
Abstract
Calpainopathy, or limb–girdle muscular dystrophy type R1/2A (LGMDR1/2A), is the most prevalent form of LGMD, comprising about 32% of all cases. The disease is caused by mutations in the CAPN3 gene, leading to dysfunction of the corresponding protein–an enzyme critical for muscle fiber cytoskeleton remodeling and protein signaling regulation through selective proteolysis. Clinical manifestations demonstrate significant phenotypic polymorphisms, ranging from oligosymptomatic forms to severe early-onset cases, with the loss of ambulation occurring 10–25 years after disease onset. A characteristic feature is predominantly symmetrical involvement of limb and trunk muscles, leading to early mobility loss, disability, and reduced work capacity. Noninvasive imaging can suggest dystrophic muscle disease but requires differentiation from other myopathies. Confirming the diagnosis involves histological, immunological, and molecular genetic studies to identify calpain-3 activity or CAPN3 gene expression alterations. Currently, no targeted or etiological therapies are available for calpainopathy. Treatment focuses on symptom management, complication prevention, and slowing disease progression. Preclinical research demands the development of an appropriate animal model that displays disease phenotypes mirroring those observed in humans. Preclinical and clinical research are also investigating therapeutic options, including the use of drugs that have proven effective in other myopathies and genome editing via transgenic CAPN3 delivery to restore protein activity. Gene therapy has shown promise in murine models, but safety concerns, particularly systemic toxicity affecting the heart and other organs, remain significant. This review comprehensively analyzes the clinical features, diagnostic approaches, and advancements in modeling and therapeutic development for calpainopathy.
Keywords
Introduction
Calpainopathy (LGMDR1/2A, OMIM# 253600; LGMDD4/1I, OMIM# 618129) is the most common form of limb–girdle muscular dystrophy. It accounts for an average of 32% (95% confidence interval (CI): 26–37) of all LGMD cases,1–6 with an estimated occurrence rate of 8.3 per 1 million people. 7 However, in certain endemic regions, such as northeastern Italy, the prevalence can reach 26.5 per 1 million. 8
LGMDR1 is predominantly inherited in an autosomal recessive (AR) manner, although rare cases of autosomal dominant (AD) LGMDD4 inheritance have been documented.9,10 The disease is caused by mutations in the CAPN3 gene, which consists of 24 exons and encodes a 94 kDa protein known as calpain-3. 11 Calpain-3 is part of the calpain superfamily and facilitates the selective proteolysis of various proteins (e.g., protein kinases A and C, rhodopsin, actin, myosin, and tropomyosin), which contributes to cytoskeletal remodeling.11–13
To increase diagnostic vigilance among clinicians, data on the phenotypic variability of calpainopathy, along with diagnostic algorithms and criteria, should be consolidated and systematized. Additionally, recent studies have explored various aspects of this disease, including advancements in genome editing and efforts to repair mutant genes in animal models.
Clinical characteristics
The age of onset of clinical symptoms in patients with calpainopathy varies widely, ranging from 2–50 years of age, and onset can be classified as early (before 12 years), typical (between 12 and 30 years), or late (after 30 years). 14 For the AD form, the average age of symptom onset is 34 years. 9
When the disease manifests in childhood, disease progression and clinical symptoms are more severe, 15 although age of onset is not an absolute predictor of disease severity. 12 Ethnic background can also influence disease severity, with more severe cases observed in Afro-Brazilian populations than in European populations. 16 Additionally, the predominant symptoms and rate of progression depend on the type of mutation (presence of two alleles associated with complete loss of calpain-3 protein 17 ) and sex (women tend to develop a more severe increase in serum creatine kinase (CK)18,19). Furthermore, interfamilial variability is predominantly observed, whereas intrafamilial variability is less pronounced.15,20
Early severe, moderate, and late mild phenotypes are distinguished on the basis of their severity and age of onset 15 (Figure 1).
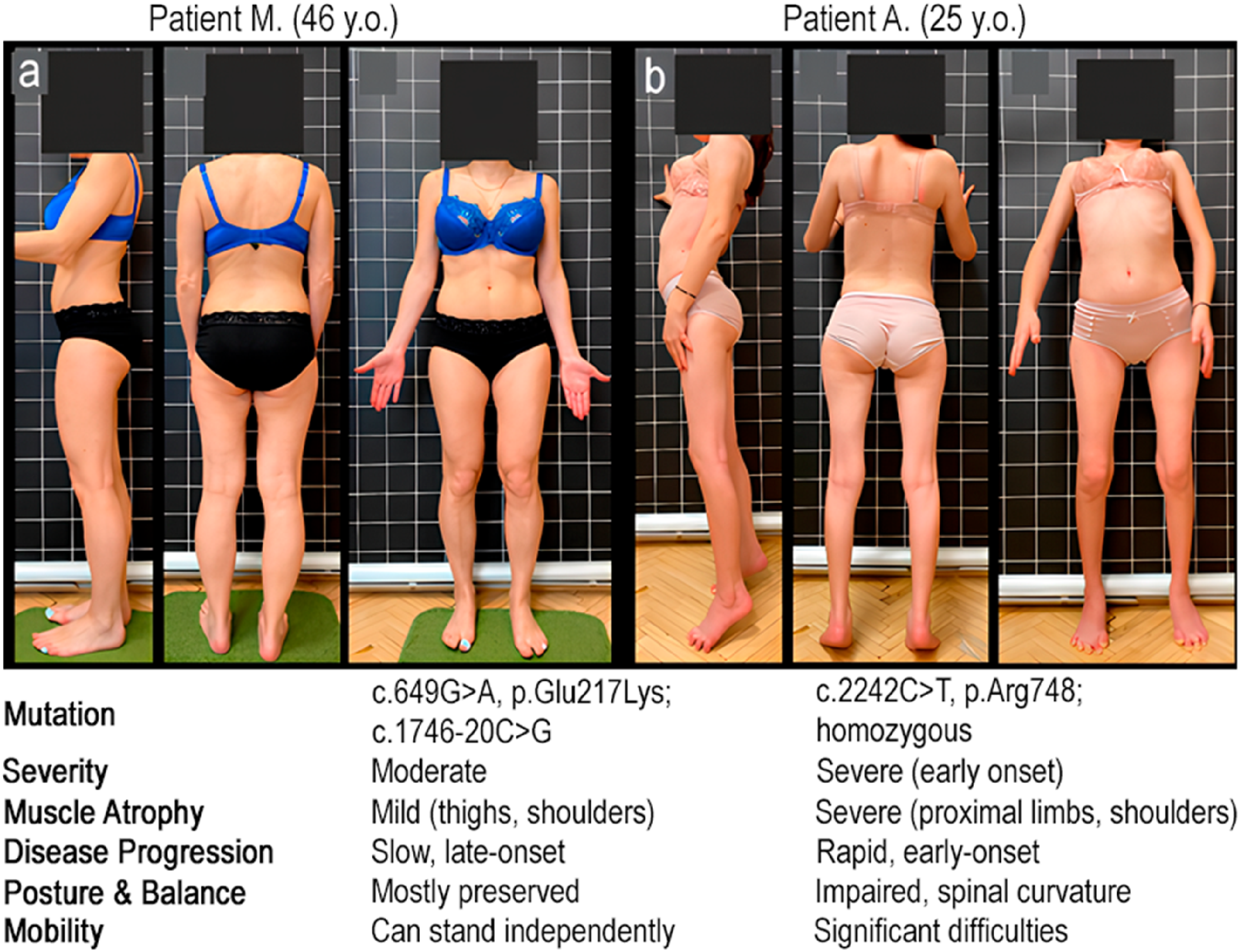
Patient phenotypes based on severity and age of onset. a – patient M., 46 years old, moderate phenotype, with c.649G > A, p.Glu217Lys; c.1746–20C > G in CAPN3; b – patient A., 25 years old, early severe course, with c.2242C > T, p.Arg748; homozygous in CAPN3.
Symptoms of the disease
The primary clinical symptom is the involvement of the skeletal muscles, primarily in the limbs, manifesting as weakness and motor deficit (inability to perform routine activities, such as running, climbing stairs, blow-drying hair, hanging curtains, etc.). Muscle dystrophy primarily manifests as atrophy and is mostly symmetrical; however, minor asymmetry can be observed in 14% (95% CI: 6–25) 17 to 21% (95% CI: 4–50) 21 of cases. A characteristic feature is the development of compensatory hypertrophy of the m. gastrocnemius (in 75% of cases) and, less commonly, the anterior thigh muscles (m. sartorius and m. obturator externus).16,21 In the case of calpainopathy, this is pseudohypertrophy, as muscle fiber enlargement is accompanied by hypotonia and reduced muscle strength.
In addition to skeletal muscle damage, contractures contractures are common, affecting the ankle (86%), hip (69%), knee (63%), and elbow (56%) joints. 22 These contractures are progressive in both severity and location. 15 As a result, they significantly impair the weight-bearing function of the lower limbs, often leading to toe walking. 19 Patients who develop early contractures of the Achilles tendons, knee flexors, elbow joints, finger flexors, and spinal rigidity are classified under the “early contracture phenotype” concept.19,23
The involvement of the respiratory muscles typically occurs only in the later stages of the disease, characterized by a reduction in forced vital capacity (FVC) to 78–80% without signs of diaphragmatic involvement.12,22 However, among European cohort patients, 11% experienced a reduction in FVC to less than 50%, and a few required noninvasive ventilation. 22 Additionally, certain endemic alleles are associated with diaphragmatic dysfunction, leading to a decrease in vital capacity to 30–50% in the late stages (Gardner–Medwin and Walton classes V–VII). Isolated cases of respiratory failure and subsequent death by the age of 19 have been reported. 15
Myocardial dysfunction is not a typical feature of calpainopathy and generally follows a benign course, presenting with conditions such as tachycardia and hypertrophic cardiomyopathy. 24 In most cases, these patients develop secondary myocardial remodeling due to valve dysfunction. 25 Meanwhile, the average left ventricular ejection fraction was 67.5%, with a reduction below 60% observed in only 16% of cases. 22 In cases of significant myocardial damage, such as dilated cardiomyopathy, arrhythmias, or cardiac conduction disorders, additional diagnostic studies should be conducted to exclude other types of LGMD, particularly those associated with the sarcoglycan complex. 26
As the disease progresses, dependence on a wheelchair typically develops 15–25 years after initial onset, 12 with the average age for the loss of ambulation (loss of independent mobility) being 31 years (95% CI: 27–33). 22
LGMDD4 is characterized by a variable and milder clinical phenotype, ranging from asymptomatic forms to moderate cases in which loss of ambulation occurs after the age of 60. One of the most common manifestations of the AD form is muscle pain in the limbs and back, observed in 50% of cases. 9
Clinical phenotypes
The clinical forms of LGMDR1 include the most common Leiden–Möbius pelvifemoral phenotype, characterized by onset and predominant muscle weakness in the pelvic girdle muscles; the Erb scapulohumeral form, with initial manifestations in the shoulder girdle; and asymptomatic hyperCKemia, which represents a presymptomatic stage that can be prolonged in some cases. 12 Previously described idiopathic eosinophilic myositis (with or without peripheral hypereosinophilia), considered an initial manifestation of calpainopathy in oligosymptomatic patients, was also identified in patients with fully developed clinical symptoms in subsequent studies.27,28
The main manifestations of the Leiden–Möbius phenotype primarily involve the thigh muscles (e.g., m. adductor magnus, m. gluteus maximus, and the posterior group, predominantly m. semimembranosus, with subsequent spread to the anterior muscles) and the calf muscles (e.g., m. gastrocnemius medialis and m. soleus, with later progression to the peroneal group).12,17 As the disease progresses, functional impairments extend to the scapular muscles (within 2.5–5 years, on average 15 ), involving m. latissimus dorsi, m. erector spinae, m. rhomboideus, m. serratus, and m. pectoralis. This leads to the development of winged scapulae (in 18–20% of cases) and hyperlordosis.12,17,22,29 Eventually, the m. triceps brachii and forearm muscles are also affected, 17 whereas the facial, extraocular, tongue, and neck muscles typically remain intact.12,15
The scapulohumeral form is characterized by a milder course and later onset (averaging 22.6 years), with predominant involvement of the periscapular muscles and m. biceps brachii.14,15,30 Over time, the disease extends to the abdominal muscles and, in later stages, affects the pelvic girdle muscles.15,30 Asymptomatic patients or those with mild clinical signs of calpainopathy may exhibit exercise-induced rhabdomyolysis and/or myoglobinuria 31 (Figure 2).
