
Abstract
Background:
Limb girdle muscular dystrophy recessive type 1 (LGMDR1, Previously LGMD2A) is characterized by inactivating mutations in CAPN3. Despite the significant burden of muscular dystrophy in India, and particularly of LGMDR1, its genetic characterization and possible phenotypic manifestations are yet unidentified.
Material and Methods:
We performed bidirectional CAPN3 sequencing in 95 LGMDR1 patient samples characterized by calpain-3 protein analysis, and these findings were correlated with clinical, biochemical and histopathological features.
Results:
We identified 84 (88.4%) cases of LGMDR1 harboring 103 CAPN3 mutations (71 novel and 32 known). At least two mutant alleles were identified in 79 (94.2%) of patients. Notably, 76% exonic variations were enriched in nine CAPN3 exons and overall, 41 variations (40%) correspond to only eight exonic and intronic mutations. Patients with two nonsense/out of frame/splice-site mutations showed significant loss of calpain-3 protein as compared to those with two missense/inframe mutations (P = 0.04). We observed a slow progression of disease and less severity in our patients compared to European population. Rarely, presenting clinical features were atypical, and mimicked other muscle diseases like FSHMD, distal myopathy and metabolic myopathies.
Conclusion:
This is first systematic study to characterize the genetic framework of LGMDR1 in the Indian population. Preliminary calpain-3 immunoblot screening serves well to direct genetic testing. Our findings prioritized nine CAPN3 exons for LGMDR1 diagnosis in our population; therefore, a targeted-sequencing panel of nine exons could serve well for genetic diagnosis, carrier testing, counseling and clinical trial feasibility study in LGMDR1 patients in India.
INTRODUCTION
Limb girdle muscular dystrophies (LGMDs) comprise a clinically and genetically heterogeneous group of degenerative muscle disorders characterized by progressive muscle wasting and weakness [1]. Presently, more than 40 specific muscular dystrophy gene loci have been identified, including nine autosomal dominant (LGMD1A to LGMD1I) and 29 autosomal recessive forms (LGMDR1 to LGMDR24, formerly LGMD2A to LGMD2X) [2]. Calpainopathy, also referred as Limb-girdle muscular dystrophy recessive type 1 (LGMDR1, previously LGMD2A), is the first characterized limb girdle muscular dystrophy, caused by mutations in the CAPN3 gene (MIM 114240) encoding for calpain-3, the muscle-specific member of a family of Ca++-activated neutral proteases. CAPN3 gene on chromosome 15q15.2 is expressed as a 3.5-kb transcript composed of 24 exons, which encodes a 94-kD muscle-specific translated protein [3]. The exact role of calpain-3 is not known; however, it is possibly involved in transcription factor-mediated regulation of survival and apoptosis genes, cytoskeletal repair mechanisms, and muscle maturation [4]. CAPN3 mutations result in loss of its autolytic and proteolytic activity, broadly leading to impaired sarcomeric remodeling.
LGMDR1 is one of the most prevalent LGMDs worldwide, particularly in Europe, Japan, Brazil, Russia, Australia, Turkey and India [1, 5]. It is clinically characterized by progressive symmetrical weakening of proximal limb-girdle muscles, with a variable age at onset ranging from 2 to 40 years [6]. The phenotype shows marked intra- and interfamilial variability, ranging from mild to severe clinical forms [5, 7–9]. Recently, heterozygous CAPN3 mutations with autosomal dominant inheritance pattern have been identified and re-classified as autosomal dominant Limb-Girdle Muscular Dystrophy Dominant type 4 (LGMDD4), with a later onset and milder clinical phenotype [10]. Geographically, the incidence of LGMDR1 varies from 10 to 50%, and in some ethnic populations it is as high as 80% [5, 9]. There are now over 496 documented distinct pathogenic CAPN3 mutations and 70 apparently neutral polymorphic/benign variant or unclassified variants listed in the Leiden muscular dystrophy database [http:www.dmd.nl]. Sparing the exception of founder effects in few isolated populations, there are no hotspot mutations [7, 12]. In most studies, mutation analysis invariably leads to the identification of novel mutations [7, 13–19]. CAPN3 mutations show a non-uniform distribution that spans almost the entire length of the gene; nevertheless, studies have found a grouping of mutations in a subset of exons in several extensively studied populations [20]. Diagnosis of LGMDR1 on clinical grounds alone is difficult due to overlapping clinical features with other LGMDs, as well as presentation with some odd features like hyperCKemia, idiopathic eosinophilic myositis and pseudometabolic myopathy [5]. Further, the clinical course is often discordant with muscle calpain-3 levels [21]. Thus, genotype-phenotype correlation is difficult due to the heterogeneity of CAPN3 mutations and varied clinical manifestations.
