
Abstract
Calpainopathy, also known as limb girdle muscular dystrophy (LGMD) type 2A (LGMD2A) or LGMD R1 Calpain3-related, is one of the most common genetically characterized forms of limb-girdle muscular dystrophy with a wide range of phenotypic severity. We evaluated a consanguineous family with a clinical phenotype consistent with calpainopathy in whom conventional sequencing did not detect any mutations in the CAPN3 gene. Using whole exome sequencing paired with haplotype analysis, we identified a homozygous deep intronic single base pair deletion in CAPN3 (c.946-29delT). Familial segregation studies were consistent with recessive inheritance. Immunoblotting of muscle tissue from the patient showed complete absence of calpain 3. In silico analysis predicted the deletion to disrupt the branch point and subsequently alter splicing of exon 7. Studies of patient fibroblasts and muscle tissue confirmed altered splicing, resulting in an inclusion of a 389-bp intronic sequence upstream of exon 7, originating from a cryptic splice acceptor site in intron 6. This out-of-frame insertion results in a premature stop codon, leading to an apparent absence of protein likely due to degradation of the transcript via nonsense-mediated decay. We then designed phosphorodiamidate morpholino oligomers (PMOs) as splice modulators to block the new splice acceptor site. This approach successfully prevented the aberrant splicing – reverting the majority of the splice to the wildtype transcript. These results confirm the pathogenicity of this novel deep intronic mutation and provide a mutation-specific therapeutic strategy. Thus, deep intronic mutations in CAPN3 may be pathogenic and should be considered in the appropriate clinical setting. The identification of mutations which may be missed by traditional Sanger sequencing is essential as they may be excellent targets for individualized therapeutic strategies using RNA-directed splice modulation.
INTRODUCTION
Limb-girdle muscular dystrophies (LGMDs) are a clinically and genetically heterogenous group of neuromuscular disorders. Calpainopathy, also known as LGMD2A, is the first to be defined at the molecular level and has since emerged as the most common genetic cause of LGMD [1]. Clinically, calpainopathy is characterized by childhood to juvenile-onset, symmetric, progressive, proximal more than distal muscle weakness that is most notable in the posterior and medial compartments of the thigh and pelvic girdle muscles. In the upper extremities, it results in significant atrophy of the periscapular muscles and biceps with relative sparing of the facial muscles. Serum creatine kinase (CK) is typically elevated (5–80 times), although it can be highly variable, and muscle pathology is dystrophic with absent or reduced calpain 3 expression [2, 3]. While the classic calpainopathy phenotype can be readily recognized by the trained neuromuscular clinician, the potential for significant phenotypic variability and atypical features can make the diagnosis challenging.
Recessive calpainopathy is caused by biallelic inactivating mutations in CAPN3 which encodes a proteolytic enzyme – calpain 3 [4–7]. Recently, a dominantly inherited form has been reported [8]. To date, about 500 different pathogenic mutations in the CAPN3 gene have been reported on the Leiden Muscular Dystrophy Database. Approximately 70% are missense mutations located in the coding region of the gene, while the remaining are loss-of-function variants caused by either deletions or insertions that result in a frameshift and premature stop codon or splice site variants [9].
Deep intronic changes that modify precursor mRNA splicing causing a pseudo-exonization of an intronic sequence are increasingly being reported as cause of disease [10–14]. Many deep intronic variants can be easily overlooked by sequencing using genomic DNA from blood or cells. Identification of such mutations in CAPN3 necessitated sequencing on the cDNA obtained from muscle tissue [15–17].
In this study, we present a consanguineous family from Sudan with a clinical diagnosis of calpainopathy. Using whole exome sequencing (WES) paired with haplotype analysis, we identified a homozygous deep intronic single base pair deletion in CAPN3 (c.946-29delT) that was not detected by conventional exon-based sequencing. This mutation is predicted to disrupt the branch point and interfere with normal splicing of exon 7. Studies of patient fibroblasts showed altered splicing of exon 7 through the inclusion of a 389-bp “exon extension” of an intronic sequence from an alternatively used cryptic splice acceptor site located upstream. Splice-altering mutations are an excellent target for mutation specific antisense-mediated splice modulation interventions. We show the successful application of a phosphorodiamidate morpholino oligomer (PMO) to block the newly created splice acceptor site, thereby preventing aberrant splicing and reverting to wildtype transcript with high efficiency in patient fibroblasts. This case illustrates the importance of clinical recognition and accurate phenotyping to direct targeted genetic testing and highlights the potential of a “precision-based” molecular therapeutic approach to splice-altering pathogenic mutations.
