
Abstract
The development of cell- and gene-based strategies for regenerative medicine offers a therapeutic option for the repair and potential regeneration of damaged cardiac tissue post-myocardial infarction (MI). Human umbilical cord subepithelial cell-derived stem cells (hUC-SECs), human bone marrow-derived mesenchymal stem cells (hBM-MSCs), and human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs), all derived from human tissue, have been shown to have in vitro and in vivo therapeutic potential. Additionally, S100a1, VEGF165, and stromal-derived factor-1α (SDF-1α) genes all have the potential to improve cardiac function and/or effect adverse remodeling. In this study, we compared the therapeutic potential of hBM-MSCs, hUC-SECs, and hiPSC-CMs along with plasmid-based genes to evaluate the in vivo potential of intramyocardially injected biologics to enhance cardiac function in a mouse MI model. Human cells derived from various tissue types were expanded under hypoxic conditions and injected intramyocardially into mice that had undergone left anterior descending (LAD) artery ligation. Similarly, plasmids were also injected into three groups of mice after LAD ligation. Seven experimental groups were studied in total: (1) control (saline), (2) hBM-MSCs, (3) hiPSC-CMs, (4) hUC-SECs, (5) S100a1 plasmid, (6) VEGF165 plasmid, and (7) SDF-1α plasmid. We evaluated echocardiography, hemodynamic catheterization measurements, and histology at 4 and 12 weeks post-biologic injection. Significant improvement was observed in cardiac function and contractility in hiPSC-CM and S100a1 groups and a significant reduction in left ventricle scar within the hUC-SEC group and a slight improvement in the SDF-1α and VEGF165 groups compared to the control group. These results demonstrate the potential for new biologic therapies to reduce scar burden and improve contractile function.
Keywords
Introduction
Myocardial infarction (MI) is characterized by extensive cardiomyocyte apoptosis and eventual functional degradation of the cardiac tissue due to a coronary occlusion. Postinfarction, apoptosis drives a cascade of dynamic biochemical and morphological changes resulting in the formation of nonfunctional scar tissue in place of the infarcted myocardium in a process referred to as cardiac remodeling 1 . To date, there are no approved therapeutic solutions available to reverse fibrotic scar formation or replace scar tissue with new cardiomyocytes. Current pharmacological and surgical measures are limited to palliative effects. Heart transplantation, the only long-term treatment available, is limited by the shortage of donor hearts, the high associated costs, and the requirement for immunosuppression 2 . The roles of both gene- and cell-based therapies to treat ischemic heart disease have been increasingly studied over the past 25 years. In 1990, Lin et al. demonstrated the feasibility of gene delivery via direct intramyocardial injection 3 . In 2001, Orlic et al. 4 demonstrated that bone marrow-derived mesenchymal stem cells (BM-MSCs) differentiated into cardiomyocytes after coronary artery ligation in a murine model. These important observations, although early and limited in their results, formed the foundation for cardiac biological therapies aimed at redirecting the heart's postinfarct response toward a more efficient myocardial repair and regeneration system5–9. On the basis of these and other important developments, we introduce the rationale for cell- and gene-based approaches for cardiac repair.
Currently, there are a number of cell types that are being investigated as therapeutic agents that may be useful for cardiac repair or regeneration 10 . These cells include embryonic stem cells (ESCs), human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs), and adult stem cells (ASCs). ESC and hiPSC-CM cell types, although highly potent and organ specific, are plagued with the potential for teratoma formation posttransplant. Thus, additional regulatory guidelines for safety have slowed these cell types in their therapeutic applications 11 . ASCs can be isolated from a number of different tissues and organs. Mesenchymal stem cells (MSCs), a type of ASC, have been utilized for cardiac cell therapy due to their immunomodulatory role that enables major histocompatibility complex (MHC)-mismatched allogeneic transplantation 12 . MSCs are present in nearly all postnatal tissues and organs, including bone marrow 13 , adipose tissue 14 , and umbilical cord tissue 15 . Their self-renewing and immunomodulatory effects as well as the capability to differentiate into cells of mesenchymal lineage, including cartilage, muscle, and vascular endothelial cells, make MSCs prime candidates for allogeneic cell-based therapeutics. BM-MSCs have been shown to aid in cardiac repair in preclinical models, with associated improvements in heart function and decreased scar formation posttransplantation16–20. The mechanisms involved in MSC-induced cardioprotective effects appear to function in concert involving: (a) limited transdifferentiation/fusion21–24, (b) decrease in adverse cardiac remodeling25–27, and (c) improved angiogenesis28–30. These cardioprotective events are believed to be a result of limited cell engraftment into the host myocardium 31 and mostly paracrine effects via secreted factors 32 .