The current strategy to diagnose LGMDR1 is primarily based on calpain-3 immunoblotting. Several laboratories also perform calpain-3 functional in-vitro assay for possible loss of autocatalytic activity in patients with normal calpain-3 protein. However, the gold standard for diagnosis of LGMDR1 is by CAPN3 mutation screening [22].
Our previous study, the first cohort of 75 Indian LGMD2A/R1 patients diagnosed using calpain-3 immunoblotting, revealed LGMDR1 to be the most frequent form of LGMD in India [1]. Despite the large disease burden of muscular dystrophies in India, [23] no comprehensive genetic studies are available on spectrum of CAPN3 mutations in our population, barring two candidate mutations in a particular community analyzed in small numbers [24, 25]. Thus, in order to validate and extend our strategy of LGMDR1 diagnosis and determine the genomic map of CAPN3 in India, we performed CAPN3 mutation analysis, and correlated genotype with clinical, histopathological and biochemical features of LGMDR1 patients.
MATERIALS AND METHODS
The present study was approved by the Institute Ethics Committee, All India Institute of Medical Sciences, New Delhi, India (A-108/80208).
Patients
All patients diagnosed as LGMDR1 based on the following criteria were identified: 1) clinical phenotype consistent with LGMD, such as weakness in proximal or distal muscles of the lower and/or upper girdle with onset in childhood or adulthood; 2) muscle histopathology consistent with dystrophic process; 3) moderate to increased serum creatine kinase (CK) level; and 4) electromyography consistent with myopathic pattern. Clinical parameters including muscle strength, loss of ambulation, age at which wheelchair-bound, family history and any other relevant features were retrieved from archives of Department of Neurology.
Patients categorized as primary calpainopathy following calpain-3 immunoblotting and loss of autocatalytic function (LOAF) assay on muscle biopsy performed as described earlier underwent CAPN3 mutation analysis [1, 21].
DNA isolation and CAPN3 sequencing
DNA was extracted from muscle biopsy specimens using Genelute mammalian DNA isolation Kit (M/s. Sigma Aldrich, St Louis, USA) as per the manufacturer’s protocol. Genomic DNA from 100 normal controls from North India was also included for CAPN3 mutation analysis to exclude any possible polymorphism/benign variant being taken as pathogenic variations. All 24 exons and exon-intron boundaries of CAPN3 gene were amplified using 50 ηg genomic DNA by Polymerase chain reaction. Supplementary Table 1 shows primer sequences and PCR conditions. Sequencing reaction PCR was carried out using BigDyeTerminator V1.1 ABI sequencing reaction mix for forward and reverse primers separately. Sequencing was performed on ABI-3137xl genetic analyzer (Applied Biosystems, Foster City, USA). NM_000070 was used as the reference sequence for CAPN3, and analyzed by ABI sequencing analysis software 5.1.1 and DNASTAR sequencing analysis tool. Sequence variant nomenclature was verified using the Mutalyzer tool (https://mutalyzer.nl/). All the variants were classified according to 2015 American College of Medical Genetics and Genomics (ACMG) guidelines [26]. PolyPhen-2 [27], SIFT [28], and Mutation-Taster [29] were used for in silico analysis. CAPN3A mutation predicted as disease-causing by any one of the prediction tools was considered as pathogenic or potentially pathogenic in cases of novel mutations. Leiden muscular dystrophy database (http://www.dmd.nl; and HGMD) for CAPN3 mutations was referred for reporting the novelty of candidate mutations.