MATERIALS AND METHODS
Patients and data
This study was approved by the Institutional Review Board of the National Institute of Neurological Disorders and Stroke at the National Institutes of Health (Protocol 12-N-0095). Written informed consent from the patient was obtained by a qualified investigator. Medical records were obtained and reviewed and clinical assessments were performed as part of a diagnostic evaluation. In addition, muscle MRI, muscle ultrasound, blood laboratory testing and a muscle biopsy were obtained. Genomic DNA was obtained from the patient’s dermal fibroblasts and blood while saliva samples were obtained from family members based on standard procedures.
Mutation analysis
Whole Exome Sequencing (WES) analysis was performed on genomic DNA from the patient’s blood sample at the Broad Institute using their dual-barcoded library construction followed by Illumina Rapid Capture Exome enrichment kit with 38 Mb target territory (29 Mb baited). Total RNA was isolated from the patient’s muscle biopsy using miRNeasy Kit (Qiagen Sciences). Reverse transcription reactions were performed using SuperScript III Reverse Transcriptase (ThermoFisher) following manufacturer’s protocols. Targeted familial segregation testing was performed on genomic DNA extracted from saliva samples using PCR amplification of Exon 7 of CAPN3 followed by sanger sequencing. Sequencing of the PCR products was performed on an ABI 3130×1 capillary sequencer in the forward and reverse directions.
Protein analysis – Western blot analysis
Frozen muscle tissues from the patient and a healthy control were cut in a cryostat at 10μm and quickly weighed (∼10–20 mg). After lysis in RIPA buffer and sonication, 20μg of protein in loading buffer was applied to NuPAGE 4% – 12% Bis-Tris gel and transferred onto a PVDF membrane. The primary antibody against Exon 8 of Calpain 3 (Novacastra, NCL-CALP-12A2) was applied after a 1-hour block in 5% non-fat milk/0.05% TBST and incubated at 4°C overnight. After washing, the membrane was incubated for one hour at room temperature with goat anti-mouse HRP-conjugated secondary antibody (GE Healthcare, NA9310V; 1:5,000). Detection was performed using the ECL system (GE Healthcare, RPN2106). After stripping the membrane, a loading control was performed with the same method using a mouse monoclonal primary antibody against α-Tubulin (Sigma-Aldrich; T5168).
phosphorodiamidate morpholino oligomer (PMO) design and treatment
A total of four PMOs and one scramble PMO (SPMO) (Gene Tools, LLC) were designed targeting: at the aberrant splicing site (PMO1), before the aberrant splicing site (PMO2), at the 51-bp aberrant splicing site (PMO3) and at the branch point (PMO4). Dermal fibroblasts established from patient (P1) and control were grown in Dulbecco’s modified Eagle medium with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin in a 24-well plate in 5% CO2 at 37°C for about 22 hours. Cells were treated with SPMO and PMO in the presence of a final concentration of 2.5μM with 6μM Endo-Porter and continuously cultured without antibiotics for 48 hours. Cells were then harvested and splicing evaluation was performed by PCR analysis using cDNA obtained from RNA, which was extracted from the patient’s dermal fibroblasts.
Quantification of normal splicing restoration
After 48 hours with PMO treatment, total RNA was extracted from the patient and control dermal fibroblasts using miRNeasy Kit (Qiagen Sciences). RT-PCRs were performed with 0.8 ug of RNA using SuperScript III Reverse Transcriptase (ThermoFisher). PrimeTime qPCR assays consisting of mutation-specific primers and fluorescent-labeled probes were designed for assessing the specificity and efficacy on selective inhibition of mutant CAPN3 gene expression and restoration of normal CAPN3 gene expression. Three microliters of diluted cDNA (1/1 dilution in RNase-Free water) was used in total, and 10μl of each qPCR reaction were run in quadruplicates on a QuantStudio 6 Flex instrument (Applied Biosystems/Life Technologies) using default amplification settings. Relative gene expressions were calculated using 2-ΔΔct method normalized by PGK1.
RESULTS
Clinical findings
A 37-year-old man (P1) from a consanguineous Sudanese family with childhood-onset, progressive muscle weakness presented for diagnostic evaluation. Family history was significant for a similar disease onset and progression in two brothers as shown in the pedigree (Fig. 1A). First symptoms arose at eight years of age with frequent falls and difficulty running. Over several years, these symptoms progressed, resulting in difficulty climbing stairs and rising from the floor. By his late teenage years, he also developed weakness in his upper extremities, and by his mid-to-late twenties he lost the ability to ambulate independently. Upon examination of P1 at age 37 years, muscle bulk was noted to be globally reduced with significant, bilateral biceps and quadriceps atrophy in addition to scapular winging (Fig. 1B). The affected siblings were not available for clinical evaluation. Muscle strength was significantly reduced with subgravity strength in the proximal upper extremities and in the proximal and distal lower extremities (Supplemental Table 1). Deep tendon reflexes were diffusely reduced or absent. Extraocular movements were full, and cognition was intact.