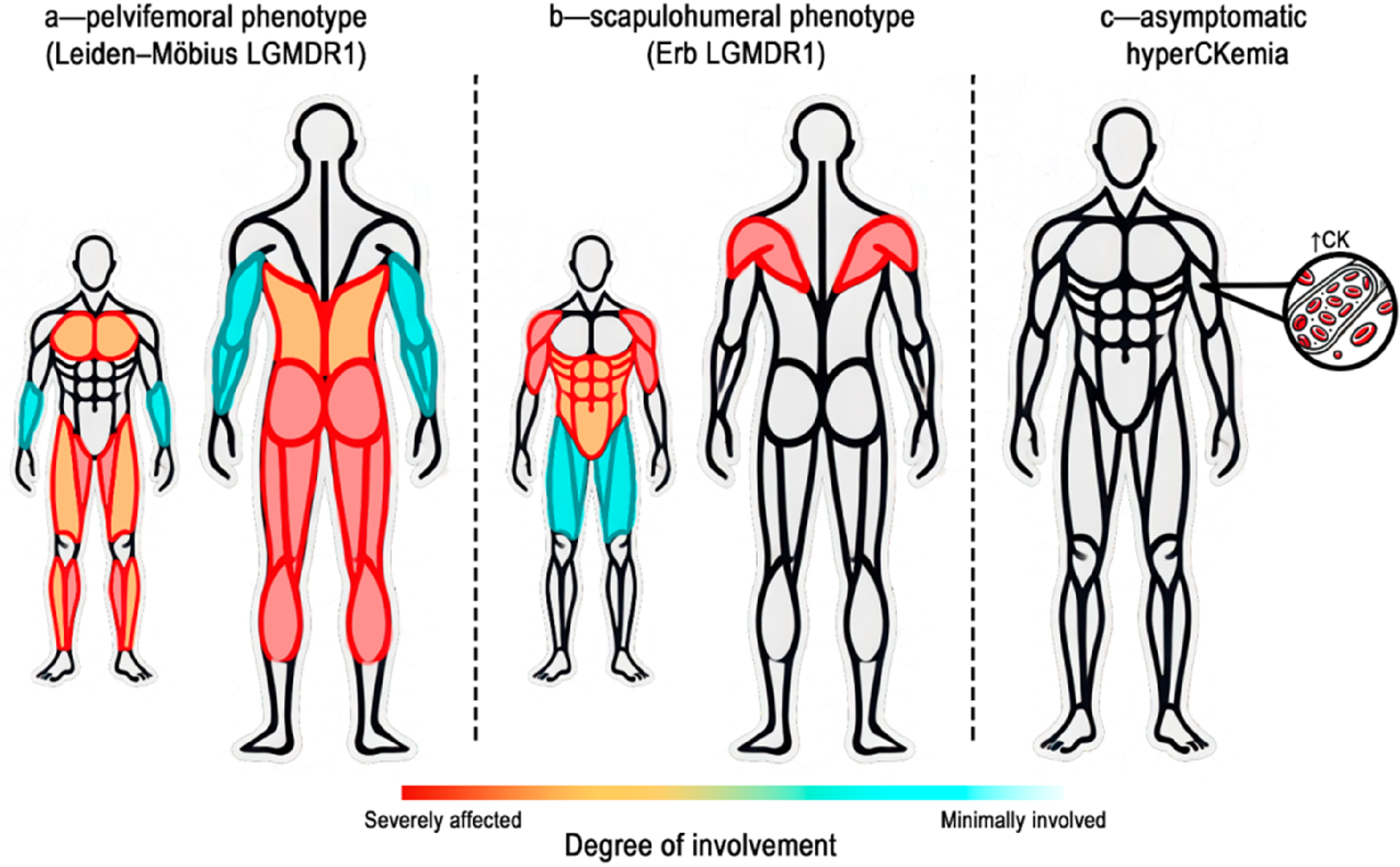
Clinical phenotypes of patients based on the topographic distribution of predominantly affected muscles. a – pelvifemoral phenotype (Leiden–Möbius LGMDR1); b – scapulohumeral phenotype (Erb LGMDR1); c – asymptomatic hyperCKemia.
Atypical variants have been identified in 2% of cases, 18 including a case of camptocormia in a carrier of a mutant allele in the CAPN3 gene 32 and a pseudometabolic type characterized by complaints of muscle stiffness, myalgia, and exercise intolerance.19,33
Therefore, the clinical manifestations of calpainopathy are nonspecific and exhibit significant phenotypic variability. The late age of onset in some cases makes it difficult to suspect a genetic disorder in the early stages, leading to diagnostic challenges.
Diagnosis of calpainopathy
Laboratory methods
The primary laboratory method for suspecting myodystrophic damage is a biochemical blood test to determine serum CK levels. In LGMDR1, the CK levels are always significantly above reference values (5–80 times higher), with a subsequent decline as the fibrofatty replacement of the affected muscles progresses. 34 Average CK levels vary widely, from 750 U/L 22 to 5048 U/L 18 in different cohorts.
Additionally, elevated levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), lactate dehydrogenase (LDH), CK-MB, and myoglobin can be noted as a result of dystrophic changes in muscle fibers. 35
Electromyography
In the clinical context, needle electromyography (EMG) can help distinguish between myogenic and neurogenic damage. Additionally, EMG can provide preliminary insights into the possible type of myopathy based on indirect signs, such as the presence of myotonic or pseudomyotonic discharges. However,in the presymptomatic stage of calpainopathy, EMG often has normal characteristics.12,27,32
Imaging methods for muscle assessment
Imaging methods, such as computed tomography and magnetic resonance imaging (MRI), for soft tissues of the trunk and limbs are considered useful components in the comprehensive evaluation and differential diagnosis of progressive myopathies. Whole-body MRI enables the assessment of the extent, severity, and symmetry of muscle pathology (the degree of fatty and connective tissue infiltration and edematous changes). This approach helps establish a specific MRI pattern and suggests a differential range of myopathies, ultimately facilitating the search for causative mutations.29,36
On the basis of the degree of connective and fatty infiltration identified through MRI, muscles can be classified into several subgroups, each assigned a severity grade – the median semiquantitative score, according to E. Mercuri (Me)17,21,27,29,36 (Table 1).
Classification of muscles by degree of involvement and semiquantitative score according to E. Mercuri (Me) on the basis of MRI findings.

*In a Russian cohort, m. gracilis was involved to almost the same extent as m. vastus lateralis and medialis. 21
As previously mentioned, there is no significant involvement of the head and neck muscles,17,36 and the back extensors are characterized by a caudal-to-cranial descending gradient of involvement. 29 Smaller, deeper muscles responsible for spinal rotation and fine postural adjustments appear relatively intact on MRI. However, spinal extensors, such as the m. erector spinae, exhibit significant fatty replacement and atrophy. These muscles are more prone to degeneration in myopathies because they play a crucial role in maintaining an upright posture, requiring prolonged, high-force contractions. This constant mechanical load makes them more susceptible to damage and fibrosis over time (Figure 3).
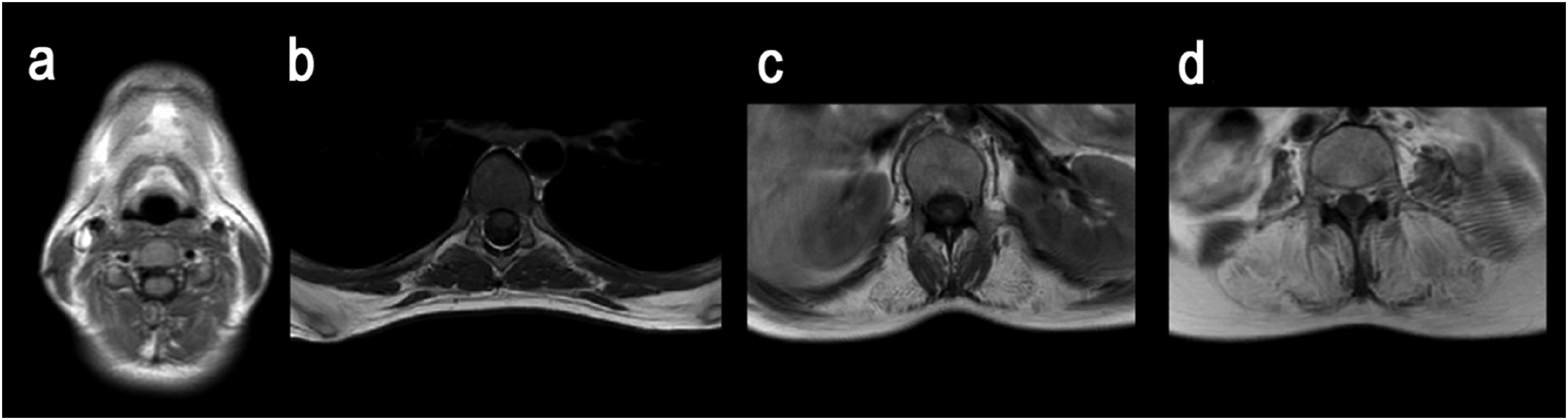
MRI of the axial musculature in patients with calpainopathy. a – undamaged neck muscles; b – thoracic region (Th6–Th7); c – lumbar region (L1) – fatty replacement of spinal extensors with preservation of rotators; d – lumbar region (L5) – total fatty replacement of back muscles during the progression of calpainopathy.
To enhance calpainopathy diagnosis, “early”, “typical/completed”, and “late” MRI patterns have been identified. The “early” pattern includes minimally affected muscles at the onset of calpainopathy; the “typical” pattern is based on the muscles most frequently involved in the fully developed stage; and the “late” pattern is characterized by the muscles that persist longest during the late stages (Figure 4).
