
Abstract
Congenital insensitivity to pain with anhidrosis (CIPA) is a rare autosomal recessive disorder, characterized by loss of algesthesis and inability to sweat. CIPA is known to be caused by mutations in the neurotrophic tyrosine kinase receptor type 1 gene (NTRK1). However, the details of NTRK1 mutations in Chinese CIPA patients remain unclear. In the present study, we recruited 36 CIPA patients from 34 unrelated families in mainland China. Blood samples from these patients and their available familial members were collected and subjected to genetic analysis. We identified 27 mutations in NTRK1 from this cohort, including 15 novel mutations. Interestingly, we discovered two forms of novel recurrent mutations: the first was a large intragenic deletion c.429–374_717 + 485del mediated by recombination between Alu elements, and the second was a deep intronic substitutions c.[851–798C > T;851–794C > G]. All probands were homozygotes or compound heterozygotes of these mutations. Current findings expand our knowledge about the mutation spectrum of NTRK1 in Chinese CIPA patients and provide more evidence for precise diagnosis of the clinically suspected patients with CIPA.
Introduction
Congenital insensitivity to pain with anhidrosis (CIPA; MIM 256800), also known as hereditary sensory and autonomic neuropathy type IV, is a rare autosomal recessive disorder. CIPA was first described by Swanson 1 in 1963 and is characterized by the absence of normal responses to painful stimuli, anhidrosis (inability to sweat), recurrent episodic hyperthermia, self-mutilating behavior, as well as mild-to-severe intellectual disabilities.2–4 CIPA patients often suffer complications due to accidental injuries, such as skin lacerations and fractures. These injuries can evolve into serious complications including osteomyelitis, septic arthritis, persistent infection, and delayed wound healing.5,6 Recessive loss-of-function mutations in a single gene, neurotrophic tyrosine kinase receptor type 1 gene (NTRK1; MIM 191315), were suggested to cause CIPA. 2 Human NTRK1 maps to chromosome 1q21–22 and contains 17 exons, spanning a genomic length of approximately 20 kb. The protein encoded by NTRK1 is tropomyosin receptor kinase A (TrkA), which is the preferred receptor for nerve growth factor (NGF). So far, over 105 mutations have been identified in NTRK1 from CIPA patients. However, only a few studies have examined CIPA patients in China, and most of these studies were case reports but lack in-depth genetic analysis.7,8 In the current study, we collected blood samples from 36 CIPA patients from 34 unrelated Han families in mainland China for genetic analysis of NTRK1. By identifying 15 novel mutations including two forms of recurrent mutations, current findings expand our knowledge about the mutation spectrum in NTRK1 associated with CIPA.
Material and methods
Subjects
A total of 36 CIPA patients from 34 unrelated Han families living in mainland China were recruited for this study between December 2008 and December 2017. These patients showed different levels of clinical manifestations of CIPA. All patients started to show symptoms of sensory and autonomic neuropathy from their infancies or early childhoods and were given a preliminary diagnosis of CIPA. After obtaining institutional review board (IRB) approval from the Peking Union Medical College IRB and receiving the informed consent from all participants, we collected peripheral blood samples from these patients and their family members.
Genetic analysis
Genomic DNA was extracted from blood samples using the standard sodium dodecyl sulfate-proteinase K-phenol/chloroform extraction method. 9 The coding regions and exon–intron boundaries of NTRK1 (NM_001012331.1) were amplified by polymerase chain reaction (PCR) and then subjected to automated Sanger DNA sequencing. Exon–intron boundaries were determined based on the reference sequence from the University of California at Santa Cruz (UCSC) Genome Browser website (http://genome.ucsc.edu/). The mutations found in each proband were further confirmed by PCR and the sequencing of candidate mutation region. In case that Sanger sequencing did not identify disease-causing variants in both alleles, real-time quantitative PCR (Q-PCR) was used to detect any large intragenic deletion. Gap-PCR was used to ascertain the presence of deletions, and Sanger DNA sequencing was further used to identify the breakpoints of gross deletions. The primers used for PCR amplification, DNA sequencing analysis, Q-PCR, and Gap-PCR were shown in Supplemental Table S1.