RESULTS
Clinicopathological features
We received 728 muscle biopsies for various muscle-related disorders in a period of two years, of which 204 patients showed clinical phenotype of LGMD and histopathological features of muscular dystrophy. Based on calpain-3 immunoblot and LOAF assay, 124 (60.7%) patients were categorized as primary calpainopathy. Ninety-five of these were further examined for CAPN3 mutations, while 29 were excluded due to lack of either sufficient tissue or good quality of DNA. All patients included were sporadic and non-consanguineous. Family history was positive in 17.8 % (15/84). All 95 cases showed normal immunostaining for dystrophins, α-, β-, γ-, and δ-sarcoglycans, merosin, emerin, lamin A/C, caveolin 3, collagen VI and α & β-dystroglycans, accompanied by either partial or total loss of calpain-3 protein on immunoblot. Four patients showed calpain-3 LOAF. Twelve patients (12.7%) also showed secondary partial dysferlin loss. NADH-TR oxidative stain revealed lobulated fibers in 8.5% (8/94) cases. One patient with clinical features suggestive of LGMDR1 could not be tested by immunoblotting or LOAF. The clinicopathological features of the cohort are summarized in Table 1.
Demographic, clinical, biochemical and molecular features of 84 LGMDR1 patients
*Also includes cases of Wheelchair bound and non-ambulatory.
Mutation analysis of CAPN3 gene
Eighty-four of the 95 patients (88.4%) showed pathogenic or potentially pathogenic mutations in CAPN3. Remaining 11 patients (11.6%) with calpain-3 total loss (3 cases), partial loss (6 cases) and LOAF (2 cases) did not show any CAPN3 genetic alterations with disease-causing potential. Among these 84 patients, 79 (94%) presented with at least two mutations, and 5 (6%) had a mutation in one allele only. In addition, nine of 79 patients with 2 mutations harbored a third mutation too. A total of 172 variations (including both alleles) were identified, including missense, frameshift deletion or insertion, splicing, and synonymous mutations with in-silico predicted disease-causing effect. They encompassed 103 different mutations, including 71 novel and potentially pathogenic mutations and 32 previously reported pathogenic mutations (Table 2). We also found 38 different polymorphic/benign variants among all the patients analyzed (Supplementary Table 2).
Pathogenic CAPN3 mutations identified in 84 LGMDR1 patients
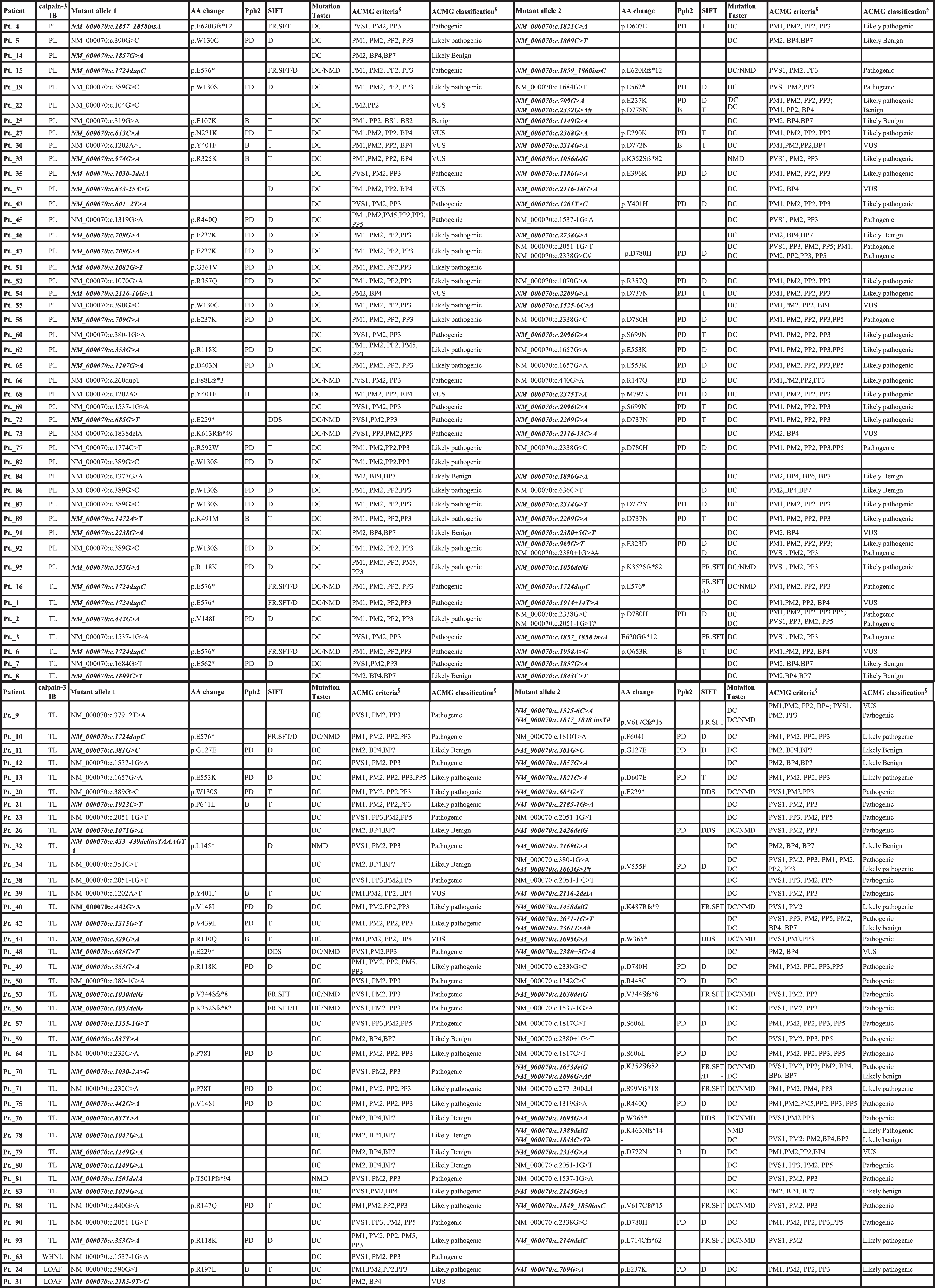
