire LV was used to estimate the total infarct volume (13). To quantify the granulation tissue in the border zone, we obtained one 100x magnification image from the border zone on both sides of the infarction (Fig. 2). Granulation tissue was defined as loose connective tissue, rich in blood vessels. A morphometric analysis was performed on a randomly superimposed 150 × 150 μm grid, dividing intersections with granulation tissue (hits) by the total number of myocardial intersections (hits + nonhits) on the grid. Cardiomyocytes and collagenized scar tissue were excluded from the granulation tissue. The pericardial zone and the right ventricle were not included in the analysis.
Tissue sections stained with Masson Goldener without (A) and with (B) a superimposed grid for calculation of infarct size (magnification: x12.5). Viable myocardium is stained red, and the hits are marked with green small circles. Scar tissue is stained green, and the hits are marked with blue small circles. Representative images (magnification: x100) from the border zone in the mesenchymal stem cell (MSC) group (A) and placebo group (B). Hematoxylin staining (blue) and anti-vascular endothelial (VE)-cadherin with peroxidase (brown).
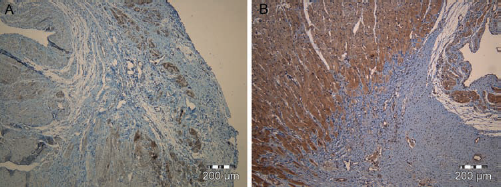
To assess vascular density, sections were stained with goat anti-mouse vascular endothelial (VE) cadherin primary antibodies (R&D Systems, Minneapolis, MN), biotinylated rabbit anti-goat secondary antibodies (Dako, Glostrup, Denmark), and streptavidin peroxidase (Dako). Hematoxylin was used for contrast staining. Morphometry was performed on images collected at 400x magnification on a 15 × 15 μm randomly superimposed grid. Two representative fields in the infarct zone (scar tissue), border zone, and remote myocardial segments were counted on six representative sections from 10 random animals in each group. The number of intersections on vessel lumen and wall was divided by the total number of intersections on the relevant tissue on the section to obtain the density of VE cadherin-positive vessels. As larger vessels (diameter > 50 μm) were rare events within the myocardium and the scar tissue, these were not included in the analysis.
For identification of the transplanted cells, we performed FISH with a FITC-conjugated probe specific for the human Alu sequence (Alu-positive control probe, Ventana, Tucson, AZ) on sections incubated in a Ventana Discovery machine according to the manufacturer's instructions. The sections were subsequently qualitatively evaluated by fluorescence microscopy.
To investigate the possibility of in situ differentiation of transplanted cells, tissue sections with Alu-positive cells were counterstained with myocardium-, smooth muscle-, and endothelium-specific antibodies. Primary mouse anti-human Mabs specific for anti-smooth muscle actin (SMA, Dako), anti-desmin (Dako), anti-CD31 (Dako), and anti-Nkx 2.5 (R&D Systems) and Alexa 555-conjugated anti-mouse secondary antibodies (Molecular Probes, Eugene, OR) were used. We also applied rabbit anti-Troponin I primary antibody (Abcam, Cambridge, UK) with biotinylated goat anti-rabbit (Vector Labs, Burlingame, CA) and Alexa 594-conjugated streptavidin (Molecular Probes). Nuclei were stained with DAPI. Sections from human hearts and hearts from rats in the placebo group were used as positive and negative controls, respectively.
An Olympus microscope with AnalySIS software (OlympusSIS, Münster, Germany) was used for morphometry. Multiplane fluorescence microscopy was performed on a Zeiss Axioplan 2 microscope (Göttingen, Germany) with ISIS software (Metasystems, Altlussheim, Germany). All image analyses were performed blinded regarding treatment allocation.
Statistics
Data are presented as mean ± SD. Values are presented for all animals included at baseline and all animals surviving to follow-up, respectively. All continuous data were analyzed by one-way ANOVA with Bonferroni correction for multiple comparisons. Categorical variables were analyzed by chi-square tests. SPSS version 16.0 software was used. Statistical significance was assigned if p < 0.05.