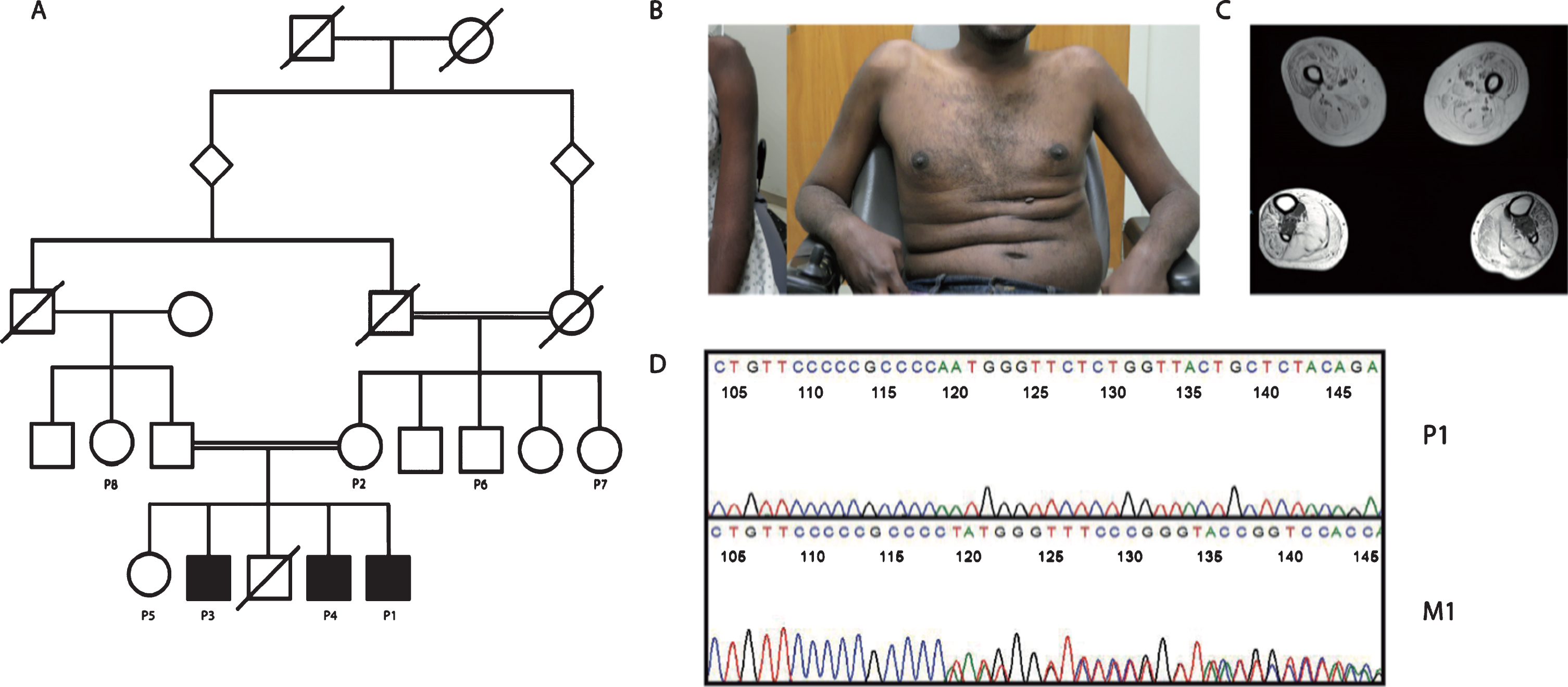
Genetic and phenotypic characterization of a family with a deep-intronic mutation in CAPN3. (A) Pedigree of a 37-year – old man (P1) from a consanguineous family. (B) Globally reduced muscle bulk with significand biceps atrophy. (C) Muscle MRI of lower extremities. (D) Sanger sequencing of CAPN3 in patient (P1) and unaffected mother of P1 (M1).