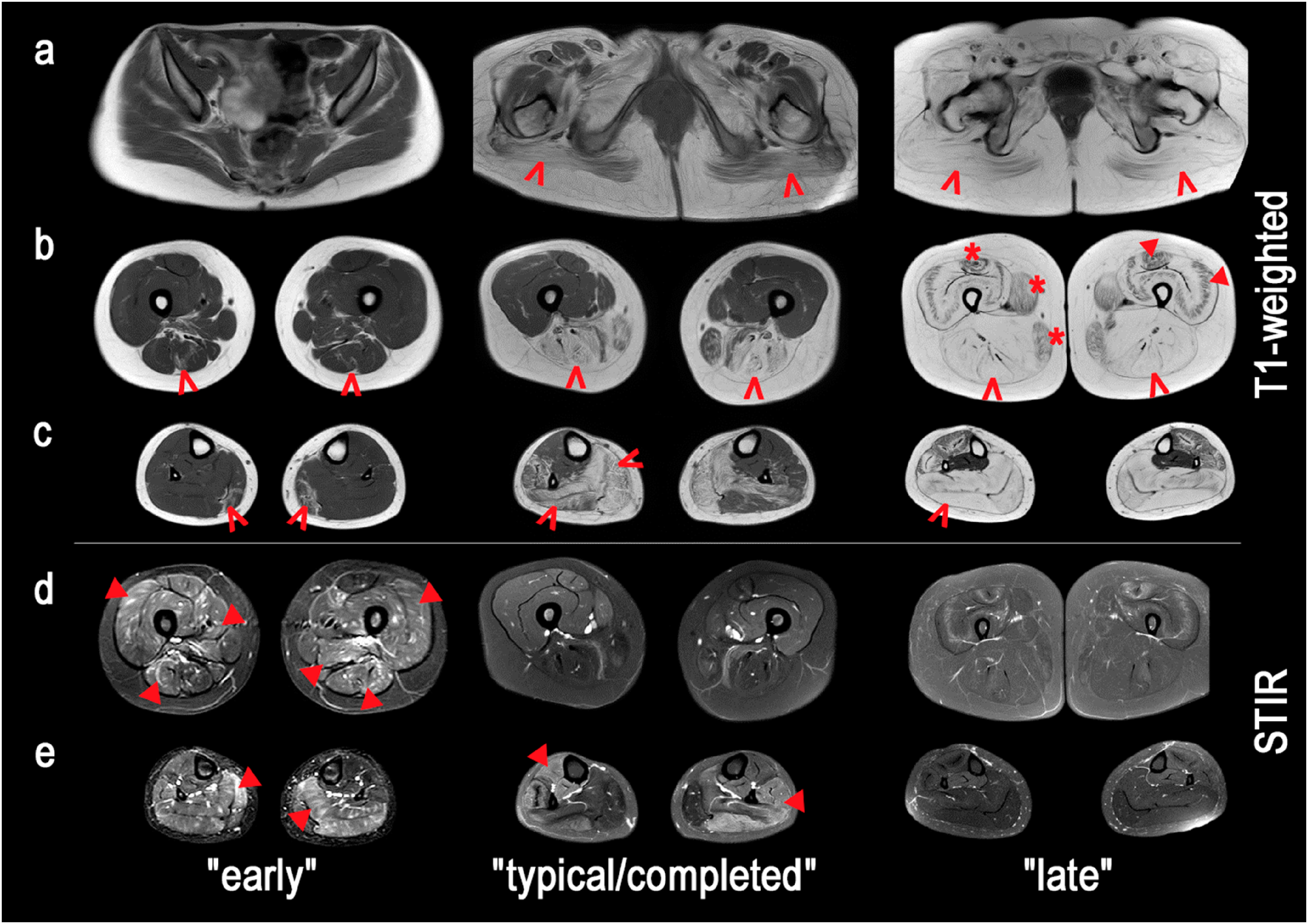
MRI patterns of muscle involvement at various disease stages in patients with LGMDR1. a, b, c – pelvic region, mid-thigh, and calf compartments on T1-weighted images – fat replacement of mm. glutei, m. vastus lateralis and intermedius, m. gastrocnemius, and m. soleus (arrows), with relative sparing of m. gracilis, m. sartorius, and m. rectus femoris (asterisks), and a “collagen-like” pattern of the anterior thigh compartment (triangles); d, e – mid-thigh and calf muscles on short tau inversion recovery (STIR) sequences, highlighting areas of active muscle inflammation and edema, which appear as bright hyperintense signals and decrease in later disease stages (triangles).
Although inflammatory myopathies are sometimes falsely diagnosed in cases of LGMDR1, the edematous changes observed in the STIR protocol are mainly observed in the semitendinosus and rectus femoris. 21
A distinctive MRI pattern, termed the “collagen-like” pattern (also referred to as“pseudocollagen sign,” “U-sign,” “central shadow,” “target,” and “sandwich” signs), is noted.17,37 It is characterized by significant fatty replacement in perifascial areas, most commonly involving m. vastus lateralis, m. vastus intermedius, m. rectus femoris, and the caput laterale m. triceps brachii. This concept also includes the “fan sign”, which refers to the splitting of the m. deltoideus by band-like areas of fat infiltration. 17 In a Russian cohort, this pattern was further defined by specific changes in the calf muscles (peripheral involvement of the m. soleus, central involvement of the anterior lateral muscle group, or “central shadow” pattern). 21
The “collagen-like” pattern was found in 21% of a Russian cohort (n = 14), 21 50% of a European cohort (n = 57), 17 and 72% of a Brazilian cohort (n = 18). 29 This is typical for patients with a longer and more severe disease course and is associated with the presence of two “null” alleles (i.e., the absence of the calpain-3 protein).17,21,22
This pattern in calpainopathy should be distinguished from collagen VI-related dystrophies (COL6-RDs), which are characterized by an earlier onset, involvement of both proximal and distal muscles, and a “tiger-like” pattern of muscle involvement, with selective sparing of certain muscle regions. 38 In addition, fat infiltration in calpainopathy is mosaic-like and asymmetrical, with more pronounced manifestations in the posterior thigh compartment (Figure 5).
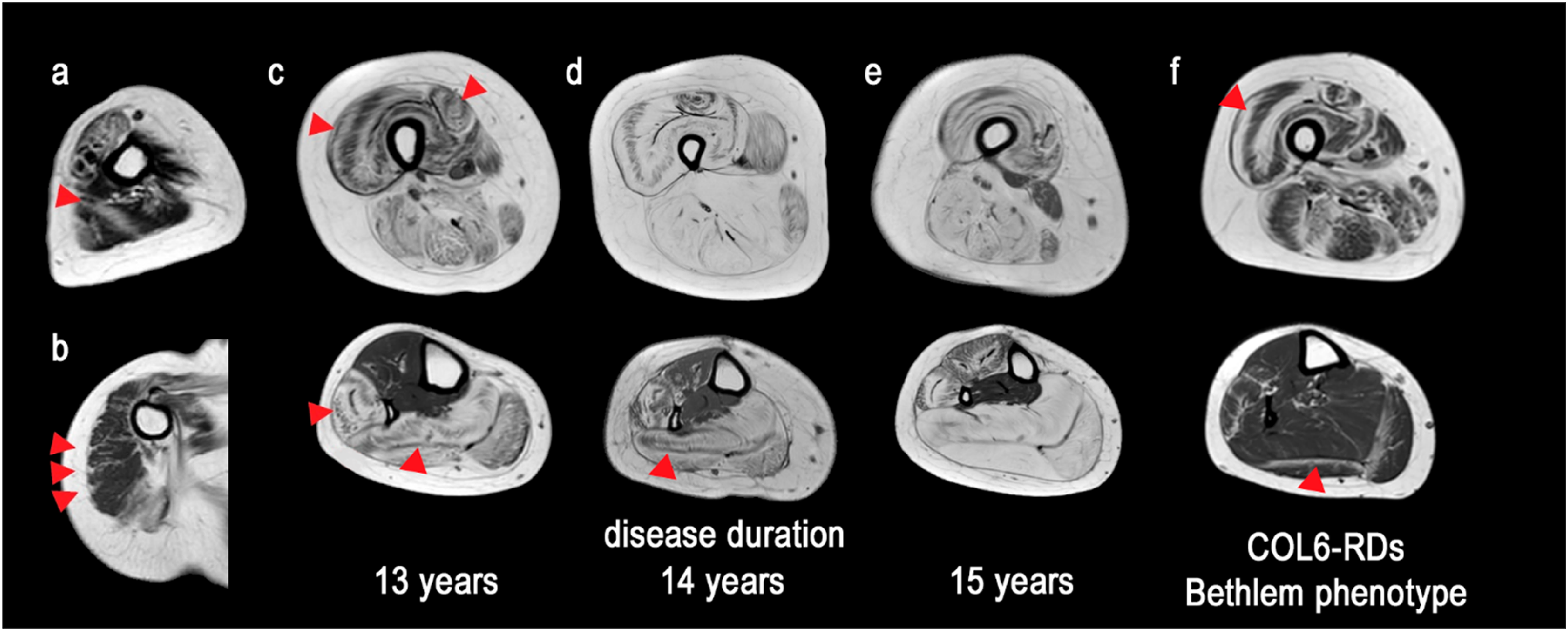
“Collagen-like” MRI pattern in patients with calpainopathy at various disease durations. a – caput laterale m. triceps brachii, showing pronounced fibrous and fatty infiltration, creating a characteristic “collagen-like” pattern (triangles); b – m. deltoideus, marked replacement of muscle tissue by connective tissue structures (triangles); c, d, e – "central shadow” sign in m. quadriceps femoris and mm. peronei – a dark area in the center of a muscle surrounded by a hyperintensive zone (triangles); f – "tiger-like” fat replacement in patients with COL6-RDs (triangles).
Molecular genetics
Next-generation sequencing (NGS) is the first-line approach for patients with suspected calpainopathy, enabling the identification of LGMD-related pathogenic variants. To identify mutations in the relatively large CAPN3 gene, which consists of 24 exons, a multigene panel, whole-exome sequencing, and whole-genome sequencing can be used. However, for certain intronic mutations, complementary deoxyribonucleic acid (DNA) sequencing obtained from muscle or blood tissue may be needed. 39
Pathogenic and likely pathogenic homozygous and compound heterozygous allele variants can cause the autosomal recessive form LGMDR1, whereas heterozygous mutations result in the autosomal dominant variant LGMDD4.9,10,40 Approximately 45% of the identified mutations lead to the absence of the protein, indicating that LGMDR1 is due to calpain-3 functional deficiency. 41 LGMDR1 has been described in many countries across several continents, but certain regions exhibit a higher prevalence of specific mutations. The most common CAPN3 mutations in the Russian population are c.550delA and c.598-612del15 (up to 35%). 42 These mutations are also prevalent in other countries, including Croatia, Turkey, the Czech Republic, Bulgaria, Germany, Italy, specifically in Northeastern Italy, and Poland, likely due to a founder effect from the Eastern Mediterranean region. 33 Additionally, several enclaves worldwide have been described with highly prevalent CAPN3 mutations, as shown in Figure 6.