We have previously identified a novel MSC population isolated from the subepithelial lining of umbilical cord tissue (UC-SECs) with unique immunomodulatory properties, which may be useful in the treatment of ischemic heart disease 15 . In order to better understand the potential of novel adult cells for treating ischemic heart disease, we investigated the effect of direct transplantation of BM-MSCs, hiPSC-CMs, and UC-SECs in an MI model in immunodeficient mice as previously reported 33 . In addition to cell therapy options for cardiac repair, genes have also been studied for this application. A number of gene-based approaches, both viral and nonviral, are reported for use in cardiac repair with limited but positive results34,35. As to limit the potential immunogenicity related to viral-based clinical translation, our goal has been to utilize plasmid-based approaches for gene delivery36,37.
Vascular endothelial growth factor (VEGF) has many isoforms that have been used for preclinical and clinical programs to promote angiogenesis36,37. The most interesting isoform is VEGF165, which when administered in an adenoviral construct was found to have clinical benefit in patients with myocardial ischemia36,37. Along with angiogenesis, the potential for improving intracellular calcium function for inotropic response is very relevant in ischemic heart failure36,37.
The use of sarcoplasmic endoplasmic reticulum calcium (Ca2+) ATPase (SERCA-2a) has been translated into patients but has failed to have a clinically meaningful response in a large randomized trial 38 . Other genes such as S100a1, which is also involved in calcium signaling, has had more promising data in studies with large animal models of ischemic heart failure 39 . However, the S100a1 gene has not been tested as a stand-alone plasmid. Gene-based approaches are more targeted than cell-based therapies and can be used to recruit cells to the site of myocardial injury. The use of stromal-derived factor-1α (SDF-1α) to recruit CXCR4+ cells, its natural receptor, has been demonstrated in preclinical and clinical trials to have benefit in ischemic heart disease and heart failure40,41. To date, multiple human cells and human genes have been utilized in preclinical and clinical therapies but have not been compared in terms of safety and efficacy. Our goal is to simultaneously compare the cells and genes in a preclinical ischemic heart model.
Materials and Methods
Umbilical Cord Tissue and Cell Procurement
Human umbilical cord tissues and peripheral blood (PB) were obtained from the University of Utah with institutional review board (IRB) approval. Human bone marrow mesenchymal stem cells (hBM-MSCs) were obtained from iBiologics (Phoenix, AZ, USA).
Umbilical Cord Tissue Processing
Umbilical cord tissue was washed with Hanks' balanced salt solution (HBSS) (GE Healthcare Life Sciences, Logan, UT, USA). The cords were dissected; care was taken to remove the artery, veins, and Wharton's jelly (gelatinous material). The remaining subepithelial layer was then placed into Dulbecco's phosphate-buffered saline (DPBS) containing 10% XcytePLUS Supplement (iBiologics).
Derivation of MSCs From Umbilical Cord Tissue
The subepithelial layer was cut into 3 × 3-mm sections and placed into six-well dishes. A 22 × 22-mm sterile coverslip (Thermo Fisher Scientific, Pittsburgh, PA, USA) was placed over the subepithelial layer. Explants were cultured in Dulbecco's modified Eagle's medium (DMEM) (Life Technologies, Carlsbad, CA, USA), 10% XcytePLUS Supplement (iBiologics), 1× GlutaMAX, and 1× minimum essential medium (MEM) nonessential amino acids (NEAA) (Life Technologies) and cultured in 5% CO2/5% O2 at 37°C 15 .
Cell Culture of MSCs From Umbilical Cord Tissue
After 2 days, the cells were 70% confluent and passaged using trypLE (Life Technologies). Cells were passaged into T225 flasks (Thermo Fisher Scientific) at a density of 1,000 cells/cm2 and cultured in 5% CO2/5% O2 at 37°C.
In Vitro Differentiation of Subepithelial-Derived MSCs
In vitro differentiation toward the cardiac lineage was performed by exposing passage 4 cells to a cardiogenic cocktail as previously described 42 . Briefly, high-glucose DMEM containing cardiogenic growth factors [2.5 ng/ml transforming growth factor-β (TGF-β), 5 ng/ml bone morphogenetic protein 4 (BMP4), 5 ng/ml activin A, 10 ng/ml fibroblast growth factor 2 (FGF2), 100 ng/ml cardiotropin, and 1 U/ml α-thrombin] was used to differentiate the cells toward a cardiac lineage for 15 days in 5% CO2/5% O2 at 37°C.
Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes
Using the modified technique described recently by the Fukuda and Wu laboratories in Japan and the US, respectively43,44, our team has reproduced hiPSCs utilizing PB cells 45 . We have evolved this technique to use a non-integrating Sendai virus to reprogram CD71+ PB iPSCs from healthy donors and then differentiate them into functional cardiomyocytes. Briefly, CD71+ cells were obtained from PB mononuclear cells (PB-MNCs), in the presence of Fms-like tyrosine kinase 3 (FLT-3), interleukin-6 (IL-6), stem cell factor (SCF), and thrombopoietin (TPO) according to Thermo Fisher Scientific guidelines for PB-MNC reprogramming using the CytoTune-iPS Sendai Kit (Thermo Fisher Scientific). All somatic cell samples were tested and cleared for the presence of mycoplasma and endotoxin. Cells were transitioned to Essential 6 medium (Life Technologies) containing basic fibroblast growth factor (bFGF; also known as FGF2) on day 7 and to Essential 8 medium (Life Technologies) on day 10. After successful reprogramming, colonies were observed by day 10 and subsequently picked on days 18–25. A minimum of six colonies were picked from each line, and one colony per well was cultured on Matrigel-coated plates (Corning Inc., Corning, NY, USA) in the presence of Essential 8 media. Reprogramming efficiency ranged between 0.05 and 0.1%. On the basis of cell morphology and growth kinetics, one clone was further expanded for each parental sample. All lines tested negative for the presence of mycoplasma. Characterization was carried out for pluripotency by immunostaining for SSEA4 (1:250; Abcam, Cambridge, MA, USA) and Tra-1-60 (1:500; Abcam) as extracellular markers, and Nanog (1:250; Abcam) and octamer-binding transcription factor 4 (Oct-4; 1:250; Abcam) as intracellular markers prior to expansion and cryopreservation of all iPSC lines followed by differentiation into cardiomyocytes, which were troponin positive.
Plasmid Construction
Plasmids for VEGF165 code for the 165-amino acid isoform of the human form of VEGF. It is derived from the 191-amino acid construct and cleaved in vivo to form the functional VEGF165. Our group identified that with our plasmid constructs for cardiac models that CAG (CMV + chicken β-actin gene + rabbit β globin) is the best promoter in terms of transfection efficiency into human cardiac cells and protein expression (data not shown). The stromal-derived factors, SDF-1α and S100a1 plasmid constructs, were constructed in a similar manner to VEGF165. Each plasmid was propagated in E. coli from New England Biolabs (Ipswich, MA, USA). The plasmids were purified using NucleoBond Xtra Midi Plus EF kits according to the manufacturer's instructions (Macherey-Nagel GmbH, Düren, Germany).
Animal Groups
All experiments were conducted using 12-week-old female severe combined immunodeficiency (SCID) mice purchased from Charles River Laboratories (Wilmington, MA, USA). Mice were injected with hUC-SECs, hiPSC-CMs, hBM-MSCs in the cell therapy groups, and naked plasmid DNA of S100a1, VEGF165, or SDF-1α was used in the gene therapy groups. Saline injections were used in the control group. There were 18 mice in each treatment group. The present study was approved and performed in accordance with the Guide for the Care and Use of Laboratory Animals (Department of Health and Human Services, Publication No. [NIH] 86-23), and with the guidelines of the Institutional Animal Care and Use Committee (IACUC) at the University of Utah.
MI Model
MI was surgically induced as previously described 33 . Briefly, all mice underwent complete permanent ligation of the left anterior descending (LAD) artery. For this procedure, animals were first anesthetized with isoflurane (Sigma-Aldrich, St. Louis, MO, USA), shaved, and intubated. With a left intercostal thoracotomy between the fourth and fifth ribs, the heart was exposed, the pericardium was incised, and dissection was performed to identify the LAD. Using an 8-0 Prolene suture (Ethicon, Somerville, NJ, USA), the LAD was permanently ligated to produce the MI.
Biologic Delivery
After LAD ligation, the mice were closely monitored until the heart rate stabilized (approximately 20 min). Upon stabilization, 5 × 105 cells or 50 μg of plasmid for each cohort were delivered in PBS (Gibco, Thermo Fisher Scientific) by four intramyocardial injections of 10 μl each using a Hamilton syringe (Hamilton, Reno, NV, USA) attached to a 30-gauge needle (n = 18) based on prior data from our team. A control group (n = 18) was subjected to the same surgical procedure, although they were injected with saline only. A total of four injections (10 μl each) were made in the peri-infarct region in a circular pattern at the border between infarcted and noninfarcted myocardium as seen by blanching of the tissue in order to maintain consistently among the large number of groups.