Muscle MRI of the lower extremities performed at age 37 years revealed diffuse muscle atrophy and fatty replacement of all upper and lower leg muscles except for isolated sparing of the tibialis posterior muscles (Fig. 1C). Pulmonary function testing revealed a forced vital capacity (FVC) of 95% while sitting with no significant decline in FVC with transition to the supine position. Select laboratory test results showed mildly elevated serum creatine kinase levels of 608 units/L at age 32 years old (normal range 52–386 units/L) and 266 units/L at age 37 years (normal range 39–308 units/L). No significant cardiac abnormalities were found on echocardiogram or electrocardiogram. Review of muscle biopsy obtained from the quadriceps performed at age 32 years showed moderate to severe fiber size variability, fatty infiltration and fibrosis. There were several degenerating myofibers with no evidence of inflammation, vacuoles or inclusions. Prior testing using next-generation sequencing analysis was negative for any mutations in the dystrophin (DMD) gene. Deletion/duplication testing for targeted DMD exons was negative.
WES analysis and confirmatory studies
Initial review of WES with standard filtering for known disease-causing genes was negative. Following careful neuromuscular assessment, which revealed a clinical phenotype suggestive of calpainopathy, a targeted re-analysis of the BAM files specifically for CAPN3 revealed a homozygous deep intronic single base pair deletion in intron 6 of CAPN3 (c.946-29delT; NM_000070). Targeted familial segregation studies confirmed that all three affected siblings (P1 and two affected brothers) were homozygous for the CAPN3 mutation, and unaffected relatives were found to be heterozygous for this mutation (Fig. 1D). This mutation had not been previously reported in patients with calpainopathy nor was it found in databases of healthy individuals (Gnomad). In silico analysis predicted that the deletion disrupts the branch point, which then interferes with splicing of exon 7. Studies performed on cDNA from RNA extracted from P1’s muscle tissue