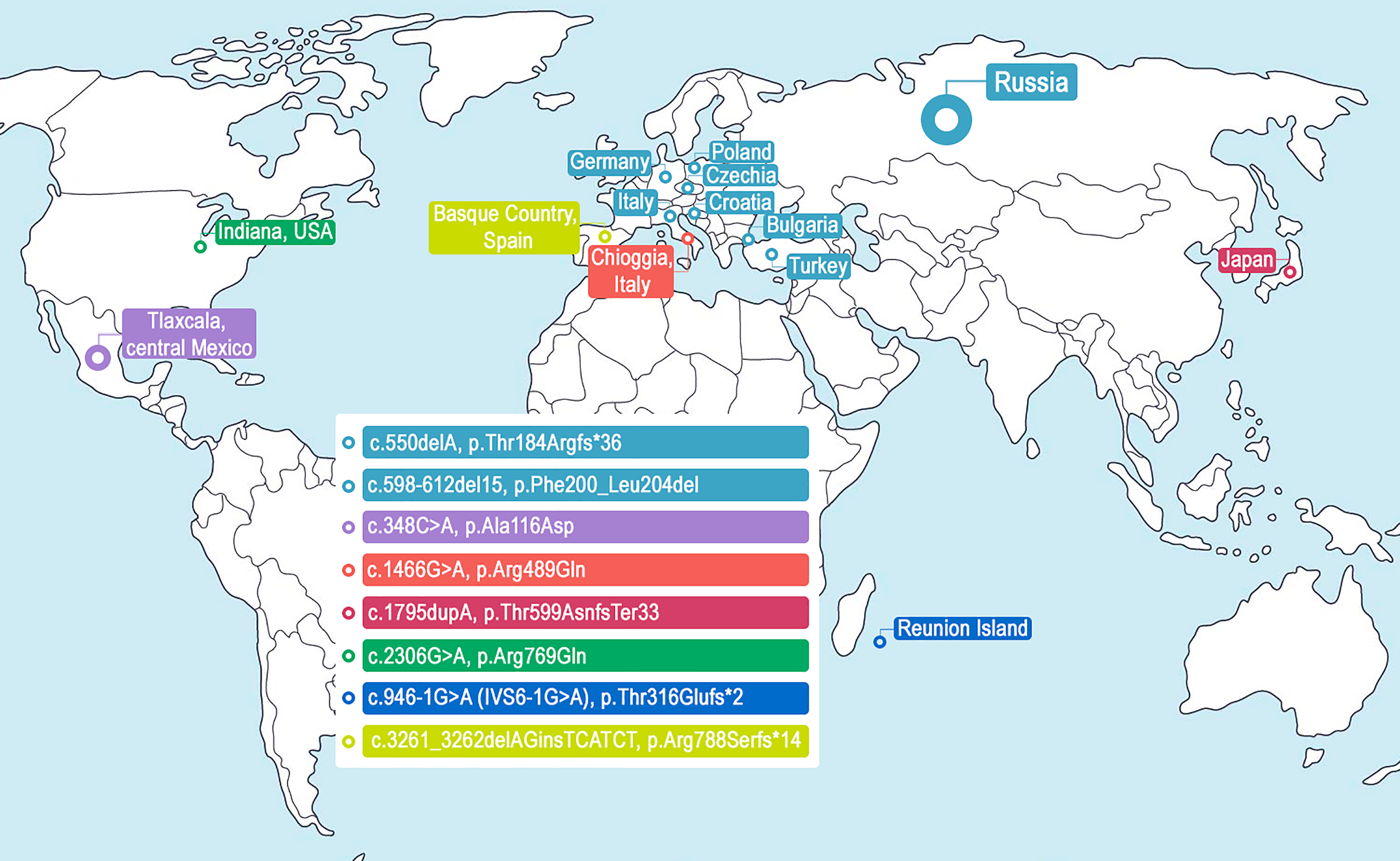
The most common mutations in the CAPN3 gene. c.550delA, p.Thr184Argfs*36 and c.598-612del15, p.Phe200_Leu204del (Eastern Mediterranean region); 33 c.348C > A, p.Ala116Asp (Tlaxcala, central Mexico); 43 c.1466G > A, p.Arg489Gln (the village of Chioggia in the Venetian Lagoon, Italy); 8 c.1795dupA, p.Thr599AsnfsTer33 (Japan); 44 c.2306G > A, p.Arg769Gln (Amish community, northern Indiana, USA); 45 c.946-1G > A (IVS6-1G > A), p.Thr316Glufs*2 (Reunion Island); 15 and c.3261_3262delAGinsTCATCT, p.Arg788Serfs*14 (Basque Country, Spain). 46
Mutations leading to LGMDD4 are typically located near the calmodulin-binding site in the CAPN3 gene, disrupting the proteolytic activity of calpain-3. 10 Mutations in CAPN3 leading to LGMDD4, such as c.643_663del21, have a dominant-negative effect. 9 Since active calpain-3 forms a homodimer, an aberrant protein can polymerize with the wild-type protein, rendering the complex inactive.9,40 Almost full penetrance is observed in most patients by adulthood. 40
There are no absolute, consistent genotype–phenotype correlations in calpainopathy, although “null” homozygous variants (indicating the complete absence of protein expression) are typically associated with a severe phenotype, early onset, and relatively high levels of serum CK.18,22,47
Mutation analysis at the messenger ribonucleic acid (mRNA) level is a rapid and informative method and is less costly than genomic analysis. 47 However, the diagnosis of LGMDR1 through CAPN3 mRNA analysis should be confirmed via DNA investigation, as the results may vary. In cases with two “null” mutations, transcripts produced by both alleles degrade, but polymerase chain reaction (PCR) is sufficiently sensitive to amplify residual amounts from both alleles. In cases involving a “null” mutation combined with a missense mutation, the transcript of the allele with the null mutation degrades, whereas the PCR fragment of the allele with the missense mutation is amplified and often interpreted as homozygous. 12 Importantly, mRNA analysis should be conducted via skeletal muscle biopsy rather than peripheral blood, as leukocytes express four different transcripts (generated by alternative splicing of exons 6, 15, and 16), all of which lack exon 15. 39
Immunoblotting
If molecular methods fail to identify a pathogenic LGMD variant, a percutaneous muscle biopsy should be performed on the most affected muscles, guided by clinical characteristics and MRI findings, followed by protein immunoanalysis. One of the important diagnostic methods for calpainopathy is immunoblotting/Western blotting via monoclonal antibodies against calpain-3 (Calp3–2C4, which recognizes exon 1). 48 This antibody allows for the detection of the absence of or a reduction in the protein,12,49 and helps distinguish most true defects in calpain-3 from secondary reductions in the protein. 48
In approximately 80% of cases with pathogenic variants in the CAPN3 gene, a complete absence or significant reduction in calpain-3 protein (over 50%) is observed, whereas in 20% of cases, a normal quantity of the protein is found due to mutations that impair its catalytic function. 50 Cases of secondary reductions in calpain-3 levels have been reported in other forms of LGMD, such as dysferlinopathy 51 and titinopathy. 52 Conversely, decreased dysferlin expression has been noted in muscle biopsies of patients with LGMDR1. 47 It has been suggested that calpain and dysferlin may interact and directly participate in sarcolemma repair 53 , and that the presence of titin stabilizes calpain-3, preventing autolytic degradation. 54
Conversely, muscle biopsy analysis revealed that approximately 20–30% of patients exhibit normal calpain-3 expression via Western blotting.34,55,56 These patients typically have missense alleles that result in impaired autolytic activity of the protein or, in 32% of cases, interdomain disintegration of the protein or reduced sensitivity to Ca2+ without functional impairment.56–58
Thus, Western blotting serves as an initial screening test for LGMDR1, where a normal level of calpain-3 does not exclude the diagnosis of calpainopathy and necessitates more complex analysis of autocatalytic activity. The probability that a patient has calpainopathy is very high (up to 84%) only when a complete absence of calpain-3 is detected in the muscle biopsy via Western blotting. 12 Moreover, the secondary reduction of the protein in LGMDR2 and titinopathy accompanied by diminished or absent bands on Western blot analysis and shows a normal IHC pattern with Calp3–2C4 antibodies. 48 A reduction in calpain-3 in immunoblotting can also result from partial degradation due to inadequate preparation of the muscle biopsy sample (e.g., partial thawing or moisture exposure). 56
Myopathological data
In certain cases, when there is an incomplete correlation between clinical symptoms and MRI findings, when identified CAPN3 gene mutations are of uncertain significance, or when calpain-3 levels are normal according to Western blot analysis, an in vivo myopathological examination of muscle biopsy samples is conducted. It refers to the microscopic analysis of muscle tissue taken from a living patient, usually from the m. biceps brachii or m. quadriceps, to diagnose or monitor muscle diseases. In equivocal cases, hematoxylin and eosin staining supports a chronic dystrophic process and helps to exclude mimics, such as inflammatory myopathies, neurogenic atrophy, and metabolic or mitochondrial myopathies. Also, certain histopathological features can raise suspicion for calpainopathy specifically, and immunohistochemistry might show abnormal localization or patchy expression not detected by Western blot.
Histologically, it typically reveals characteristic signs of an active, nonspecific myodystrophic process in most patients with calpainopathy. These signs include polymorphisms of individual muscle fibers (round, atrophied, and hypertrophied on cross-sections), numerous regenerating fibers amidst occasional necrosis, and endomysial and perimysial lipomatosis and fibrosis. 12 In patients with an autosomal dominant inheritance pattern, myopathic changes are less pronounced.9,10
Histoenzymatic reactions for ATPase at various pH levels reveal a normal ratio and diameter of type I and type II fibers in patients with a mild course of the disease, while more severe cases show a predominance of type I fibers and a reduction in the size of type II fibers. 15
A characteristic histological feature is the presence of lobulated (trabecular) fibers, which were initially considered typical for this pathology. Their number correlates with disease progression, increasing in the late stages and being more commonly found in proximal muscles.28,59 However, in some cases, a significant number of lobulated fibers are also detected in the early stages of calpainopathy. 59 In 1999, B. Weller et al. introduced the term “myopathy with trabecular muscle fibers,” referring to a group of diseases characterized by a high frequency (20–90%) of lobulated fibers as the dominant pathology. 60 Subsequent research demonstrated that trabecular fibers, with a unique pattern of oxidative enzymatic reactions in NADH–tetrazolium reductase histochemical staining, reflect an abnormal distribution of the intermyofibrillar mitochondrial network. 61 These fibers were found to be nonspecific muscle changes and were also observed in Duchenne muscular dystrophy, α-sarcoglycanopathy, and facioscapulohumeral dystrophy, where their presence did not correlate with disease severity. 59
Notably, microarray analysis of muscle biopsies from patients with a high frequency of lobulated fibers and calpainopathy revealed the upregulation of actin-related genes, which is consistent with the disorganization of myofibrils described earlier. 62 Patients with no detectable calpain on Western blot showed more lobulated fibers (11.6%) compared to those with reduced calpain-3 levels (7.9%). 18
In immunohistochemical (IHC) analysis using NCL-CALP-2C4 (exon 1) and NCL-CALP-12A2 (exon 8) antibodies, a homogeneous cytoplasmic reaction with calpain-3 is observed in control human muscle, which is more pronounced with NCL-CALP-2C4 and, to a lesser extent, with 12A2 antibodies. 48 At higher magnification in longitudinal muscle fiber sections, calpain-3 is detected in the sarcomere region, with the reaction being more pronounced at myotendinous junctions when 12A2 antibodies are used. 63 The IHC reaction is more positive in immature muscle fibers, whereas in biopsy areas with a significant number of necrotic changes, variability in the reaction intensity is observed, ranging from complete absence to weak homogeneous or granular cytoplasmic patterns. 48 However, due to the structure of calpain-3 and its existence in multiple isoforms, no reliable antibodies are currently available for its detection. 64 Developing antibodies that can distinguish these isoforms is technically challenging, as many may cross-react with other members of the calpain family or unrelated proteins, leading to false-positive results and complicating data interpretation.