Column 1 and 2: Patient divided based on protein loss. Abbreviations: calpain-3 IB: calpain-3 immunoblot; TL: Total loss of protein; PL: Partial loss of protein; LOAF: Loss of calpain-3 autolytic function activity; FR.SFT: Frameshift mutation; D: Damaging; DC: Disease causing; NMD: Nonsense mediated decay; PD: Probably damaging; B: Benign; T: Tolerable; #Mutant allele 3; Mutation highlighted in bold and Italics are novel variation identified in this study, AA, amino acid; VUS, variant of unknown significance; §American College of Medical Genetics and Genomics criteria, Richards et al, 2015.
Among 84 patients harboring CAPN3 mutations, 130 variations were identified in exons, and 38 in introns. One mutation (-c.104G>C) was in the promoter region of CAPN3. Among the 103 CAPN3 mutations, 75.6% were enriched in nine exons (exon 2, 3, 5, 8, 10, 11, 13, 16 and 22) (Fig. 1A). Notably, exon 16 was most frequently mutated, and 45% of exonic mutations were in exons 3, 8, 16, and 22 only. Exons 12, 15, 18, 23 and 24 did not show any mutation. Among intronic variations, 79.2% were encompassed by eight introns (intron 2, 7, 11, 12, 17, 19, 20, and 22), and intron 17, 19 and 22 mutations were the most frequent intronic variation (Fig. 1 B, C). Screening of exon 2, 3, 5, 8, 10, 11, 13, 16 and 22 that also covers exon-intron boundaries, identified 67% of all the mutations. The genetic variations identified included 80% single base substitutions, 13% deletions, 3% insertion, 2% delins and 2% duplication (Fig. 1D). There were 35 (34%) missense mutations, 46 (45%) null mutations including frameshift deletion/insertion and splicing mutations, and 22 (21%) variations with synonymous mutation but predicted as probable splicing defects or likely benign variants (Fig. 1E). The most recurrent mutation found in our patients was c.2051-1G>T (7 times), a splice site change and novel mutation. c.389G>C, c.1537-1G>A, Ex 13_c.1724dupC and c.2338G>C were found in six patients, respectively, while c.709G>A were present in five patients. Patient 63 who was included in the study based on clinical features of LGMDR1 without calpain-3 protein analysis showed c.1537-1G>A heterozygous mutation. Two patients with LOAF showed missense mutations (Pt._24: c.590G>T and c.709G>A) and a splicing mutation in only one allele (Pt._31: c.2185-9 T> G). Among polymorphic/benign variant changes, the most frequent mutation found in our patients was c.96C>T (13 times). This is a known polymorphism/benign variant with unknown pathogenicity in homozygous state. However, we also found nine of our patients with heterozygous state for this variant. Another most recurrent polymorphic/benign variant genetic variation was c = -c.243C>A (5 times) in promoter region of CAPN3.