confirmed that the mutation disrupts normal splicing and redirects splicing to an alternative upstream intronic splice acceptor site, resulting in an inclusion of a 389-bp exon extension in intron 6 (Fig. 2A). This out-of-frame insertion ultimately results in a premature stop codon. Gel electrophoresis of the PCR amplification of cDNA from P1’s muscle tissue confirmed aberrant splicing of CAPN3 in P1 with an inclusion of the 389-bp exon extension, which is absent in control tissue (Fig. 2B), and results in an out-of-frame mRNA product with a premature truncation. Western blot of muscle tissue from P1 and control using an antibody against Exon 8 of calpain 3 (NCL-CALP-12A2 predictive of bands 94 and ∼60 kDa) showed complete absence of calpain 3 protein in P1 (Fig. 2C). Tubulin detection in the same lane was used as an internal loading control.
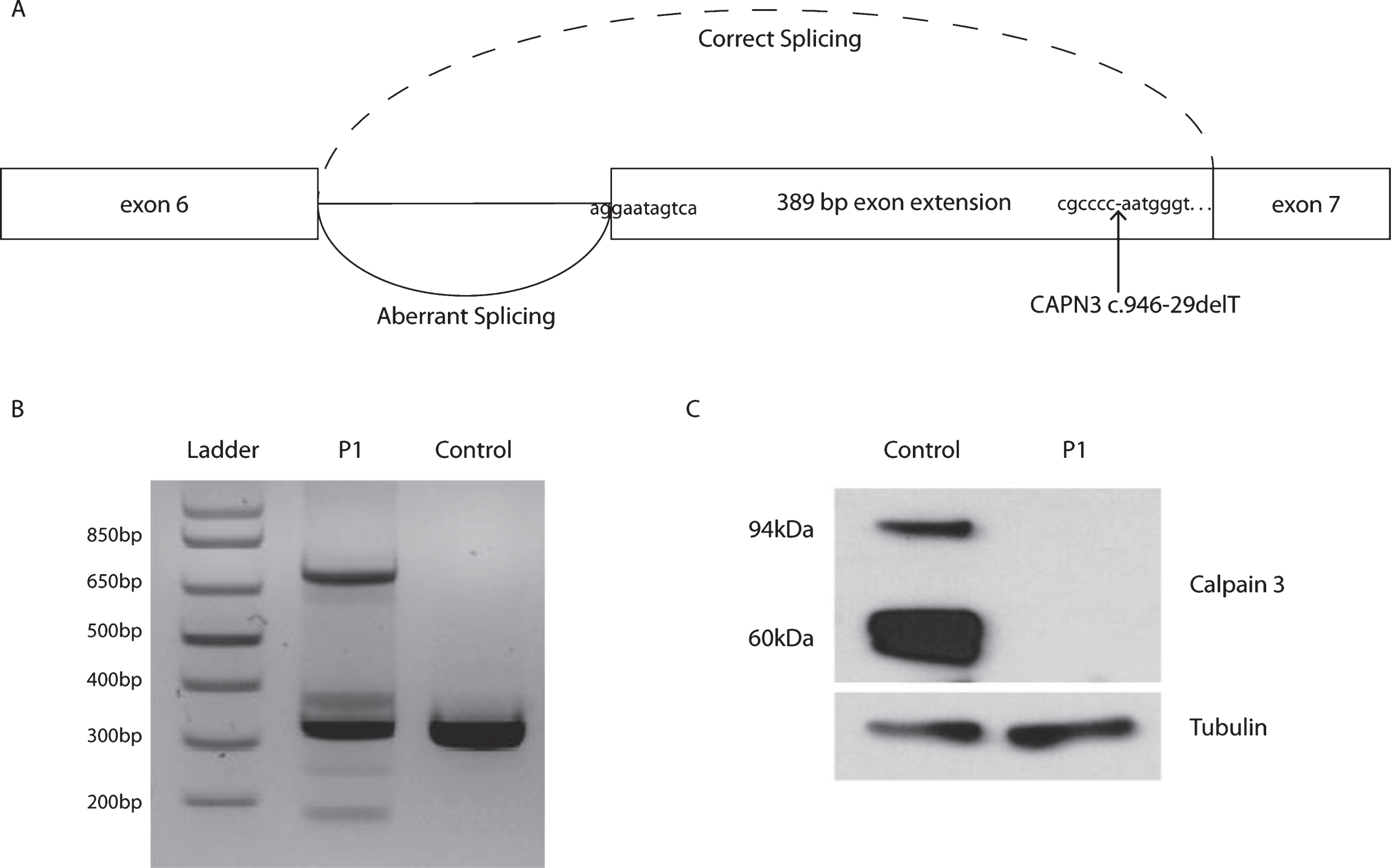
Confirmation of aberrand splicing of CAPN3 in muscle tissue from P1. (A) Model of the disrupted branch point preventing normal splicing resulting in an inclusion of a 389-bp exon extension in intron 6. (B) Representative gel electrophoresis of endpoint PCR using cDNA from muscle tissue. (C) Western blot of control and patient (P1) using a calpain 3 antibody on muscle tissue.
Quantitative analysis of PMO treatment on fibroblasts
We designed phosphorodiamidate morpholino oligomers (PMOs) as splice modulators to block the new splice acceptor site (Fig. 3A). Representative gel electrophoresis of the PCR from fibroblasts after 48 hours of PMO1 treatment showed that the aberrant splicing was suppressed with high efficiency and splicing directed back to the normal splice site, despite of the still present mutant branch point (Fig. 3B, Lane 5). This splice modulating treatment using one PMO was thus able to restore the majority of correct splicing of the full-length CAPN3. However, a 51-bp exon extension remained after the PMO1 treatment, resulting from a small amount of newly abnormal splice redirection, 51 bp upstream of the natural splice. To address this, an additional treatment using PMO4 was designed