The inflammatory cell infiltrate in calpainopathy within the endomysium contains between 0 and 8.2 clusters (an average of 0.7 clusters per cm²) and includes PU.1+ (macrophages), CD3+, CD4+, and CD8+ lymphocytes. The expression of major histocompatibility complex class I and the deposition of complement C5b-9 are either absent or focal.28,65 Notably, a significant number of eosinophils are present in both children and adults with fully developed clinical symptoms.28,66 Eosinophilia in the infiltrate occurs not only in calpainopathy but also in γ-sarcoglycanopathy, which, in some cases, has been misdiagnosed as idiopathic eosinophilic myositis.67,68 In LGMDR1 patients, eosinophilia can be observed not only in the inflammatory cell infiltrate of the muscle but also in the periphery, 66 especially during the subclinical and early symptomatic stages. 27 The exact cause of this increase is unknown, although an atypical T-lymphocyte response, characterized by high calpain-3 expression, 69 is suspected to develop in response to muscle fiber necrosis. Upon activation, eosinophils release eosinophil cationic proteins and major basic proteins, which cause damage to the sarcolemma of muscle fibers. 70 The dystrophic phenotype and IHC reactions of muscle biopsies from patients with calpainopathy are shown in Figure 7.
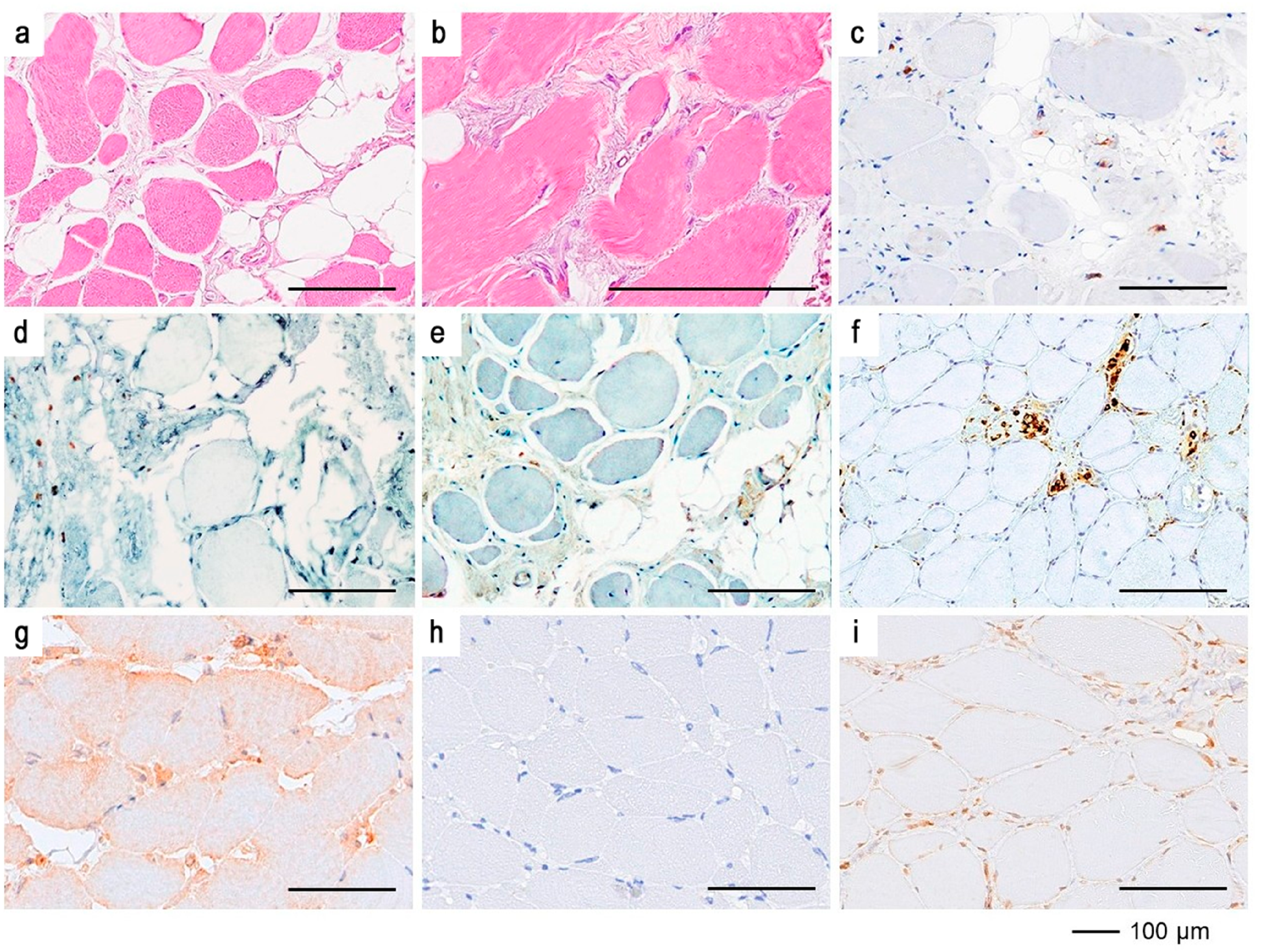
Myopathological examination of the m. vastus lateralis and IHC reaction. a – lobulation and polymorphism of muscle fibers with areas of fibrosis and fatty replacement; b – eosinophils in the endomysium of muscle fibers; c, d, e – clusters of CD45+, CD3+, and CD4+ T cells, respectively, in the fibrotic region; f – invasion of CD68 + macrophages into muscle fibers; g – cytoplasmic pattern of calpain-3 distribution in muscle fibers with normal CAPN3 expression (positive control); h – absence of specific calpain-3 staining in muscle fibers with normal protein levels when primary antibodies are replaced with buffer solution (negative control); i – absence of calpain-3 protein in the cytoplasm of muscle fibers in a patient with calpainopathy. Staining: a, b – hematoxylin and eosin.
Electron microscopy reveals the disorganization of myofibrils against a background of nonspecific changes, 15 which does not necessitate its performance in clinical practice.
The rational diagnostic algorithm for LGMDR1 is based on identifying genetic mutation variants through NGS. If molecular genetic testing results are inconclusive, a quantitative analysis of muscle proteins is performed, including Western blotting and immunohistochemical reactions using Calp3–2C4 antibodies against calpain-3. Early and timely diagnosis is a significant factor in monitoring the rate and specific features of disease progression, with the potential to slow the progression of symptoms.
Treatment of calpainopathy
Similar to many hereditary diseases, there are currently no approved or implemented etiological or pathogenetic therapies for LGMDR1 that can alter the course of the disease. 71 The primary management strategy for these patients involves symptom control, rehabilitation measures, and treatment of complications, necessitating a multidisciplinary approach. The prognosis varies widely depending on the mutation location within the CAPN3 gene, the type of mutation, and the homozygosity/heterozygosity status. 16 Nonetheless, new technologies aimed at improving the condition and quality of life of patients with this pathology are under development. 72
Symptomatic therapy and rehabilitation
Muscle involvement in LGMD leads to decreased muscle strength, reduced joint range of motion, and orthopedic issues (such as foot deformities, scoliosis, and Achilles tendon contractures).40,73 Accordingly, it is recommended that a combination of aerobic exercises (e.g., swimming and/or cycling) be incorporated with a controlled strength training program. 74 Despite the risk of muscle damage, moderate physical activity is relatively safe, improves cardiovascular function, enhances muscle efficiency, and reduces fatigue.73,75 It is important to carefully monitor the overall condition during exercise and for 24–48 h afterward, assessing for excessive muscle soreness, muscle spasms, limb heaviness, and prolonged shortness of breath, as well as regularly checking muscle necrosis markers and episodes of myoglobinuria.76,77
After diagnosis, therapeutic physical exercise and physiotherapy, such as neurodynamic rehabilitation (Bobath therapy), which helps improve balance and coordination, stabilize gait, and prolong walking duration, are also recommended. 78 Stretching exercises should be applied to the muscle groups most prone to contracture development. 79
Orthopedic correction methods include the use of orthoses, 79 mobility aids (canes, walkers, wheelchairs), scapular stabilization devices, 71 and positioning and seating aids. 80
Supportive measures include respiratory support for individuals with nocturnal hypoventilation and/or respiratory failure, timely treatment of respiratory tract infections and cardiac complications, 81 vaccinations (COVID-19, Influenza, Streptococcus pneumoniae), effective pain management, 82 and social adaptation and daily care assistance when necessary73,83 (Figure 8).
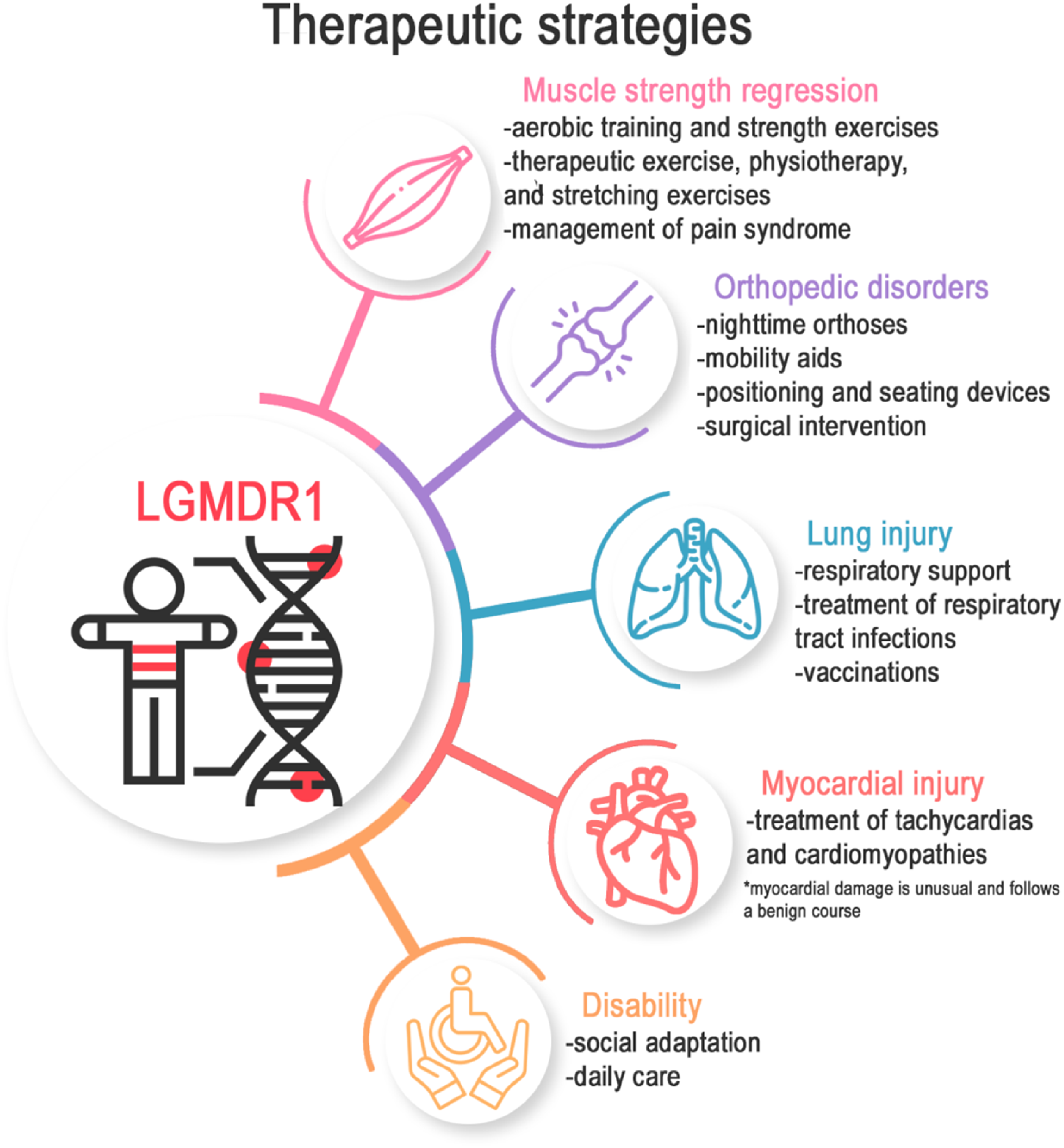
Main directions for symptomatic therapy, methods of complication management, and rehabilitation in patients with calpainopathy.