Results
Animals and Cells
The three groups of rats were well matched for physiological characteristics except for weight at entry into the study, where the animals in the placebo group had marginally higher body weight (Table 1). No significant difference in weight change or mortality was found during the study. The verification of the CD56- phenotype of the SM-MSCs is shown in Figure 3. Both ADSCs (7) and SM-MSCs were verified as bona fide MSCs by expression of CD105, CD73, CD90, and HLA class I and failure to express CD34, HLA DR, and hematopoietic markers CD45, CD14, CD3, and CD19 prior to injection (Fig. 3) (7). Also, both cell populations from all donors showed robust differentiation toward adipogenic and osteogenic lineages (Fig. 4). For unknown reasons, the staining for mineralization following osteogenic differentiation of SM-MSCs from donor 1 was only patchy, but the upregulation of osteomodulin following differentiation of these cells was robust and similar to that observed for the other SM-MSC donors (Fig. 4A).
Characterization of the skeletal muscle (SM)-MSCs by flow cytometry. Histograms in one column represent results from each one of the three donors. Tracings represented by thick lines; no fill are from cells incubated with irrelevant control antibodies. Tracings represented by thin lines; gray fill are from cells incubated with antigen-specific antibodies. HLA, human leukocyte antigen. Assays for osteogenic and adipogenic differentiation of SM-MSCs and adipose tissue-derived MSCs (ADSCs) performed after 4 weeks in differentiation medium. Quantitative RT-PCR for osteomodulin (OMD) (A) and peroxisome proliferator-activated receptor γ (PPARG) (B) were performed on undifferentiated and differentiated cells from three donors (D1–D3) each for SM-MSC and ADSC (different donors for the two cell populations). All results presented are scaled relative to the expression level of glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Staining assays (C) were for calcium deposition by Alizarin Red S to show osteogenic differentiation (top) and lipid droplets by Oil-Red O to show adipogenic differentiation (bottom) on cells from the same donors. The 200-μm scale bar in the lower right image is representative for all images in the panel. Animal Characteristics Values are mean ± SD or number (percentage). p Values by one-way ANOVA, except for mortality where the chi-square method was used. SM-MSCs, skeletal muscle-derived mesenchymal stem cells; ADSCs, adipose tissue-derived stem cells.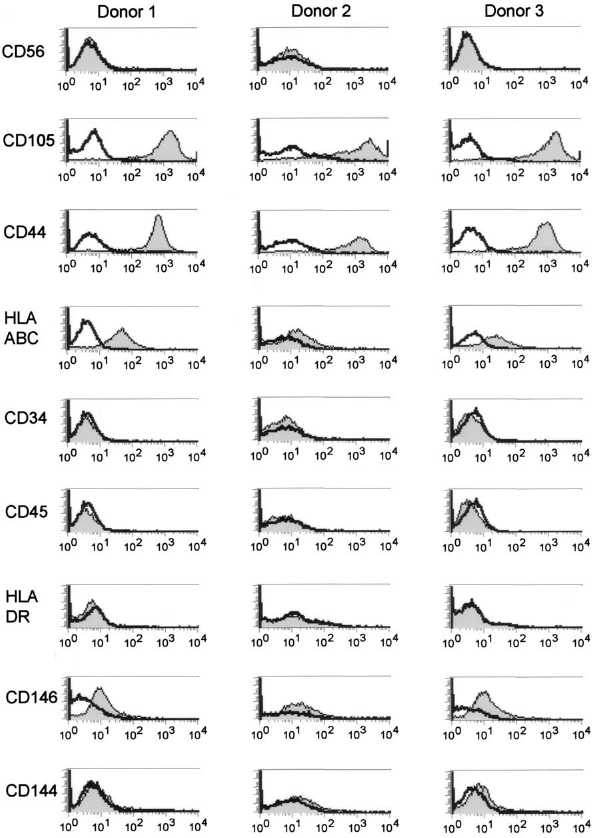


Echocardiography
LV dimensions and function were similar between groups at baseline, with mean LVEF 33 ± 10% 1 week after ligation of LAD indicating substantial myocardial infarctions (Table 2). During 4 weeks of follow-up, we observed significant remodeling with 1.3 ± 0.8 mm (p < 0.001) increase in left ventricular end-diastolic diameter (LVEDd). LV systolic function as measured by FS improved by 1.9 percentage points (%) in the ADSC-treated group (p = 0.08 vs. placebo) and by 2.8% in the SM-MSC group (p = 0.01 vs. placebo), compared to a 1.2% decrease in the placebo group (Table 2, Fig. 5). LVEF increased 2.5% in the ADSC group (p = 0.03 vs. placebo) and 4.9% in the SM-MSC group (p = 0.002 vs. placebo), compared to a 3.9% decrease in the placebo group. There was also a trend (p = 0.09) suggesting improved WMSI in the groups receiving cell therapy. The relative improvement in LV function tended to be better in the SM-MSC group compared to the ADSC group, but the difference did not reach statistical significance.