to target both the new predicted branch point next to the mutation-induced intronic abnormal splice site as well as a nearby enhancer. Still, the PMO4 treatment did not show a reduction of the 51-bp insertion (not shown). Overall, the approach prevented the aberrant splicing with high efficiency, reverting to a majority wildtype transcript and a residual smaller amount of wildtype transcript containing a 51-bp in-frame insertion resulting from the remaining exon extension.
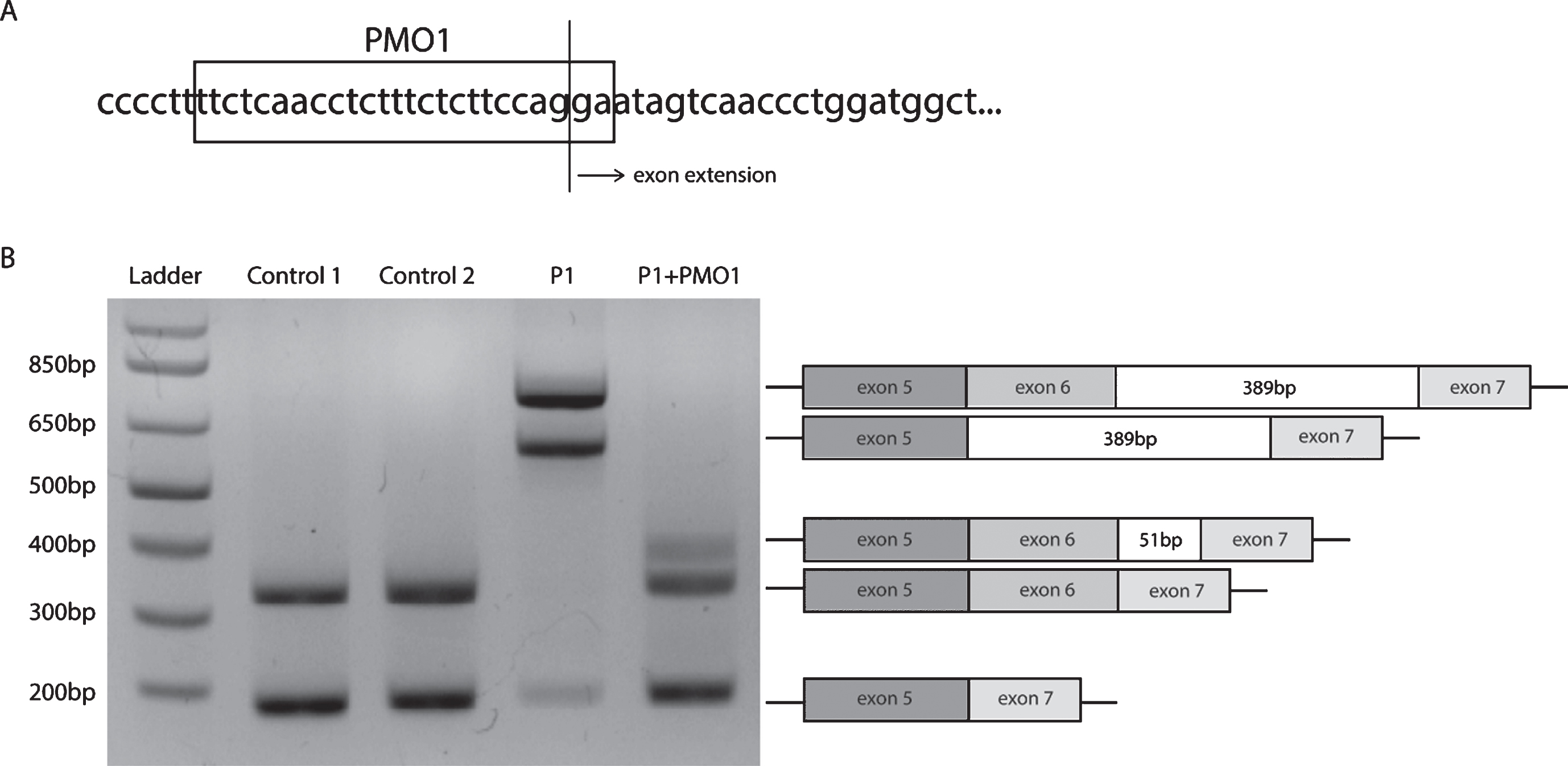
PMO treatment corrects the aberrant splicing in CAPN3. (A) PMO1 target sequence at the abrrant 3’ splice acceptor. (B) Patient-derived fibroblasts (P1) and control fibroblasts (Control) were treated for 48 hours with PMO1. Representative gel electrophoresis of endpoint PCR after PMO1 treatment using primers located in exons 5 and 7, demonstrating suppression of the aberrand splice products and also a remaning 51-bp exon extension.
To analyze the degree of suppression on the expression of mutation at the transcriptional level, we generated two sets of primers specific to the correct and aberrant splice sites (Fig. 4A). The E6I6 primers would work to only amplify the mutant splice product while the E6E7 primers would only amplify the correct splice product that would then be used for quantitative PCR (qPCR). P1’s fibroblasts and control fibroblasts were treated with PMO1 at doses ranging from 0.02 to 10μM. Representative gel electrophoresis of endpoint qPCR showed splice products (Fig. 4B) while clearly demonstrating the presence of the aberrant splice product detected with primers E6I6 (Fig. 4A, below) and of the correct splice product with primers E6E7 (Fig. 4A, above). Dose response expression of aberrant splice product (Primers E6I6) showed that more than 50% of aberrant splicing expression was suppressed using the PMO1 treatment at a concentration of 1.25μm relative to the mock-treated patient cells (Fig. 4C). Additionally, over 90% of the normal splicing product (Primers E6E7) was recovered after treatment with PMO1 at the same concentration of 1.25μm (Fig. 4D).