CIPA is an inherited autosomal recessive disorder that involves mutations in the NTRK1 gene. Accordingly, CIPA patients would carry a pair of mutated alleles, either homozygotes or compound heterozygotes. Among 36 probands, we found that four probands from families 4, 22, 23, and 32 carried only one mutant allele. We postulate that some intronic causative mutations may be responsible for CIPA in these patients, after excluding the possibility of any causative mutation in the coding and promoter regions in these patients by conducting Sanger sequencing and quantitative real-time PCR. In order to identify deep intronic mutation in these patients, we conducted commercial whole-genome sequencing (WGS) using the Illumina Hiseq X Ten platform (the service provided by Annoroad Gene Technology Co. Ltd.) and used Sanger sequencing to verify findings from WGS. The genome coverage and physical read depth of WGS were shown in Supplemental Figure S1.
Validation of splicing mutations
RNA analysis was used to confirm if deep intronic mutation c.[851–798C > T;851–794C > G] affects RNA splicing. Briefly, total RNA from the blood sample was isolated using Trizol reagent (Invitrogen, Cat No.15596018). Reverse transcriptase-PCR (RT-PCR) was performed using oligo dT (Promega, Cat. No. A5001). Nest-PCR was used to amplify the target cDNA fragments. T-clones (Pmd19-T Vector Cloning Kit, Takara) were used to analyze the sequence of the amplicons.
A minigene assay was used to determine the pathogenic severity of splice mutation c.575–19G > A. Briefly, DNA fragments containing the candidate splicing site and flanking regions (including two exons and one intron in each side) were generated by PCR amplification using primers NTRK1-pCAS2-F and NTRK1-pCAS2-R. The PCR products were then cloned into the pCAS2 plasmid using the In-Fusion HD Cloning kit (Clontech, Code No. 639642). Clones with wild-type or mutant genomic inserts were selected and verified by sequencing of the cloned DNA fragments. The recombinant plasmids were transfected into 293T cells using Lipofectamine™ 3000 reagent (Invitrogen, Cat No. L3000–015). For RT-PCR, total RNA was isolated from the transfected cells using Trizol reagent (Invitrogen, Cat No.15596018), and reverse transcription was performed using the GoScript™ Reverse Transcription System (Promega, Cat. No. A5001). PCR amplification was performed using the pCAS2-RT-F and pCAS2-RT-R primers, and the products were sequenced using pCAS2-RT-F. Insertion induced by a splicing mutation c.575–19G > A in NTRK1 was confirmed by RT-PCR and agarose gel electrophoresis.
In silico analysis
The pathogenicity of the candidate mutations was predicted by three mutation tolerance prediction approaches, PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), Sorting Intolerant From Tolerant (SIFT, http://sift.jcvi.org/), and MutationTaster (http://www.mutationtaster.org/). Variant frequencies were determined in the 1000 Genomes Project and ExAC (http://exac.broadinstitute.org/) database. Informatics analyses provided further ancillary support including conservation of the amino acid across species, variant predicted to be damaging in more than one in silico analyses, and mutation lying in the functional disease-related domains. Splice scores of wild-type and cryptic donor and acceptor sites were calculated in the Splice Site Score Calculation website (http://rulai.cshl.edu/new_alt_exon_db2/HTML/score.html).
Results
Clinical assessment
We ascertained 24 male and 12 female CIPA patients from 34 unrelated Han families (Figure S2). None of these patients has consanguineous parents. Except for probands 6 and 7, other 32 probands do not have affected sibling(s). The ages of these patients ranged from 10 months to 15 years old. All patients developed typical symptoms of CIPA, including anhidrosis, recurrent fever, absence of reaction to noxious stimuli, and self-mutilating behaviors. The skin of these patients was extremely dry with hyperkeratosis and cracking, especially in palm and sole (Figure 1(a)). These patients also had self-mutilating behavior, evident by the damaged tongues and fingers (Figure 1(b) and (c)). They exhibited slow wound healing (Figure 1(d)), and most (27/34) had fractures (Figure 1(e)). Deep site infections such as osteomyelitis (Figure 1(f) to (h)) were found in five patients (Table 1). Most patients (30 of 34) also have intellectual disabilities, learning disabilities, language barrier, irritable temper, and exhibited delays in motor developmental milestones, such as sitting, standing, walking, and talking (Table 1). Blepharoptosis was found in a subset of patients, with five showed obvious eyelid ptosis (Figure 1(i)).