Left ventricle (LV) function by echocardiography. FS, fractional shortening; LVEF, left ventricular ejection fraction. *See Table 2 for details. Left Ventricular Function Measured by Echocardiography Values are mean ± SD. p Value by ANOVA. LVEDd, left ventricular end-diastolic diameter; LVESd, left ventricular end-systolic diameter; FS, fractional shortening; LVEF, left ventricular ejection fraction; WMSI, wall motion score index. p = 0.01 between SM-MSCs and placebo, p = 0.08 between ADSCs and placebo. p = 0.002 between SM-MSCs and placebo, p = 0.03 between ADSCs and placebo.

Morphometry
Tissue Morphometrics

Values are mean ± SD.
p < 0.001 between SM-MSCs and placebo, p < 0.001 between ADSCs and placebo, and p = 1.00 between SM-MSCs and ADSCs.
p = 0.02 between SM-MSCs and placebo, p < 0.001 between ADSCs and placebo, and p = 0.05 between SM-MSCs and ADSCs.
Vascular Density

Values are mean ± SD.
p = 0.01 for difference between SM-MSCs and placebo, p = 0.03 for difference between ADSCs and placebo. For SM-MSCs versus ADSCs, p = n.s.
In Situ Hybridization and Immunohistochemistry
In the transplanted hearts, a small number of Alu-positive human cells were retrieved in clusters, mainly in the peripheral region of the infarct zone and in the border zone (Fig. 6). No donor-derived cells were found within the preserved myocardium. The Alu-positive cells did not stain positive for SMA, desmin, CD31, TnI, and Nkx 2.5, indicating that differentiation to cardiomyocyte, smooth muscle cell, or endothelial cell phenotypes had not occurred.
Alu sequence-positive cells by fluorescence in situ hybridization (FISH). (A) Cluster of Alu-positive cells in the infarct zone. Blue represents nuclei stained with DAPI. Green and blue merged (bright green) represents transplanted cells stained with DAPI and Alu probe. Magnification: x200. (B) Same as in (A). Magnification: x600.
Discussion
In this study, intramyocardial injection of both ADSCs and SM-MSCs 1 week after AMI led to a substantial decrease in infarct size and a significant improvement in LVEF compared with injections of cell culture medium only. The proportion of granulation tissue within the border zone was significantly increased in both treatment groups. There was a trend for better functional improvements in the SM-MSC-treated group compared to the ADSC group, but this did not reach significance. A small number of transplanted cells were still present after 4 weeks, but they did not express markers of differentiation toward cardiomyocyte, endothelial, or smooth muscle lineages.
Our study confirms the favorable effect of MSC injections after AMI reported by other groups (9,19,20,27). We also show that the reason why the cell-treated animals maintained greater myocardial contractile power was that, following induction of the same-size anterior wall myocardial infarctions, the MSC-treated rats developed a much smaller scar area at 4 weeks than the placebo-treated animals. In fact, the difference in infarct size between the cell-treated and placebo-treated rats was even higher than expected from the differences in LV function. Our data suggest that this may in part be explained by a larger amount of granulation tissue in the border zone in groups receiving MSCs. The granulation tissue was immature, as the content of fibrous elements was low. Cardiomyocytes were not observed in the granulation tissue. The granulation tissue was not included in the parameter “infarct size,” as it stained Masson Goldener negative, but we believe that this tissue would eventually have turned into fibrous scar tissue. Even so, the injection of human MSCs must have led to a considerable reduction in the size of the infarct.