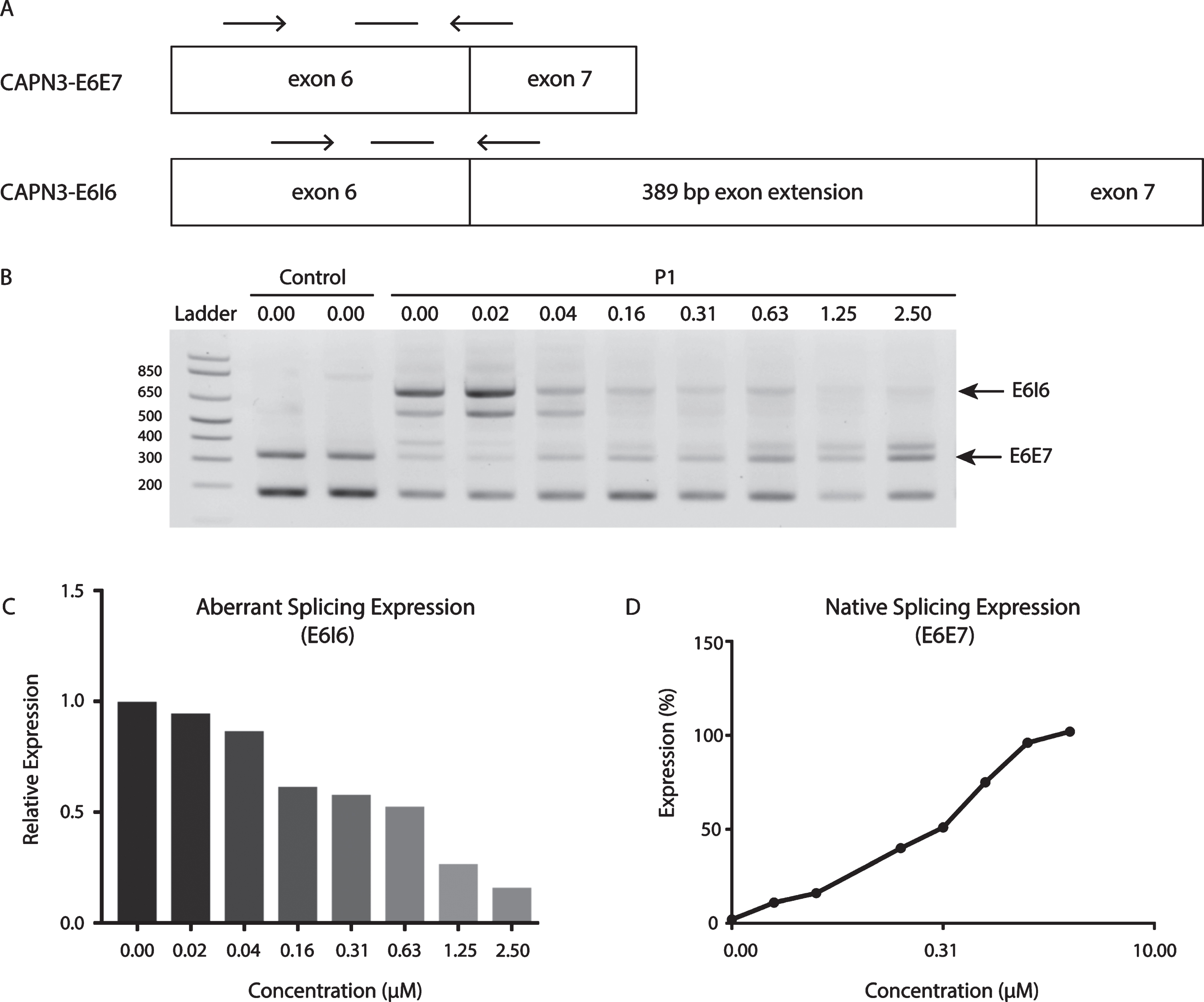
Dosage effect of PMO1 on abrrant and native CAPN3 splicing. (A) Location of primers (arrows) and probe (line with no arrowheads) in CAPN3-E6E7 and CAPN3 E616. (B) Representative gel electrophoresis of endpoint RT-PCR following PMO treatments ranging from 0–2.50μM. (C) Expression of the aberrant splice product (E616) relative to mock-treatede patient cells. (D) Expression of the native splice product (E6E7) relative to normal control cells.
DISCUSSION
Despite the recent advances in next-generation based sequencing technologies, including multi-gene panels, whole-exome sequencing and whole-genome sequencing, the rate of successfully arriving at a genetic diagnoses remains between 25–50% for an array of rare diseases [18–21]. Deep intronic mutations that alter normal splicing are an emerging class of disease-causing mutations that may be ‘hidden’ from the commonly used exon-based genetic testing strategies [14, 23]. However, due to their frequent interference with splicing, these mutations may be readily targetable using current RNA splice modulators such as PMOs, which are already FDA approved as a class for use in patients [12]. Thus, identification of these deep intronic mutations may have direct therapeutic implications, more so than exonic mutations. The therapeutic design will have to be highly individualized and targeted to the specific mutation, as a precision treatment approach.