Echocardiography
Transthoracic echocardiography was performed under anesthesia and visualized with the use of a Vevo660 (Visual Sonics, Toronto, ON, Canada) equipped with a 30-MHz transducer as previously described 46 . Echocardiograms were obtained at baseline before LAD ligation, and again 4 and 12 weeks post-MI to assess left ventricular (LV) function and remodeling, as immediately after ligation the LV function has not stabilized. Also, the goal of the study was to compare between the groups as opposed to each biologic compared to their own baseline left ventricular ejection fraction (LVEF), end-systolic volume (ESV), end-diastolic volume (EDV), and fractional shortening (FS) were measured. Three cycles were measured for each assessment, and an average was used. Echocardiography and data analysis were performed blinded.
Hemodynamic Pressure Monitoring
Pressure data including LV pressures at systole and diastole, dP/dtmax and dP/dtmin, were obtained at 4 and 12 weeks post-MI prior to euthanization. LV function was studied by measuring hemodynamics and pressure. Data were acquired by 1.0 F (Millar, Houston, TX, USA) catheterization using a previously described technique 46 . Measurements were recorded and analyzed using Notocord-hem 4.2 software (Notocord Systems, Croissysur-Seine, France).
Tissue Analysis
Mice were sacrificed at 4 and 12 weeks postinjection. Tissues were analyzed by two blinded reviewers as previously described47,48. The hearts were arrested in diastole with potassium chloride (Sigma-Aldrich), followed by perfusion and fixation with 10% formalin (Sigma-Aldrich). Tissue was then embedded in paraffin (Sigma-Aldrich) and cut into 5-mm sections. Infarct size assessment was performed by staining paraffin sections with modified Masson's trichrome staining, which distinguishes fibrotic and cellular tissues. Immunostaining of heart sections was performed as follows. After antigen recovery, sections were permeabilized with 0.2% Triton X-100 (Sigma-Aldrich) for 5 min and incubated for 1 h in 4% fetal bovne serum (FBS)/1% bovine serum albumin (BSA) in PBS for blocking unspecific staining. This was followed by incubation in primary antibody solutions against CD31 (1:250; Thermo Fisher Scientific), VEGF (1:250; Thermo Fisher Scientific), connexin 43 (Cx43) (1:500; Thermo Fisher Scientific), α-smooth muscle actin (α-SMA) (1:250; Thermo Fisher Scientific), and von Willebrand factor (vWF) (1:250; Thermo Fisher Scientific). Alexa Fluor 488- or Alexa Fluor 594-conjugated secondary antibodies at 1:1,000 were applied to the samples for 30 min at room temperature (RT) and then counterstained with TO-PRO-3 or 4′,6-diamidino-2-phenylindole (DAPI) (Thermo Fisher Scientific). All sections were analyzed and documented using bright-field microscopy or a Leica DM IRB inverted fluorescence microscope (Leica, Buffalo Grove, IL, USA).
Assessment of Cardiac Fibrosis
Fibrosis assessment was performed by staining paraffin sections with modified Masson's trichrome stain, resulting in grayish blue-stained, collagen-rich (fibrotic) areas and red-stained cellular elements. The fibrotic area (blue color) was expressed as a percentage of total LV area and quantified by computerized planimetry using MetaMorph image analysis software (Molecular Devices, Sunnyvale, CA, USA) as previously described 38 .
Statistical Analysis
Statistical analysis was performed by using Student's t-test, chi-square analysis, or analysis of variance (ANOVA) with Tukey's multiple comparison tests using SAS Version 9.3 (Cary, NC, USA). Values of p < 0.05 were defined as statistically significant.
Results
Successful creation of the LAD infarcted mouse model assigned to one of seven treatment groups (control, hBM-MSC, hUC-SEC, hiPSC-CM, S100a1, SDF-1α, or VEGF) was achieved with limited procedure-related death (Fig. 1). Echocardiography was used to evaluate EDV, ESV, LVEF, and FS in all groups, and results are shown in Table 1 and Figure 2. Each group was individually compared to the control group, and there was also a grouped analysis that compared the cell therapy arm to controls and the gene therapy arm to controls.

Study overview. In each of the seven arms, 18 mice received the specified therapy. Mice were studied with echocardiography and left heart catheterization at 4 weeks. Eight mice were sacrificed at 4 weeks, and the remaining mice were followed to 12 weeks posttherapy, at which time repeat echo and hemodynamic data were reobtained.
Echocardiography Findings at 4 and 12 Weeks Comparing Cell Types and Genes to Control

EDV, end-diastolic volume; ESV, end-systolic volume; LVEF, left ventricular ejection fraction; FS, fractional shortening.