As could be predicted, the density of small blood vessels in the granulation tissue was much higher than that observed for the healthy myocardium, with no difference observed between the granulation tissue in the three groups. However, the amount of granulation tissue was significantly higher in the borderzone in cell-treated hearts. The MSCs were injected 1 week after induction of the myocardial infarction, at a time when highly vascular granulation tissue had presumably already been established in parts of the infarct zone. Within and also bordering this granulation tissue, there may have been cardiomyocytes affected by ischemia that were not yet dead and endogenous cardiac progenitor cells that might have promoted cardiac regeneration. MSCs are known to secrete a large number of bioactive compounds. Some of these may have acted on ischemic cardiomyocytes to promote their survival and on progenitor cells to stimulate cell division and differentiation. In the process, the formation of scar tissue also seems to have been inhibited. Such a scenario would lead to the survival of more contractile myocardial tissue, to a larger amount of persisting granulation tissue, and to smaller areas of scar tissue in the cell-treated hearts. This line of events would explain our observations. Some of the molecules known to be secreted by MSCs that might contribute in this model are prostaglandin E2, known to inhibit fibrosis in other systems (32), and the anti-inflammatory factor tumor necrosis factor (TNF)-α-induced protein 6, which is thought to mediate the effect on myocardial infarction induced by intravenous injection of MSCs (17). One observation that might speak against our hypothesis is the small number of Alu-positive cells noted in the rat hearts after 4 weeks. Functional and morphometric differences on the scale observed in the current study would intuitively seem to require a large number of surviving cells. However, similar effects on LV function was reported in the study by Imanishi et al., although virtually all the transplanted cells were lost within 4 weeks (15). One possible explanation to this observation could be that a much larger number had survived initially and that these cells gradually succumbed during the following period, as indicated in cell-tracking studies (15,34,36). If so, enough cells may have been present to secrete bioactive compounds for a sufficient period of time to affect the surrounding tissues. This hypothesis is supported by the observation that, when MSC survival was enhanced by the transduction of the anti-apoptotic akt gene, the effects of intramyocardial injection of MSCs on cardiac function and infarct size were also enhanced (11). Several factors may influence the number of cells observed. The cells must be handled properly before injection, as detachment from the plastic surface rapidly changes the MSC morphology and may trigger anoikis. As the heart is small and contracting at 3–400 beats/min, some cells are probably also lost by myocardial perforation and endoventricular injections. Also, the intramural pressure during systole is high, which may facilitate extrusion of injected cells (34). Although MSCs are immunoprivileged, alloimmune responses may influence cell survival. Grinnemo et al. reported lymphocyte infiltration and rapid loss of cells after MSC therapy in nude rats, suggesting alloimmune rejection (12). Aggregation of lymphocytes was not observed in proximity to Alu-positive cells in our study, and cell survival may have been favored by the use of young animals, as nude rats improve cellular immune competence with age. However, we believe that our observation of surviving cells at 4 weeks is reliable. Membrane markers used to identify human cells, like DiI or PKH 26, may persist in the tissue after the cells have died because membrane fragments may be phagocytosed by cells in the vicinity or fragments of the stained membrane may fuse with the membrane of neighboring cells. Similarly, following transplantation of cells transfected with green fluorescence protein, the fluorescent proteins may leak from the transfected cells to be taken up by nearby host cells, which may then give a false impression of surviving cells long after the transplanted cells have actually died. In contrast, no study has to date shown a false-positive signal for surviving cells using the human-specific Alu probe employed in this study.
Based on very early studies of transplantation of cells between organs, the hypothesis has been advocated that committed cells from one organ might transdifferentiate to a phenotype typical of another, when placed into the microenvironment of that organ, particularly in the context of organ damage (10,14). We did not see evidence that the transplanted MSCs transdifferentiated to become human-derived cardiomyocytes or other cardiac cells that might have contributed to the functional and tissue structural effects observed here. Only a limited number of studies have reported post-transplant expression of cardiomyocyte markers, CD 31 or SMA in vivo. The ex -tent of cell engraftment and survival has been low, and only a small fraction of engrafted cells have differentiated toward other lineages (23,30,33). Thus, although we cannot exclude that transdifferentiation may take place in rare individual cells or after longer observation periods, this is clearly not the mechanism behind the functional and structural improvements following MSC injection observed in the present study.
Finally, this study was also undertaken to determine if MSCs isolated from different organs would result in different functional outcomes. We and others have shown that this is the case when MSCs are used to generate cartilage in vitro (16). Unfortunately, this question remains unanswered after the current study. Although there was a trend toward better results in the functional tests for the animals treated with SM-MSC, this did not reach statistical significance upon direct comparison and was not reflected in the morphometric data. Other questions left unanswered are the optimal time delay between the onset of acute myocardial infarction and the injection of the MSCs and whether there is a functional difference between using MSCs from isogenic or genetically different individuals. Still, at this time, MSCs appear as a potentially interesting adjuvant treatment modality for selected patients following acute myocardial infarction.
Footnotes
Acknowledgments
The authors would like to thank Linda Dorg for valuable assistance in histological staining procedures, and Klaus Beiske, M.D. for providing and supervising the use of the Zeiss microscope and ISIS software. J. O. Beitnes was funded by a research fellowship from the Norwegian Council on Cardiovascular Diseases. The study was further supported by a Storforsk grant from the Norwegian Research Council and by a grant from Rikshospitalet Research Council. The authors declare no conflicts of interest.