Clinical trials
Given the absence of established pharmacological treatments for LGMD, clinical trials are currently underway to evaluate the efficacy and safety of potential drugs in patients with calpainopathy. Myostatin – an endogenous negative regulator of muscle growth – is considered a therapeutic target in LGMD, to suppress its biological activity. 84 The use of myostatin inhibitors is based on reducing muscle atrophy and increasing muscle strength in conditions where muscle degeneration is a key feature. In 2008, a double-blind, placebo-controlled, randomized study involving 116 participants, including 38 with LGMD, was conducted. The therapeutic agent used was the monoclonal antibody MYO-029 (Stamulumab), an antagonist of myostatin with high affinity (NCT00104078). This study assessed the safety and efficacy of the drug via functional tests, dual-energy X-ray absorptiometry (DEXA), MRI, enzyme-linked immunosorbent assay (ELISA), and muscle biopsies. The treatment did not produce statistically significant improvements in muscle strength or function; therefore, it was considered ineffective. 85
Glucocorticoids (GCs) have been successfully used as a pharmacological therapy for various forms of muscular dystrophy, including DMD,86,87 and BMD, 88 as well as several LGMD subtypes (R2, 89 R3, 90 R4, 90 and R9 91 ). It is believed that the immunosuppressive action of steroids can reduce the inflammation caused by immune cell invasion into mechanically damaged muscles (with a membrane defect being the main hypothesis for the development of myopathies).92,93 Studies from 2022 to 2024 examined the effects of weekly oral GC administration (specifically prednisolone) in 20 patients with myopathies, including 9 with LGMDR1 (NCT04054375).94,95 Pulse therapy with prednisolone is associated with a lower frequency of side effects (reduced likelihood of developing obesity and insulin resistance) and metabolic reprogramming in dystrophic muscles, leading to improved cellular energy production. Weekly prednisone intake led to increased levels of histone 3 lysine 27 acetylation, a marker of transcriptional activation at enhancers and promoters, within KLF-responsive elements and MEF2C-binding sites, which are involved in regulating muscle metabolism and growth.96,97
A placebo-controlled study on the efficacy and safety of daily prednisolone use in 80 patients with calpainopathy and dysferlinopathy was also registered in India (CTRI/2021/02/031000), although the results have not yet been published. 98 The results of the patient evaluations are presented in Table 2.
Results of clinical studies of MYO-029 (Stamulumab) and prednisolone involving patients with LGMDR1.

Potential therapeutic targets
Preclinical studies focus on evaluating potential drugs that could have a targeted effect on the course of the disease. Developing effective therapeutic strategies requires a deep understanding of the pathogenetic molecular mechanisms underlying myopathies. To facilitate drug discovery and the study of their mechanisms of action, it is essential to develop an animal model that accurately replicates the disease phenotype. Studies on animal models of calpainopathy have clarified the mechanisms underlying muscle atrophy and provided insights into the proteolytic and nonenzymatic functions of calpain-3.
In vitro and in vivo models for preclinical evaluation
The first murine model with CAPN3 knockout was created in 2000 by gene inactivation using a vector that replaces key exons. The subsequent cloning of cells with mutant alleles was achieved through homologous recombination. This led to the development of Capn3-null (Capn3−/−) mice, which are characterized by significantly reduced CAPN3 mRNA activity and the absence of the calpain-3 protein, as confirmed by quantitative PCR and Western blotting. Laboratory tests (elevated CK levels in blood) and muscle biopsy results (central nucleation, zones of necrosis/regeneration, lobulated fibers, and mononuclear infiltration on hematoxylin and eosin staining) were similar to those observed in patients with the corresponding disease. 57 Further functional testing (treadmill, escape test, and hold on a horizontal wire), mechanical analysis of isolated muscles, and histological examinations were conducted on this model, with topographic diagnostics performed across different age groups. No differences were detected in the endurance test results, whereas dystrophic changes were most pronounced in the m. soleus and m. psoas major, indicating a progressive nature over time. 99
In the same year, a transgenic murine model in which a proteolytically inactive p94 protein (calpain-3) was expressed through the substitution of Cys129 with Ser (p94 (Capn3CS/CS)) was developed. Motor function changes were observed starting at the 20th week, whereas histological muscle fiber damage was only noticeable in 106-week-old mice. 100 A comparison of Capn3−/− and Capn3CS/CS mice has led to the conclusion that both the proteolytic and non-proteolytic activities of calpain-3 contribute to the pathogenesis of muscular dystrophy. First, the loss of proteolytic activity results in a delayed dynamic distribution of calpain-3 between the M-line and N2A (titin-binding region), as well as dysregulation and induction of myofibril degeneration in response to exercise. Second, studies have shown that the protein is a structural component of the endoplasmic reticulum (ER) and myofibrils, playing a role in Ca2+ release and stabilization of sarcomere components.101–103
Another model (mice with a complete knockout of the CAPN3 gene – C3KO/CAPN3-KO) was developed in 2004 via a gene-trap retroviral vector that introduces premature stop signals during protein translation. In C3KO mice, CAPN3 gene activity and the calpain-3 protein, as well as any potential isoforms or products of alternative splicing, were absent. The histological characteristics of the muscles were similar to those observed in patients with presymptomatic or mild forms of LGMDR1.
104
This model was subsequently used to study various pathogenetic mechanisms of the disease that could become targets for pharmacotherapy, as follows:
Investigation of the contribution of calpain family proteins as proteolytic systems in sarcomere remodeling
105
; Characterization of myotubes in the absence of CAPN3 and its role in myogenesis
106
; Assessment of the adaptive response of skeletal muscles to physical exercise and its association with the activity of the Ca-calmodulin protein kinase II (CaMKII) pathway107–110; The analysis of differential gene expression involved in the regulation of muscle cytoskeleton homeostasis
111
; The evaluation of mitochondria's function and their role as a central regulatory mechanism involved in muscle fiber growth, regeneration, and maintenance.112,113
In 2009, a model – C3-null, Capn3tm2.1Gnt – was developed114,115 and crossbred in 2019 with dysferlin-deficient mice, creating a severe double-knockout (dKO) line (B6.129–DysfprmdCapn3tm2.1Gnt) – for a more detailed investigation of calpain-3 role in disease pathology. 116 In 2022, the Jackson Laboratory created four new CAPN3-deficient mouse lines across four different mouse strains (FVB, 129, CC041, and DBA) via CRISPR/Cas9 genome editing. 117 However, all murine models showed signs of the disease (elevated CK levels, fibrotic lesions, and morphometric changes) only at late stages of development, with minimal differences from the wild type.
A study of the mutant phenotype of zebrafish as a potential model for LGMDR1 was first conducted in 2023 by inactivating the capn3b gene, which is active in the intestine, liver, and skeletal muscles.118,119 Using the CRISPR/Cas9 strategy, mutants with complete RNA absence (capn3b−/−) and partial gene deletion (capn3bmut1/mut1 and capn3bmut73/mut73) were created. Abnormal muscle structure was observed after incubation in methylcellulose and azinphos-methyl, which stimulated muscle activity and induced damage. However, Evans blue staining did not reveal sarcolemma damage in both cases. Zebrafish are versatile models for investigating disease pathogenesis 120 and studying drug libraries 121 to increase drug discovery efficiency. 122
In 2022, the CRISPR/Cas9 gene editing of pig embryos was replicated by deleting the start codon and causing a frameshift mutation. RT‒PCR and electrophoresis revealed a high percentage of CAPN3 KO embryos with at least one mutation, suggesting their potential use for generating calpain-3-deficient individuals. 123
In addition to animal disease modeling, creating human skeletal muscles in vitro is a promising approach. In 2018, a new functional model deficient in dystrophin and calpain-3 proteins was developed based on the immortalized myogenic cell line LHCN-M2. An LGMDR1 cellular model with suppressed gene expression – shCapn3 – was created by infecting the resulting myoblasts with a lentivirus. 124 In 2022, more efficient myogenic lines – 8220, KM900, and 918 – were subsequently used. 125
Using induced pluripotent stem cells (iPSCs) reprogrammed from fibroblast samples of three patients with LGMDR1, calpain-3-deficient myotubes were differentiated in 2019. The use of iPSCs is particularly promising for muscle disorders, 126 as it enables the replacement of dystrophic muscles with stem cells through autologous transplantation. 127 In 2024, myogenic precursors derived from iPSCs were generated via 3D tissue engineering, followed by the delivery of lentiviral particles. 128 All the generated in vivo and in vitro models are presented in Figure 9.