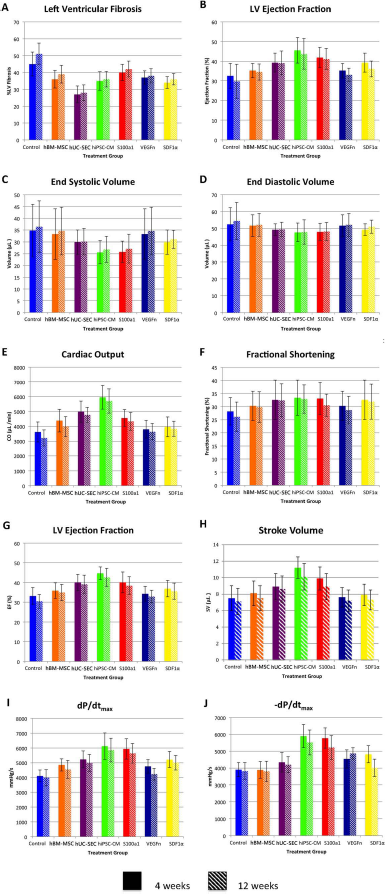
Echocardiographic and hemodynamic measurements. (A–F) Measurements from transthoracic echocardiography performed at 4 and 12 weeks. (G–J) Parameters obtained from pressure volume loops completed at 4 and 12 weeks. The p values are not depicted on this figure, as they are shown in Table 2 with full details.
Analysis of Direct Comparison of p Values for Each Treatment Group Versus Each Other at 4 and 12 Weeks
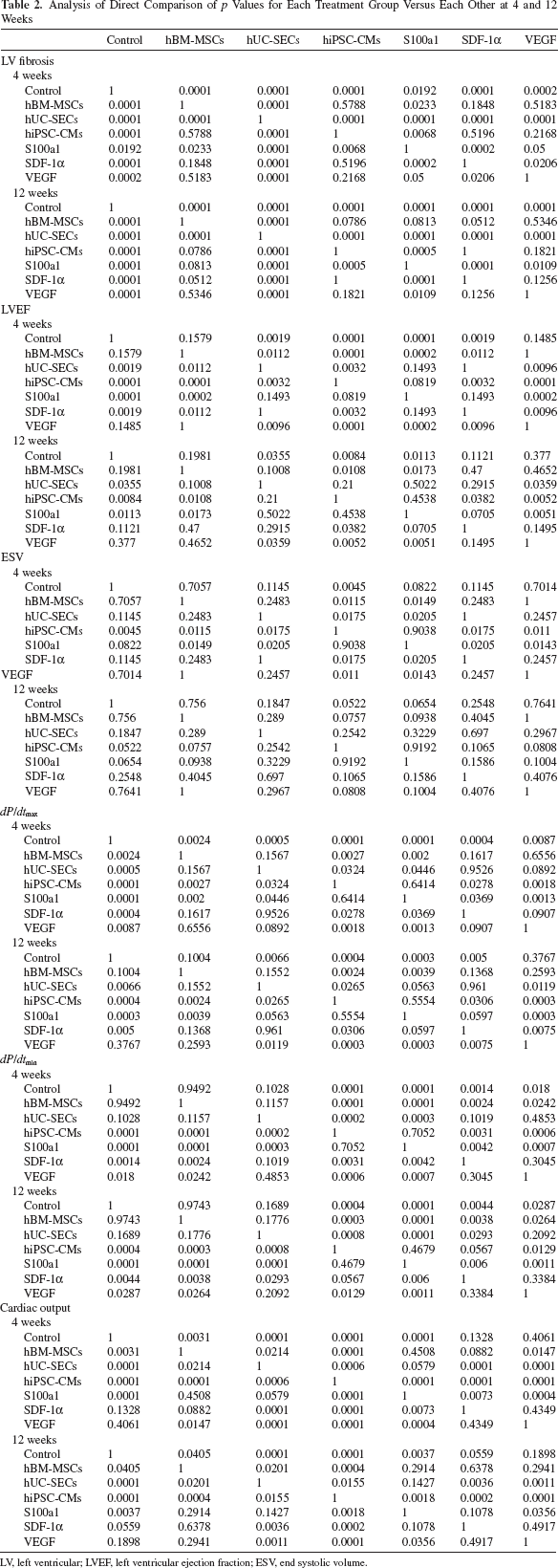
LV, left ventricular; LVEF, left ventricular ejection fraction; ESV, end systolic volume.