Example images of clinical symptoms and X-ray finding in CIPA patients. (a) Dry and hyperkeratotic plantar skin with significant fissuring; (b, c) damaged tongue and hand; (d) skin lesions with slow wound healing; (e) X-ray image showing a femoral fracture caused by failing to react to painful stimuli; (f) ulcerated lesions and deformities on the foot; (g, h) joint destruction induced by osteomyelitis; and (i) eyelid ptosis.
Mutations and clinical manifestations of the CIPA patients.
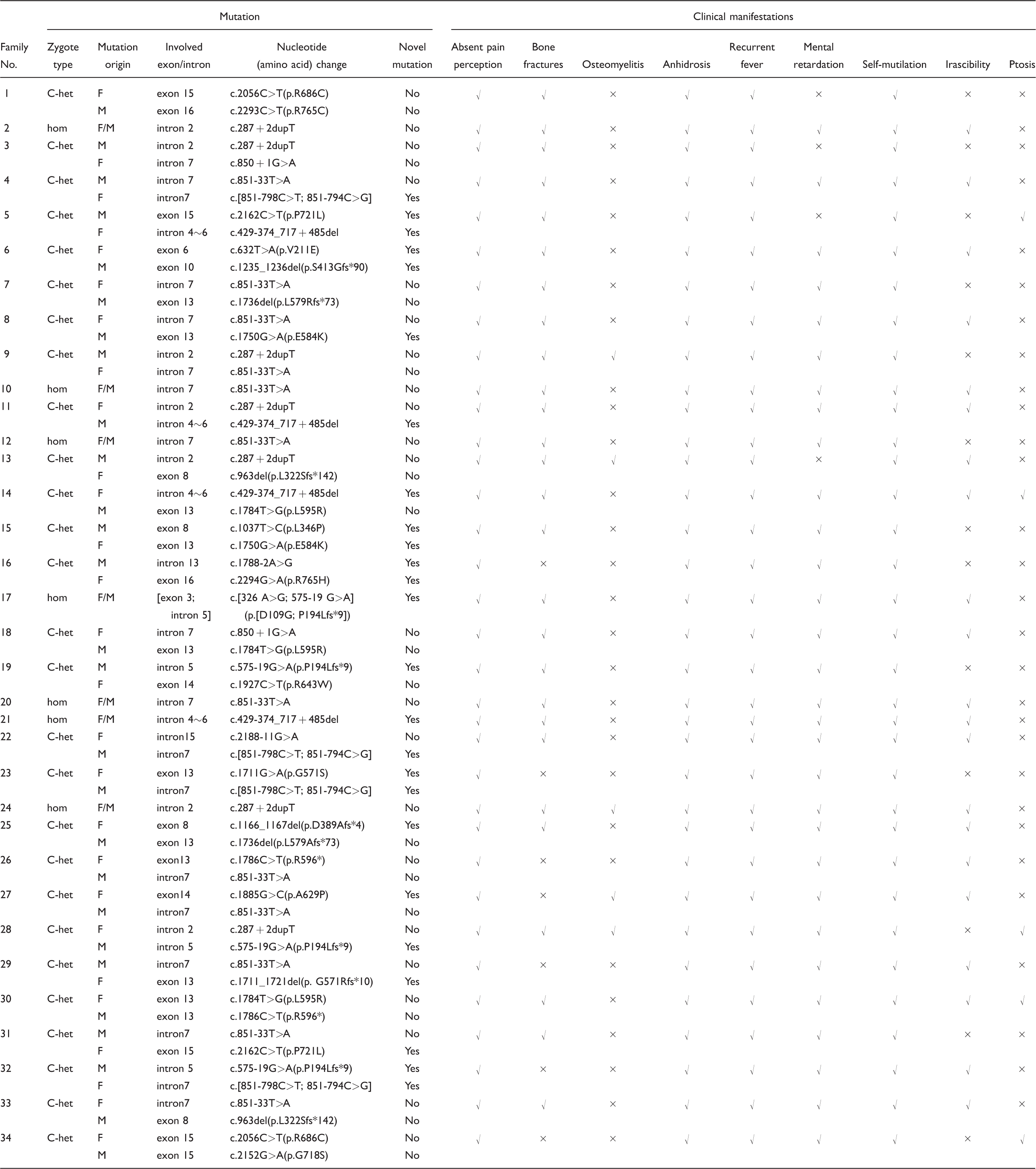
CIPA: congenital insensitivity to pain with anhidrosis; C-Het: compound heterozygote; Hom: homozygote; NA: not available.