Here we present a consanguineous family with a clinical diagnosis of calpainopathy in whom we identified a homozygous c.946-29delT mutation in CAPN3 by re-analysis of existing whole exome sequencing data, guided by strong clinical phenotyping. Manual targeted review of the CAPN3 BAM files revealed the homozygous intronic mutation, which was then confirmed by Sanger sequencing and further validation studies. Large-scale transcriptome sequencing such as RNA-sequencing would have been an alternative approach to obtain a confirmed genetic diagnosis for our patient [19]. Unfortunately, due to the significant fatty replacement of the muscle tissue at the time of the biopsy, we did not have enough muscle tissue available for sufficient RNA extraction. We were able to complete western blot analysis, which showed a clear absence of calpain 3 and further supported the diagnosis of calpainopathy. In our case the detailed clinical phenotyping, imaging data and direct targeted genetic testing strategies ultimately led to the identification of this novel, ‘hidden’ deep intronic CAPN3 mutation which was present and yet not recognized in the exome sequencing data using standard filtering approaches [24].
We demonstrate that this mutation leads to disruption of exon 7 splicing through activation of a cryptic upstream splice acceptor site, which results in an inclusion of a 389-bp out-of-frame “exon-extension” that either results in a truncated protein product or degradation through nonsense-mediated decay. Specifically, the mutation identified in this patient disrupted the splice branch point, which then prevented normal splicing of exon 7, redirecting splicing to a normally weaker alternate deep intronic splice acceptor and branch point, resulting in an out-of-frame exon-extension. Steric hinderance of this new branch point and splice acceptor using targeted PMOs masked this pathological alternative splice suffiently to direct splicing back to the original branch point and splice acceptor, largely restoring the normal splicing pattern. Interestingly, as a specific result of the treatment with the splice restoring PMO1, a new quantitatively minor misplicing was identified, resulting in a 51-bp in-frame exon extension due to partial utilization of a third intronic splice acceptor. These findings point to the active competition of the splicing machinery for different branch points and splice acceptor sites, which is highly influenced by the primary sequence of critical regions in the pre-mRNA. Given that most of the splice modulation results in restored wildtype transcript from both alleles, the splice restoration by PMO1 would be expected to be therapeutically meaningful as the contribution of the new residual misplicing event is small and by definition 50% of normal transcripts or less would be expected to be phenotypically non consequential in this recessive disease.
Thus, we show that RNA-directed splice modulation can be a viable therapeutic strategy to restore normal splicing of this mutation, and we would surmise by extension also in other, similar mutations discoverable by RNA and genome-based diagnostic strategies in various genetic diseases. With the experiments using PMOs specifically designed to restore wildtype transcript, we demonstrate the feasibility and potential for highly individualized therapies guided by modern sequencing technologies. Thus, precision diagnostics might lead to precision therapeutics. However, further evaluation is still needed for testing the efficacy of PMO treatments on splice modulation of CAPN3 in skeletal muscle, especially given the challenges for PMO delivery to specific tissues. In addition, even though this splice modulation approach is likely to increase calpain 3 levels would be capable of doing from both alleles because of the homozygous state of the mutation, it remains to be determined what amount of normal protein production can actually be achieved in vivo and how much would be sufficient to rescue the clinical phenotype as well as the optimal timing for maximizing functional benefits to patients.
We conclude that deep intronic mutations in CAPN3 may be pathogenic and should be carefully investigated in patients with clinical presentations consistent with calpainopathy for the mutation reported here and other disease-causing splice-altering mutations. In particular, patients for whom only one pathogenic mutation has been identified to date could be revisited and screened for a splice-altering, potentially deeper intronic mutation as the second mutation. This may require RNA analysis in conjunction with genome sequencing to complement exon based sequencing approaches. In addition to confirming a molecular diagnosis in those patients, uncovering additional splice-altering mutations would broaden the spectrum of pathogenic mutations in CAPN3 and provide additional personalized therapeutic targets. Also, strong phenotyping can significantly facilitate and guide molecular diagnostics in this and other genetic conditions. A precise molecular diagnosis in turn can have therapeutic implications as a viable therapeutic strategy may be conceivable for mutations similar to the one reported here.
CONFLICT OF INTEREST
All other authors declare no conflict of interest.
Footnotes
ACKNOWLEDGMENTS INCLUDING SOURCES OF SUPPORT
The authors thank the patients and their caregivers for participating in the study. This study was funded through intramural support by the NINDS/NIH. We thank Christopher Mendoza and Gilberto ‘Mike’ Averion for their help in coordinating patient visits and patient samples. Sequencing was provided by the Broad Institute of MIT and Harvard Center for Mendelian Genomics (Broad CMG) and was funded by the National Human Genome Research Institute, the National Eye Institute and the National Heart, Lung and Blood Institute grant UM1 HG008900 to Daniel MacArthur and Heidi Rehm.