Echocardiographic Parameters
LVEF was significantly better at 4 weeks in the hUC-SEC, hiPSC-CM, S100a1, and SDF-1α groups compared to the control, and remained significant at 12 weeks in the hUC-SEC, hiPSC-CM, and S100a1 groups. This change was most significant in the hiPSC-CM (45.6 ± 6.5, p = 0.0001, 43.7 ± 7.8, p = 0.0084) and S100a1 groups (41.9 ± 5.2, p = 0.0001, 41.1 ± 5.5, p = 0.0113) compared to control (32.5 ± 6.4, and 29.7 ± 8.8) at 4 and 12 weeks, respectively. ESV was better in all cell and gene treatment groups compared to the control; however, it only reached significance in the hiPSC-CM group at 4 weeks with ESV of 25.6 ± 4.9 versus 34.8 ± 11.2 μl (p = 0.0045). Echocardiographic data are shown is Tables 1 and 2.
Hemodynamic Results
Cardiac output (CO) was greater in all treatment groups compared to the control, with significant improvements in the hBM-MSC, hUC-SEC, hiPSC-CM, and S100a1 groups at both 4 and 12 weeks. At 4 weeks, the groups with the most significant changes were hiPSC-CM (5,954 ± 799 μl/min, p = 0.0001), hUC-SEC (4,987 ± 721 μl/min, p = 0.0001), and S100a1 (4,561 ± 573 μl/min, p = 0.0001) compared to the control (3,622 ± 678 μl/min). Similarly, at 12 weeks, the change in CO was most significant in the hiPSC-CM (5,706 ± 815 μl/min, p = 0.0001), hUC-SEC (4,769 ± 512 μl/min, p = 0.0001), and S100a1 (4,335 ± 602 μl/min, p = 0.0037) groups compared to the control (3,622 ± 678 μl/min). The change in dP/dtmax compared to the control was significant at 4 weeks in all of the six groups; however, hiPSC-CM (6,125 ± 897 mmHg/s, p = 0.0001) and S100a1 (5,934 ± 695 mmHg/s, p = 0.0001) were the most significant compared to the control (4,106 ± 396 mmHg/s). This finding remained significant at 12 weeks in the hUC-SEC, hiPSC-CM, S100a1, and SDF-1α groups. Data are presented in Table 3 and Figure 2.
Left Heart Catheterization Pressure–Volume Loop Findings at 4 and 12 Weeks Comparing Cell Types and Genes to Control

CO, cardiac output; EF, ejection fraction; SV, stroke volume.
Histological Results
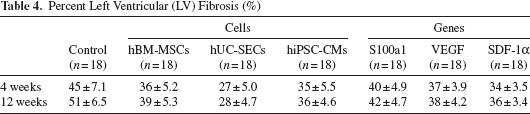
LV fibrosis was evaluated in all treatments groups at 4 and 12 weeks as seen in Table 4 and Figure 3. All cell and gene therapy arms showed significantly less LV fibrosis at both 4 and 12 weeks compared to the control group at 12 weeks (p = 0.0001). Histological evaluation via immunofluorescence staining for cell engraftment demonstrated that at 12 weeks postimplant, overall engraftment was low as would be expected. Relative engraftment by cell type was hiPSC-CMs > hUC-SECs > hBM-MSCs and is shown in Figure 4. Furthermore, Cx43 and human nuclei were identified at the infarct border (Fig. 5). Positive staining of Cx43 among transplanted cells indicates the expression of gap junctions between neighboring cells, which is essential for the conduction of cardiac action potentials. Cx43 was seen in hiPSC-CMs and hUC-SECs but not in hBM-MSCs.
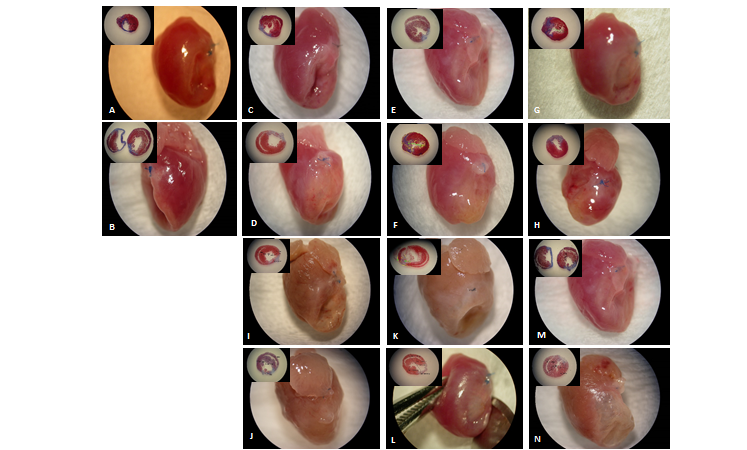
(A) Gross picture of severe combined immunodeficiency (SCID) mouse left anterior descending (LAD) infarct at 4 weeks. (B) Twelve weeks of control (saline) injections with inset of trichrome staining for scar. Similarly, (C) and (D) demonstrate human bone marrow-derived mesenchymal stem cells (hBM-MSCs), (E) and (F) demonstrate human umbilical cord subepithelial cell-derived stem cells (hUC-SECs), (G) and (H) demonstrate human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs), (I) and (J) demonstrate S100a1, (K) and (L) demonstrate stromal-derived factor-1α (SDF-1α), and (M) and (N) demonstrate vascular endothelial growth factor (VEGF).