Mutation analysis of NTRK1 in CIPA patients
Pathogenic variants were detected in both alleles of NTRK1 in these patients (Table 1). Their parents were confirmed to be carriers of one pathogenic allele. We identified 27 mutations from these patients (Figure 2(a)) including 15 novel mutations (Table 1). The 12 known mutations include five missense mutations (c.1784T > G, c.1927C > T, c.2056C > T, c.2152G > A, and c.2293C > T) resulting in amino acid changes (L595R, R643W, R686C, G718S, and R765C, respectively), one nonsense mutation (c.1786C > T, R596*), two frameshift mutations (c.963delG, c.1736delT), and four intronic splicing mutations (c.851–33T > A, c.287 + 2dupT, c.850 + 1G > A, and c.2188–11G > A). These mutations have been previously reported as pathogenic variants and recorded in The Human Gene Mutation Database (HGMD, http://www.hgmd.cf.ac.uk/ac/index.php). The two most common mutations, c.851–33T > A and c.287 + 2dupT, are presented in 24 of 68 alleles in these patients. Restriction endonuclease analysis and polyacrylamide gel electrophoresis were used to identify the mutation c.2188–11G > A in all members of family 22 (Figure S3).

Schematic map of NTRK1 showing the distribution of mutations identified in Chinese CIPA patients. (a) NTRK1 mutation spectrum for the CIPA cohort in our study: novel mutations are marked in red, known mutations in black. (b) Various domains of the TrkA protein. Novel missense mutations are shown above the horizontal axis.
We identified 15 novel mutations in NTRK1 from CIPA patients in 19 unrelated families. In those cases whereby samples from both patients and their parents were available, a Mendelian inheritance pattern of these mutations was confirmed. These novel mutations included eight missense mutations (c.326A > G, c.632T > A, c.1037T > C, c.1711G > A, c.1750G > A, c.1885G > C, c.2162C > T, and c.2294G > A) resulting in amino acid substitutions (D109W, V211E, L346P, G571S, E584K, A629P, P721L, and R765H, respectively), three frameshift mutations (c.1166_1167del, c.1235_1236del, and c.1711_1721del), three intronic mutations (c.575–19G > A, c.[851–798C > T;851–794C > G], c.1788–2A > G), and a large fragment deletion (c.429–374_717 + 485del) (Table 1).
Molecular characterization of a large intragenic deletion
Results from real-time PCR analysis showed that the levels of exons 5 and 6 in proband of family 14 were decreased to half of that in his mother and unaffected population controls (Figure S4). Agarose gel electrophoresis indicated that this proband and his carrier father had two amplification products with different sizes (Figure 3(a)). Sanger sequencing of the smaller amplicon identified a 1403-bp deletion (Figure 3(b)), and the breakpoints were found in introns 4 and 6. Analysis of the UCSC database revealed that the breakpoint junction was located within two Alu repetitive elements, which share a 21-bp common fusion sequence (Figure 3(b)). Further examination also indicated this gross deletion in families 5, 11, 14, and 21, including a homozygote (family 21: II-1) and seven heterozygotes (family 5: I-1, II-2; family 11: II-1; family 14: I-2, II-1; family 21: I-1, I-2) (Figure 3(a)). These findings suggest that a “founder effect” may contribute to the common gross mutation in CIPA patients.