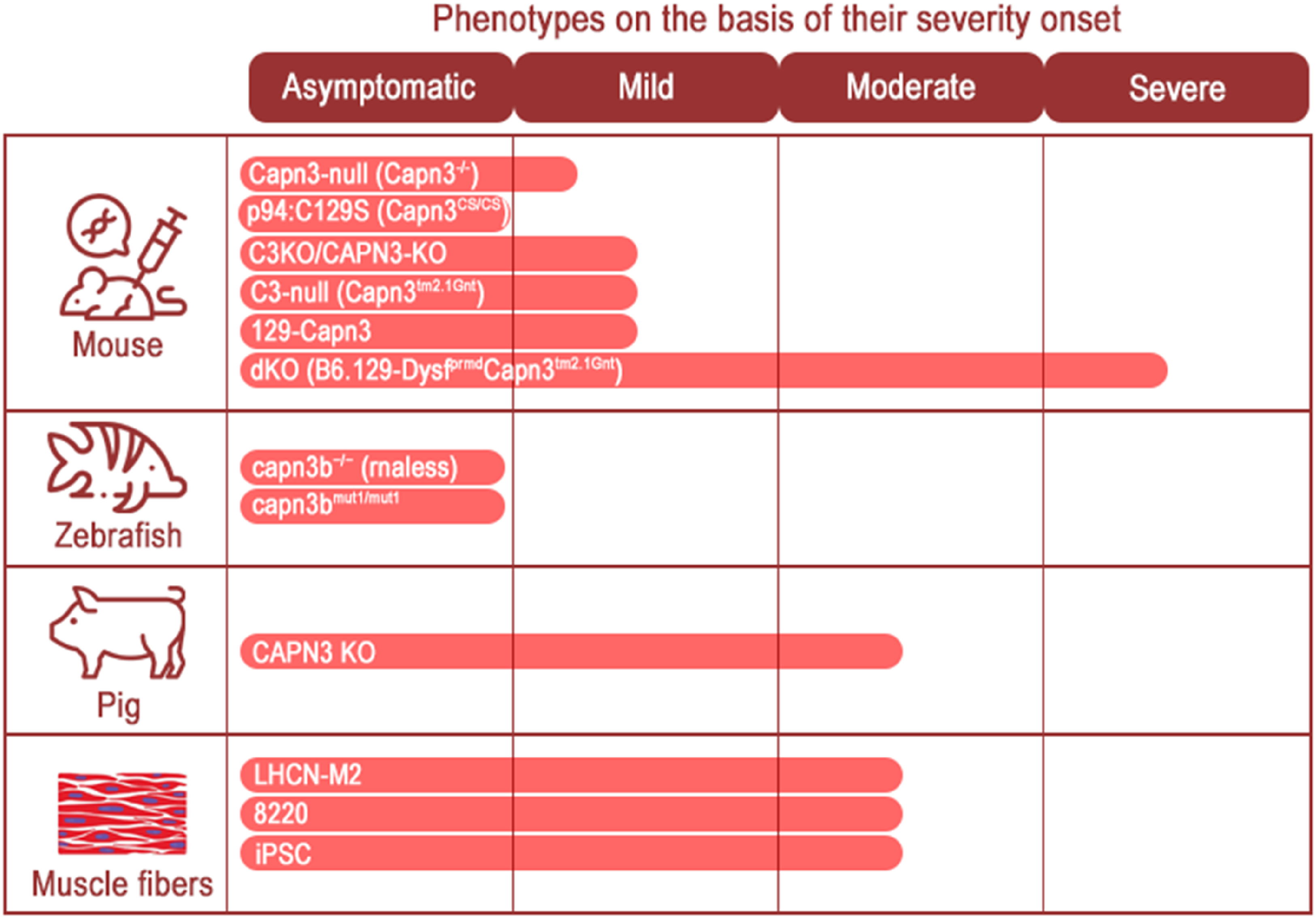
In vitro and in vivo models used to study LGMDR1 in relation to the severity of the disease in humans.
Despite successful attempts to develop murine models that exhibit some characteristics of the disease, LGMDR1 in humans is significantly more severe, with greater declines in muscle strength and endurance. In mice, physical exercise tolerance does not decrease significantly; however, muscle damage and dystrophy are indicated by elevated CK levels and histological signs of dystrophic changes. Therefore, researchers are working to develop additional animal models that are anatomically and physiologically closer to humans, aiming to enhance the understanding of potential therapeutic approaches for this pathology.
Pharmacological approaches
A significant aspect of the pathophysiology of LGMDR1 is the deficit in mitochondrial biogenesis, which leads to reduced function of the respiratory chain and adversely affects the rate of muscle fiber repair.112,113,129 Increased activity of the PPAR-δ pathway is observed during submaximal physical exertion, leading to enhanced endurance and the correction of energy imbalance between muscle protein synthesis and breakdown. 130 In this context, a potential therapeutic approach was tested in vivo in 2020 – the activation of mitochondrial activity using the PPAR-δ agonist GW501516, which simulates physical activity. 131 The RT‒PCR results revealed increased expression of Pax7 and MyoD, indicating a potential increase in the pool of satellite cells, which serve as reserves for skeletal muscle tissue. Functionally, the mice demonstrated improved coordination and physical exercise tolerance. 113
The ryanodine receptor (RyR) in skeletal muscles functions as a channel for releasing calcium from ER into the cytosol during excitation–contraction coupling. Calcium is subsequently reabsorbed via the Na+/Ca2+ exchanger via the Ca2+-ATPase pump of the ER (sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) protein). In murine models and human myotubes deficient in calpain-3, reduced expression of RyR1 and activity of the CaMKII signaling pathway were observed,101,107 along with impaired function of all SERCA protein isoforms.124,132 Diminished CaMKII signaling, in turn, leads to decreased expression of a set of genes encoding slow isoforms of sarcomeric proteins and a reduction in transcripts of genes involved in oxidative metabolism. 109 Accordingly, efforts have been made to target this described triad of proteins with small molecules to increase calcium ion release and circulation. 133
In 2020, libraries of pharmacological compounds comprising more than 2000 substances were analyzed for their potential to influence gene expression. Although this approach is quite common, specific therapeutic targets for calpainopathy have not yet been developed due to a lack of understanding of the molecular mechanisms of pathogenesis. Based on cell line data, a lead candidate, AMBMP, was selected for in vivo proof-of-concept testing. AMBMP is an activator of the Wnt signaling pathway and is hypothesized to stimulate the release of intracellular Ca2+ via CaMKII. The compound was administered intraperitoneally to C3KO mice, followed by histological examination of the lower limb muscles. Muscle biopsies revealed no significant pathological changes, such as fibrosis or inflammation. NADH staining revealed an increase in the number of oxidative fibers, as well as an increase in the cross-sectional area of both slow and fast fibers. The predominance of muscles with an oxidative phenotype was attributed to a significant improvement in mitochondrial activity and increased CaMKII signaling, as confirmed by Western blotting. 134
The absence of Wnt pathway ligands activates GSK-3β, leading to the degradation of β-catenin, which plays a key role in cellular signaling and regulates processes such as proliferation, metabolism, and apoptosis. Conversely, the inactivation of GSK-3β can cause β-catenin to accumulate, bind to T-Cell Factor/Lymphoid Enhancer Factor, and regulate gene activity. Allosteric and highly selective inhibitors of GSK-3β – tideglusib and VP0.7 – were developed and used alongside lithium treatment in 2021 on cultured cells from patients with LGMDR1. This resulted in the activation of the Wnt pathway, which was confirmed by the effective inhibition of GSK-3β in human myotubes after drug treatment. However, no such shift was observed in fibroblasts or CD56+ cells after tideglusib and VP0.7 were applied. Since GSK-3β can interact with the Akt/mTOR pathway with an inhibitory effect, the activity of mTOR and its targets were also analyzed. 135
For SERCA proteins, increased levels of ubiquitination were observed in myotubes with CAPN3 deficiency, indicating that the ubiquitin–proteasome system (UPS) is a potential therapeutic target. In 2022, the ability of bortezomib, a UPS inhibitor, to restore SERCA protein activity was investigated. 125
In 2015, the function of the Mss51 gene in skeletal muscles was studied in the context of its contribution to the pathogenesis of myopathies. It is considered one of the regulators of TGF-β superfamily proteins, which include myostatin and the cytochrome c oxidase complex of the mitochondrial respiratory chain. Mss51 expression products may modulate signaling aspects in skeletal muscles, such as fiber type determination and the level of metabolic activity. 136 Given that deletion of the gene encoding Mss51 in mice results in increased energy production and improved mitochondrial activity, researchers are exploring the deficiency of this protein as a hypothetical therapeutic target for calpainopathy. 137
The endoplasmic reticulum also plays a role in this calcium-centered mechanism, where disturbances in homeostasis can lead to a condition known as “ER stress”. The compounds studied for reducing ER stress include tauroursodeoxycholic acid, which inhibits molecules associated with ER stress.138,139 Additionally, the potential use of salubrinal is being considered, as it reduces ER protein overload by inducing the degradation of untranslated mRNAs. 140
Myostatin inhibition at preclinical stages was achieved through both pharmacological agents and gene therapy approaches. In 2020, I. Kramerova and colleagues investigated the characteristics of the CAPN3 knockout mouse line following the administration of the myostatin-specific inhibitory antibody mRK35 and the application of genetic therapy methods in a crossbred C3KO/F66 murine model, which exhibited increased expression of its endogenous inhibitor, transgenic follistatin. 141 Myostatin inactivation was also conducted in 2007 through the recombinant expression of a mutant propeptide via adeno-associated virus (AAV) (pAAV-CMV-mSeAPpropmyoD76A). 142 Overall, all attempts to modify the course of calpainopathy through myostatin inhibition proved ineffective and were characterized by increased muscle mass without improvement and/or with the regression of functional reserves. These outcomes can be explained by the multimodal pathogenesis of muscle damage in LGMD, which involves a greater number of molecules and pathways, as well as the exacerbation of myocyte membrane damage due to increased fiber size and reduced oxidative capacity.141,143
Gene therapy
The use of gene constructs to restore normal CAPN3 protein function, alongside rigorous evaluation of their safety and efficacy, presents a highly promising therapeutic approach for calpainopathy. In addition, genome editing technologies show significant potential for halting disease onset and progression, as well as enhancing outcomes in preclinical animal studies.