Evaluation of cell engraftement following intramyocardial injection by immunohistochemistry and confocal microscopy. (A) Control saline-injected heart 12 weeks following myocardial infarction. Masson's trichrome staining for scar identification. (B) Cell-treated heart 12 weeks following myocardial infarction. Masson's trichrome staining for scar identification. (C, D) Representative image of hUC-SEC-treated heart 12 weeks following myocardial infarction. Immunohistochemical detection of human nuclei by immunoperoxidase-DAB reaction at different magnifications (400× and 1,000×, respectively). No human nuclei-positive cells could be detected in the control group. (E–H) Representative engraftment of cell-treated hearts at 12 weeks following myocardial infarction was overall low but was hiPSC-CMs > hUC-SECs > hBM-MSCs. Immunofluorescence evaluation of human nuclei engraftement by confocal microscopy at different magnifications (100× and 400×, respectively). (G, H) Double stain for human nuclei and α-actin in both split channel and merged view. Of note, human antigen-positive cells could be found in proximity to vascular structures.
Percent Left Ventricular (LV) Fibrosis (%)

Coexpression of cardiac-specific marker and human antigens in hiPSC-CM > hUC-SEC-treated heart 12 weeks following myocardial infarction. Arrows show connexin 43 granulae in human antigen-positive cells. Arrow tips show nonhuman-positive cells expressing connexin 43 in the usual perimembraneous fashion. There was none seen in the hBM-MSC group.
Vascularization in the posttreated myocardium was demonstrated by immunofluorescence staining for vWF and smooth muscle actin (SMA) (Fig. 6). These, in addition to human nuclei, suggest that hUC-SECs > hBM-MSCs > hiPSC-CMs may support new vessels in vivo (Fig. 7). Confocal microscopic evaluation demonstrated revascularization in the following trend: VEGF > hUC-SECs > hBM-MSCs > SDF-1α > S100a1 > hiPSC-CMs > control.

Expression of endothelial- and vascular-specific markers von Willebrand factor (vWF) and smooth muscle actin (SMA). (A) Control saline-injected heart 12 weeks following myocardial infarction. (B–E) Coexpression of endothelial- and vascular-specific markers and human antigens in hUC-SEC > hBM-MSC > hiPSC-CM-treated hearts 12 weeks following myocardial infarction.

Muscularized arterial-like structures. (A) Control saline-injected heart 12 weeks following myocardial infarction. Masson's trichrome staining for scar identification. (B) Control saline-injected heart 12 weeks following myocardial infarction. Immunohistochemical detection of SMA (brown) and vWF (blue) by immunoperoxidase-DAB reaction. SMA could not be detected. (C) Biologic-treated heart 12 weeks following myocardial infarction. Masson's trichrome staining for scar identification in order of greater to lesser scar: Control > S100a1 > hBM-MSC > VEGF > hiPSC-CM > SDF-1α > hUC-SEC. (D) Biologic-treated heart 12 weeks following myocardial infarction. Immunohistochemical detection of SMA (brown) and vWF (blue) by immunoperoxidase-DAB reaction. (E) Control saline-injected heart 12 weeks following myocardial infarction. Immunofluorescence evaluation of vascular-specific markers by confocal microscopy. SMA could not be detected in this group. (F–H) Biologic-treated heart 12 weeks following myocardial infarction. Immunofluorescence evaluation of vascular-specific markers by confocal microscopy in order of greater to lesser marler expression: VEGF > hUC-SEC > hBM-MSC > SDF-1α> S100a1 > hiPSC-CM > control.
Discussion
The current study provides a unique approach to using multiple human cell- and gene-based therapies in an ischemic heart disease model. Our study demonstrates that depending on which aspect of cardiac recovery that is being evaluated (scar remodeling, improvement in contractile function via myogenesis or energetics, angiogenesis, or inflammation), a different biologic may be best suited. Additionally, these therapies are safe; there were no safety issues related to the biologics as demonstrated by a similar mortality in each treatment group versus control and no gross tumors or other abnormalities on histology.