Identification of gross deletion in NTRK1 using Gap-PCR and DNA sequencing. (a) Gap-PCR indicated the gross deletion covering exons 5 and 6 and introns in NTRK1 in families 14, 5, 21, and 11. The three probands (14, 5, and 11) carried a heterozygous mutation of the deletion derived from the father or mother. In family 21, the proband was a homozygote of the deletion, and both his parents were the heterozygote of the same mutation. (b) DNA sequencing of the Gap-PCR products unveiled a deletion of 1403 bp, and the breakpoint junction was located within two Alu repetitive elements with 21 bp common fusion. (c) Schematic representation of the recombination mechanism. Intrachromosomal recombination occurs between two different Alu elements which are located on the same chromosome and mediate genomic deletion. Gray and blue boxes represent Alu elements. The red box indicates homologous sequences.
Identification of intronic mutations
WGS in CIPA probands from families 4, 22, 23, and 32, identified two recurrent variations c.851–794C > G and c.851–798C > T in intron 7 (Figure 4(a)). These two variants are not present in the following public databases: 1000genomes, dbSNP141, gnomAD browser. RNA analysis showed five forms of abnormal alternative splicing between exons 7 and 8 in these patients (Figure 4(b) and (c)). Sequence analysis of aberrant splicing transcripts and bioinformatics analysis of the mutation sites suggested that the deep intronic mutation c.851–794C > G may create a cryptic donor splice site and activate three upstream pre-existing cryptic acceptor splice sites. These changes may cause pseudo-exons of different sizes to be integrated into NTRK1 mRNA (Figure 4(c)). Mutation c.851–794C > G increased the splice score of cryptic donor sites from −1.9 to 8.8 (the mean score of a 5′ ss in constitutive exons is 8.1, Figure 4(c)). Four aberrantly spliced products (1–4) utilized a cryptic splice donor in intron 7 at c.851–794 and showed inclusion of various parts of intron 7. Products 1, 2, and 3 included a part of intron 7 by using the cryptic splice acceptor sites at c.851–862, c.851–912, and c.851–931, respectively. In addition, product 3 had an exon-8 skipping. Product 4 included the same parts of intron 7 as product 2 and also included the downstream intronic region up to the splice acceptor of exon 8. Product 5 only led to an exon 7 skipping (Figure 4(c)). All these splicing patterns caused frameshift.

Identification of a deep intronic pathogenic variant in NTRK1. (a) Sequence analysis of genomic DNA of the region surrounding the variant c.[851–798C > T; 851–794C > G] from the patient and a healthy subject. (b) Primers used for nest-PCR of NTRK1 mRNA are indicated in the cartoon. (c) Cartoons of five aberrant splicing mRNAs. The blue boxes indicate exons, and the transparent boxes represent aberrant splicing events. Numbers indicate the scores for the corresponding 5′ and 3′ splice sites of wild-type and mutant-type.
Proband from family 17 was the homozygote of mutation c.[c.326A > G;575–19G > A]. Heterozygous mutation c.575–19G > A was found in three unrelated families (families 19, 28, and 32) (Figure 5(a)). Using a minigene assay, we confirmed that the c.575–19G > A substitution created a novel splicing acceptor site, resulting in the inclusion of a 17-bp intronic sequence in the mutant transcript (Figure 5(b) and (c)). In addition, this nucleotide change increased the splicing score of the pseudo-exon cryptic 3′ ss from −2.0 to 9.0 (the mean score of a 3′ ss in constitutive exons is 7.9) (Figure 5(d)). This mutation led to a premature termination of translation and a truncated protein product (P194Lfs*9).