One of the most important strategies for gene therapy in the treatment of calpainopathy is the local (intramuscular) or systemic administration of an AAV transgene carrying CAPN3. The first such construct was applied to a murine model in 2006 – AAV1-C512-CAPN3fsr – which included a muscle-specific promoter and a silent mutation to distinguish between transgenic and endogenous proteins. After intramuscular injection into the m. tibialis anterior, real-time RT‒PCR and Western blotting showed an increase in calpain-3 copy number and the restoration of its proteolytic activity. Hematoxylin and eosin staining and immunohistochemistry with CD11b (macrophage) antibodies revealed a muscle phenotype similar to that of wild-type mice, with improved morphometric parameters. Systemic administration via the femoral artery led to significant increases in the mass and tetanic isometric contraction strength of the m. extensor digitorum longus (EDL) and m. soleus. 144
Based on the concept of mitochondrial damage in LGMDR1 and the reduced oxidative capacity of muscles, 112 which leads to impaired regeneration, 145 the damage in C3KO mice was exacerbated by the administration of cardiotoxin, which triggered a cycle of necrosis/regeneration. The AAV9.MCK.CAPN3 – vector, which includes muscle-specific creatine kinase – was injected intramuscularly into the m. tibialis anterior. Subsequent succinate dehydrogenase histochemical staining demonstrated normalization of fiber size and the proportion of histochemical fiber types (slow-twitch oxidative fibers – STO/type 1) based on their metabolic activity. 146
The restoration of protein functional activity and muscle fiber regenerative capacity, and the decrease in structural changes indicate the effectiveness of therapy through the delivery of transgenic CAPN3 into cells. However, the vector-mediated overexpression of the gene (AAV2/9-desm-CAPN3), which was applied in 2013 via intravenous injection, led to the premature death of the mice before the start of the experiment. Autopsy revealed a high degree of cardiac muscle fibrosis, regardless of the dose used. Given that AAV2/9 is known for efficiently transducing cardiac muscle tissue, two other serotypes, AAV2/1 and AAV2/8, were tested. Cardiomyocyte destruction was observed in all animals at the endpoint of the experiments following injection. To confirm that cardiotoxicity is related to calpain-3 activity, the mice were injected with a vector carrying an inactive protein (mutated at cysteine 129 CAPN3 – AAV2/9-desm-CAPN3C129S). As a result, no signs of fibrosis were found. A successful strategy was subsequently developed to overcome toxicity by adding cardiac-specific microRNA-208a to the regulatory cassette to prevent CAPN3 expression in the heart. Biopsy results revealed a reduction in the number of centrally located nuclei in skeletal muscles, with no signs of cardiac fibrosis or detectable calpain-3. 147
In 2019, the therapeutic efficacy of rAAVhDes-mfaCAPN3-V5-SV40 + 2xmiR-208aT was tested in a dKO model. Quantitative gene expression analysis revealed reduced activity of muscle regeneration markers (myogenin, MyoD, and Tmem8c), macrophage infiltration markers (Cd11b), and fibrosis markers (collagen VI, collagen I, and fibronectin). The histological phenotype also demonstrated a therapeutic effect. To further explore the mechanisms of cardiotoxicity, the construct was subsequently intravenously administered to primates (Macaca fascicularis) with assessments of cardiac-specific markers (troponin T and NT-proBNP, as well as miR-208a and miR-499), serodiagnosis for anti-AAV9 IgG, echocardiography, and tissue biopsies (succinate dehydrogenase and picrosirius red staining). Predictably, the treatment increased CAPN3 expression without evidence of cardiac dysfunction or structural damage and had no toxic effects on the liver despite high concentrations. The difference in the vector's impact on cardiac tissue may be related to species-specific titin splicing, to which calpain-3 binds. 116
In 2021, Z. Sahenk et al. evaluated the efficacy of a newly developed construct – AAVrh74.tMCK.CAPN3 – a serotype with increased affinity for skeletal muscles 148 and a specific tMCK promoter. The effectiveness of the treatment was evaluated functionally (increased running endurance and contractile activity), molecularly (dose-dependent increase in CAPN3 expression and protein levels), and histologically (succinate dehydrogenase staining revealed an increase in fiber diameter with a predominance of the oxidative phenotype). In terms of safety, the drug biodistribution covered all the examined internal organs, but protein detection was limited to skeletal muscles. 149
In 2019, attempts were made to implement plasmid-mediated gene therapy by enhancing the delivery of plasmid DNA carrying the CAPN3 gene. Plasmids were injected into the m. gastrocnemius and m. quadriceps muscles of mice, followed by electroporation and quantitative measurement of the delivered therapeutic protein. 150
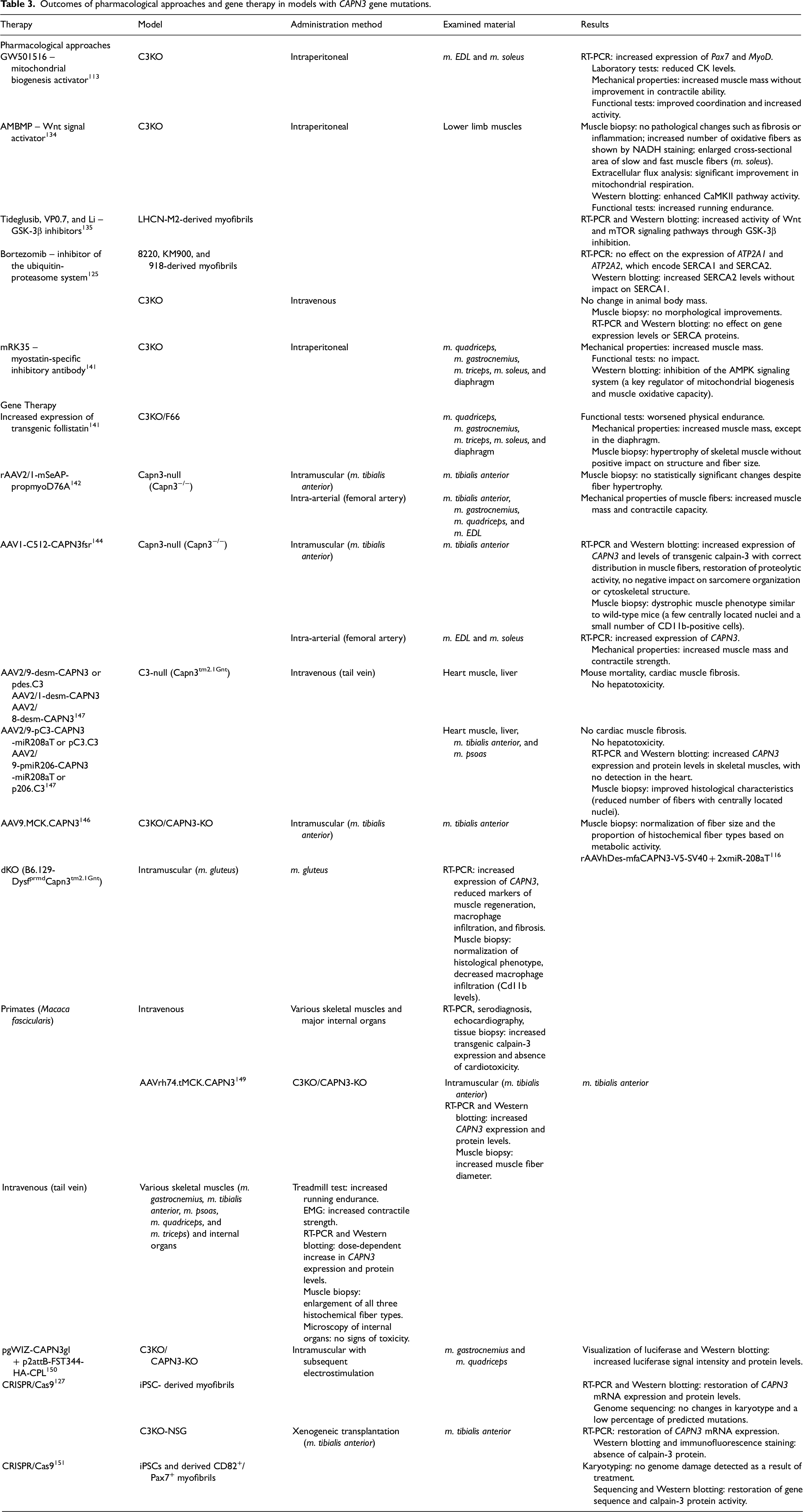
To explore the potential for autologous cell therapy, immunodeficient mice were developed in 2019 based on an existing model 104 with additional mutations in NOD/SCID and the interleukin-2 gamma receptor C3KO-NSG, which is more suitable for the heterologous transplantation of human cells. Genetically edited myofibrils were implanted into the m. tibialis anterior of mice preinjured with cardiotoxin. Immunofluorescence staining for human Dystrophin and Lamin A/C confirmed graft integration, and RT‒PCR revealed that CAPN3 mRNA expression was restored, but protein detection via Western blotting was unsuccessful. 127 Also, the CRISPR/Cas9 system was used for genome editing at the level of iPSCs and CD82+/Pax7+ myogenic precursor cells from patients with LGMDR1. 151 The results of the described therapeutic methods are summarized in Table 3.
Outcomes of pharmacological approaches and gene therapy in models with CAPN3 gene mutations.
Antisense therapy
Another potential approach for the treatment of myopathies is exon skipping, which targets mutated or adjacent exons. 152 For LGMD, the use of antisense oligonucleotides (ASOs) complementary to pre-mRNAs has been proposed as a technology for modulating splicing. ASOs specifically hybridize with the target exon, masking it from the splicing machinery, thereby increasing the size of the deletion; however, restoring the reading frame allows for the production of a shortened but partially functional protein. 153 Based on the ASO delivery method, several drugs have been developed for the treatment of Duchenne muscular dystrophy: Golodirsen, 154 Viltolarsen, 155 Casimersen, 156 and Eteplirsen. 157 Additionally, using the CRISPR system for exon skipping in vivo has proven to be an effective strategy, preventing the development of dystrophic pathology in a murine model of DMD. 158
Furthermore, to correct missense mutations in the CAPN3 gene, an alternative strategy may involve the use of mc-tRNA – engineered tRNAs that carry one amino acid but read different codons during translation. In 2024, mc-tRNA was used to correct the Arg-to-Gln mutation involved in the development of calpainopathy. 159
Conclusion
Calpainopathy, one of the most common forms of limb-girdle muscular dystrophy, is characterized by significant genotypic and phenotypic variability. The clinical presentation is nonspecific, often including progressive symmetrical weakness of skeletal muscles, leading to loss of mobility. Phenotypes include the most common pelvifemoral (Leiden–Möbius) form with early pelvic and thigh muscle weakness, and the less common scapulohumeral (Erb) form with later, milder shoulder girdle involvement. Some patients remain asymptomatic with only elevated CK levels, while rare atypical forms have also been described. These features contribute to prolonged diagnostic ability and referrals to multiple specialists before a definitive diagnosis is obtained. A rational approach to diagnosing LGMDR1 involves comprehensive clinical and laboratory evaluation, followed by quantitative analysis of calpain-3 in muscle biopsies and functional analysis of the autolytic activity of the protein. Subsequent diagnosis confirmation requires the identification of mutations in the CAPN3 gene.
Currently, there are no approved or registered drugs available for use as pathogenetic or etiological treatments for LGMDR1. Major therapeutic strategies focus on symptom management and preventing disease progression. Existing models for studying potential therapeutic approaches are viable and demonstrate disease characteristics, but disease severity does not correlate with that observed in humans. Preclinical studies of drugs affecting mitochondrial biogenesis and various calcium metabolism pathways have shown increased gene expression and/or protein levels with improvements in the muscle phenotype. Promising approaches include gene therapy methods involving the delivery of transgenic CAPN3, genome editing via the CRISPR/Cas9 system, and exon-skipping strategies.
The diverse presentation of the disease underscores the need for a thorough diagnostic framework and heightened awareness among clinicians to ensure prompt treatment and mitigate complications. Equally important is the development of better animal models to explore new pathogenic pathways and design effective therapies. Many of the molecules and genetic constructs investigated have shown promising efficacy and relative safety, highlighting the need for clinical trials to confirm their potential in slowing or halting disease progression in humans.
Footnotes
Abbreviations
Acknowledgments
The authors have no acknowledgments to report.
ORCID iDs
Authors’ contributions
SNB – literature review, data collection, and patient material with calpainopathy in clinical practice; writing sections on the clinical characteristics and diagnosis of calpainopathy. IS – literature review, writing the treatment section, text editing, and creating illustrations and tables. LAM – data collection and literature review on all experimental animal models. YSS – data collection and literature review on biotechnological treatment methods. VAT – conducting MRI studies in clinical practice for patients with calpainopathy and describing MR images. ISL – performing histological and immunohistochemical studies and describing microscopic preparations. AAI – generating the idea for the review topic, ensuring the transportation of material, and editing the text. IAY – generating the idea for the review topic, draft of the treatment section, editing the text in English, and coordination. RVD – generating the idea for the review topic, stylistic editing of the text, and ensuring the work of the research team. All authors read and approved the final manuscript.
Consent for publication
The article includes photographs and the results of imaging and histological studies conducted on patients during their diagnosis and treatment. The patients have given their consent for the publication of these results.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was carried out as part of the state assignment on the topic: “Gene-cell regulation of tissue regeneration in the musculoskeletal system and the development of pharmaceuticals based on it.” FURG-2025-0050. 1024100700005-8.
Declaration of conflicting interest
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: AAI, IAY, and RVD are shareholders in Genotarget LLC. IS, LAM, and YSS are employees at Genotarget LLC. Genotarget LLC develops gene therapy for LGMDR1. The other authors declare no conflicts of interest.