Scar remodeling is best addressed using hUC-SECs; however, both S100a1 and SDF-1α also demonstrated a significant scar reduction when compared to control and the other biologics. Contractile function is most significantly changed by injection of hiPSC-CMs or S100a1, followed by hUC-SECs. All three have different mechanisms of action; hiPSC-CMs lead to an increase in myocytes, S100a1 increases contractility via calcium signaling, and hUC-SECs work via immunomodulation to support existing cardiomyocytes. Angiogenesis was most significant in the VEGF treatment group as demonstrated by histology, but the other biologics also showed improved vascularity: hUC-SECs > hBM-MSCs > SDF-1α > S100a1 > hiPSC-CMs > control. In the case of cell-based therapies, secreted factors may be the main contributors to angiogenesis, with the exception of SDF-1α, which directly attracts CXCR4+ cells that may be either angiogenic or myogenic. We did not directly evaluate inflammation in this trial even though hUC-SECs and hBM-MSCs have been shown to have immunomodulatory effects16–30. There were no safety issues related to the biologics as demonstrated by a similar mortality in each treatment group versus control and the fact that histology did not show any gross tumors or other abnormalities.
Significant effort has been put forth to develop functional cell- and gene-based regenerative strategies to minimize or control the amount of cell loss and scarring following MI. The resulting decline in function is directly proportional to the loss of cardiomyocytes and downstream LV remodeling. The use of cell- and gene-based therapeutics to preserve cardiac function and prevent tissue loss post-MI has made considerable advancements over the past several years13,49–52. Many cells types and genes have been proposed as therapeutic options including adult-derived cells, such as skeletal myoblasts53,54, fetal stem cells55,56, pluripotent stem cells57,58, and BM-MSCs13,58. In recent years, stem cells derived from various areas of umbilical cord tissue have also been studied and characterized59–62. These cells are attractive as they possess multipotency and therapeutic potential in various in vivo models63–67. A large amount of research has been performed on cell-based therapeutics; however, challenges ranging from immune, ethical, tumorigenic potential, arrhythmogenic, or scalability have limited these technologies. Our findings are consistent with previous studies in MI models where ESCs 67 , MSCs31,68, and somatic cells 69 or genes 37 help preserve cardiac function post-MI or attenuate adverse remodeling. Previous studies have demonstrated that long-term retention and engraftment of transplanted cells in the infarct region remain a challenge in the treatment of MI, which was consistent with our data 70 .
Our study is limited by the fact that there was fixed dosing of cells and genes within each group to standardize the treatment, and therefore dose response was not evaluated. We did not use reporter genes such as luciferase or green fluorescent protein (GFP) to track cells or genes due to the fact that all biologics in this study were designed to be directly translatable into larger trials and possible clinical studies. However, we have performed labeling trials on these cells and genes in a patch for our othe studies. We evaluated only one route of delivery, direct myocardial injection, and not others such as intravenous, endocardial, intracoronary, and/or a patch70,71. Some of these would not be feasible in the current mouse model. We could have performed more immune studies to further complete the analysis of possible mechanisms related to the cells as opposed to the genes that have direct, defined mechanisms. We also did not evaluate these biologics in combination, which is an area that we are going to explore. The combination of multiple cells or genes or both could potentially have positive inotropic effects along with possible positive remodeling. We evaluated only Cx43 expression after biologics were injected. Other connexin isoforms can be expressed during cellular differentiation after implantation and may or may not be able to communicate with native Cx43 expressed mostly in ventricular myocytes, or be related to the formation of endothelial tissue (Cx43 and Cx40) 72 .
In conclusion, the current study demonstrated that multiple cell types and genes injected into a mouse model of MI can positively alter various aspects of cardiac function and scaring for up to 12 weeks post-MI. Future studies evaluating dose and combination therapies have great potential for the understanding of their possible clinical utility and translation.
Footnotes
Acknowledgment
The study was funded by internal funds. A.A.W., S.B.-G., H.T., J.A.H., D.L.A., C.E.B., F.J.S., and A.N.P. performed experiments and collected data. A.N.P. conceived the study. A.A.W., S.B.-G., H.T., J.A.H., D.L.A., C.E.B., F.J.S., E.W.H., A.P.M., D.A.G., and A.N.P. helped to draft the manuscript and were involved in the data analysis and revisions. All authors read and approved the final manuscript. A.N.P. has a patent filed on UC-SECs. A.N.P. is a section editor of Cell Transplantation. Neither A.N.P. nor any member of the editorial board or editorial office affiliated with the authors' institutions were involved with the review process and/or decision making on this article. The authors declare no conflicts of interest.