Pathogenic analysis for a novel intronic mutation in NTRK1. (a) Heterozygous mutation c.575–19G > A was found in probands 17, 19, and 28. (b, c) The minigene analysis of mutation c.575–19G > A, which led to an insertion of 17 nt in the transcript of NTRK1. (d) Scheme of NTRK1 that contains the c.575–19G > A mutation, showing the aberrant splicing in patients. Numbers indicate the scores for the 3′ splice sites.
Discussion
All patients examined in this study exhibited clinical manifestations of CIPA that are consistent with the characteristic symptoms caused by either homozygous or compound heterozygous mutations in NTRK1. 10 Most patients had fractures or joint dislocation. Moderate to severe irascibility was observed in 22 patients from 21 families. Five patients also had severe osteomyelitis in the limbs. Unilateral or bilateral eyelid ptosis was found in five patients, which may be due to neurogenic damages caused by the mutant NTRK1. According to the detailed investigation, we found that there was no significant correlation between genotype and phenotype in the Chinese CIPA cohort, but it should be noticed that home care may have a certain relevance to the children’s phenotypes. The patients who were taken care by experienced parents of raising and caring for a child showed less severe clinical symptoms of CIPA than others.
TrkA receptor is encoded by NTRK1 and has three functional domains. The extracellular domain is encoded by exons 1 to 8 and includes the first and second immunoglobulin-like (Ig-like) domains, which are post-translationally glycosylated and important to NGF binding. The transmembrane domain is encoded by exon 11. The intracellular tyrosine kinase domain is encoded by exons 13 to 17 and is crucial for signal transduction. 11 Mutations in NTRK1 may result in the production of aberrant proteins that cannot be activated, and hence cannot transmit signals that are important to cell growth and survival.12,13 Our genetic analysis of this cohort of Chinese CIPA patients identified 15 novel mutations in NTRK1, including seven (D109W, V211E, L346P, D389Afs*4, c. 429–374_717 + 485del, and c.575–19G > A, c.[851–798C > T;851–794C > G]) located in the extracellular domain, seven (G571Rfs*10, G571S, E584K, A629P, P721L, R771H, and c.1788–2A > G) located in the intracellular tyrosine domain, and one (S413Gfs*90) situated in the transmembrane domain of TrkA (Figure 2(b)). The finding that most mutations occurred in the extracellular domain and in the intracellular tyrosine domain suggests the functional significance of these domains. Intriguingly, a form of large genomic rearrangement (c.429–374_717 + 485del) in NTRK1 was found in four patients from families 5, 11, 14, and 21. Further sequence examination revealed a 1403-bp deletion, which spans the region from exon 5 to exon 6, and includes a partial deletion of introns 4 and 6. To our knowledge, this is the second gross deletion that has ever been identified in NTRK1. This deletion may cause premature termination of translation resulting in a truncated protein (V144Nfs*10). It remains possible that the truncated proteins may not be produced due to a nonsense-mediated mRNA decay pathway, which targets mRNAs harboring premature termination codons for degradation.14–16 It remains to be determined whether this gross deletion may lead to a loss of TrkA function.
Using the UCSC Genome Browser, in silico analysis revealed that both 5′ and 3′ deletion boundaries were located within Alu elements. The breakpoint in intron 4 was within the AluY region (chr1:156837449–156837726), while the breakpoint in intron 6 was within the AluSg (chr1:156838885–156839168) region. The overall similarity of these two Alu repeats was 63%, suggesting that an intragenic homologous recombination event may be the primary mutational mechanism.17,18 In humans, Alu elements have been reported to be associated with genomic deletion events by promoting nonallelic homologous recombination (NAHR).19,20 NAHR occurs between two DNA sequences that are not alleles but share a high-sequence similarity. Alu elements are the major NAHR hotspots resulting in human diseases. 21 During meiosis, misalignment of Alu elements on different alleles may occur. The subsequent crossover event leads to genetic rearrangement which causes deletions, duplications, or translocations.22,23 NAHR can be induced either by interchromosomal recombination between two different chromosomes or by intrachromosomal recombination within the same chromosome.19,24,25 In the current study, the breakpoint junction of CIPA patients was located within two Alu repetitive elements with a 21-bp common fusion segment, suggesting that the gross deletion identified in these patients may be caused by intrachromosomal recombination events. This 21-bp common fusion segment may be a common core sequence that could facilitate the recombination event (Figure 3(c)). Our study provides compelling evidence that repeat sequences, such as Alu elements, may lead to cryptic NTRK1 intragenic deletions.
Deep intronic mutation can be another possible cause of human disease, but this mechanism has been largely ignored in previous studies. Here, we show for the first time that deep intronic mutations occurred in NTRK1 in CIPA patients (families 4, 22, 23, and 32), which may be a novel genetic mechanism for CIPA. As reported previously, the deep intronic mutation could lead to the appearance of more than one aberrantly spliced mRNA isoforms. 26 Meanwhile, this mutation may create a novel donor splice site and activate three different upstream pre-existing cryptic acceptor splice sites, leading to exonization of sequences in intron 7. The mutation also prevents the recognition of natural splice sites, resulting in either skipping or inclusion of the cryptic donor splice site. All these abnormal splicing products can lead to a consecutive shift of the reading frame. It has become increasingly clear that intron exonization may be an important reason that causes diseases. 27 Findings from in silico analysis suggest that the deep intronic mutations occurred at an AluY (chr1:156842468–156842768) repetitive element. Genomic insertion of Alu sequences into coding regions can lead to mis-splicing. 28 Point mutations in Alu elements are also common reasons for exonization. 29 Current study also shows for the first time a point mutation within a pre-existing Alu element that induced a mis-splicing in CIPA patients. Attentions were paid to the exons which contain 90% of pathogenic mutations, but disease-causing mutations in deep intron are rarely reported. Therefore, future studies of other single gene diseases should also use targeted genomic sequencing to examine whether intronic variants may also underlie the disease.
Based on the in silico analysis (Table 2), the mutational predictions of the three tools were concordant for six (c.1037T > C, c.1711G > A, c.1750G > A, c.1885G > C, c.2162C > T, and c.2294G > A) of eight missense mutations. In contrast, the predictions for mutations c.326A > G and c.632T > A were not consistent among the three programs. The mutation c.575–19G > A was predicted to be benign by MutationTaster. Our minigene analysis confirmed that this mutation led to abnormal splicing process. Moreover, the proband 17 was homozygous for mutations at two different nucleotides (c.[326A > G;575–19G > A]). Accordingly, we do not need to determine the pathogenicity of mutation c.326A> G. The other five novel mutations (c.429–374_717 +485del, c.[851–798C > T;851–794C > G], c.1235_1236del, c.1166_1167del, and c.1788–2A > G) were predicted to be pathogenic by the MutationTaster tool. In addition, the minor allele frequency of the aforementioned mutations was low or absent in the two databases. The most common mutations c.851–33T > A and c.287 + 2dupT had been reported in Japanese and Korean CIPA patients, suggesting that these mutations may be common in East Asian population.30,31
Allele frequency and pathogenic prediction for novel mutations in this study.

D: damaging; P: possibly damaging; T: tolerated; B: benign.
In conclusion, we performed a genetic analysis of NTRK1 in a cohort of Chinese CIPA patients and found 15 novel mutations of NTRK1, including the gross deletion and deep intronic mutation. Current findings expand the spectrum of NTRK1 mutation associated with CIPA, which will help to improve genetic diagnosis of this disorder.
Supplemental Material
Supplementary figures and tables -Supplemental material for Novel NTRK1 mutations in Chinese patients with congenital insensitivity to pain with anhidrosis
Supplemental material, Supplementary figures and tables for Novel NTRK1 mutations in Chinese patients with congenital insensitivity to pain with anhidrosis by Xingzhu Geng, Yanshan Liu, XiuZhi Ren, Yun Guan, Yanzhou Wang, Bin Mao, Xiuli Zhao and Xue Zhang in Molecular Pain
Footnotes
Acknowledgments
The authors thank the family members for their participation in this study and thank Claire F. Levine, MS, ELS (scientific editor, Department of Anesthesiology/CCM, Johns Hopkins University), and Yuanxiang Tao for editing the manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.