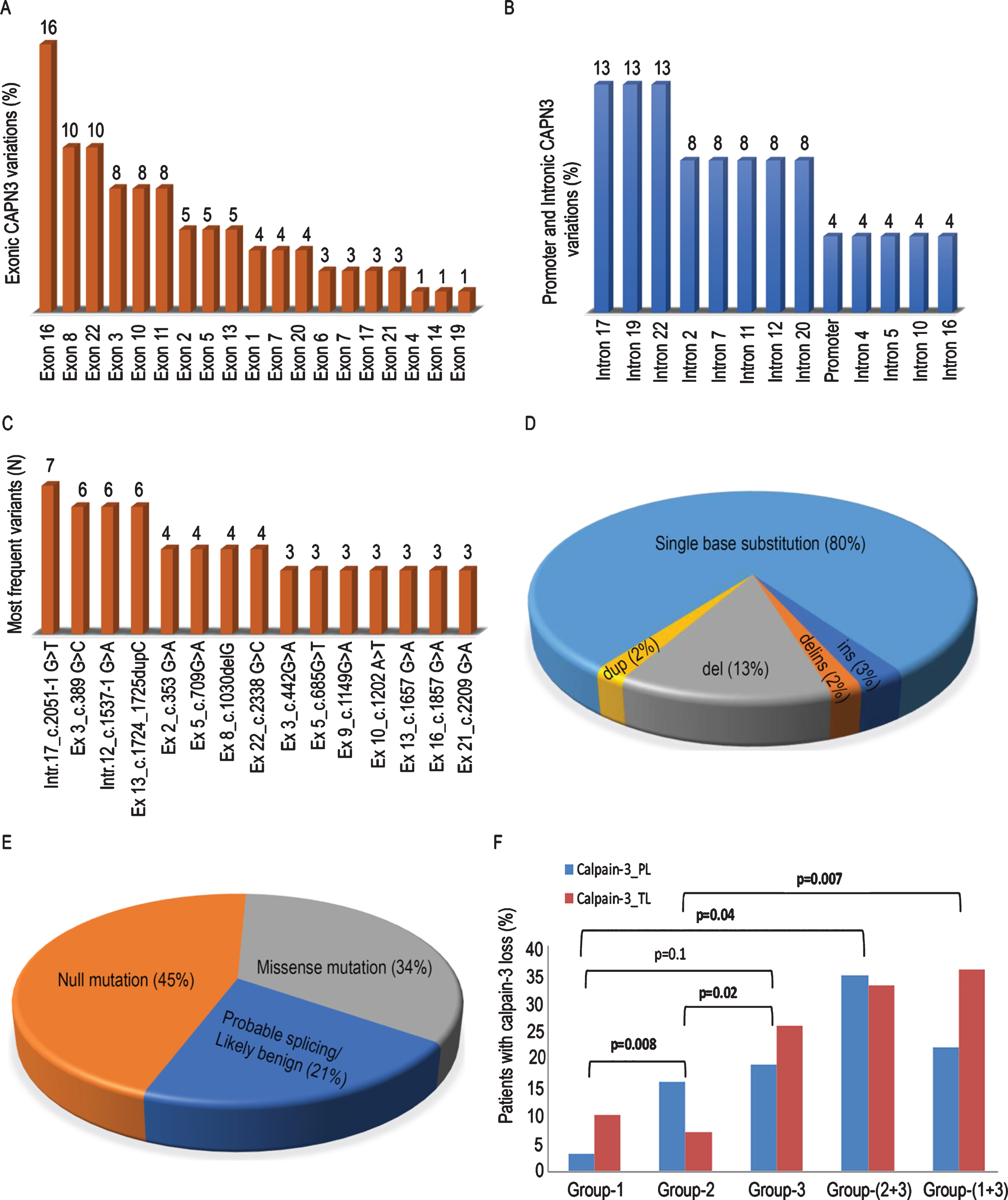
(A) Bar graph showing the distribution of all exonic and (B) intronic variations found in CAPN3 in 84 cases. (C) CAPN3 mutations identified in more than one LGMDR1 patient. (D) Pie chart showing the distribution of all variations by impact on protein sequences. (E) Distribution of mutation by type of all structural variations found in CAPN3. (F) Genotype-protein phenotype correlation between different genetic subgroups (vertical bars illustrating percentage of patients with calpain-3 total loss (calpain-3_TL) and partial loss (calpain-3_PL)). Group-1: Patients with 2 nonsense/out of frame/splice-site mutation, Group-2: 2 missense/inframe mutation, Group-3: One missense/in-frame mutations or one nonsense/out-of-frame/splice-site mutation or both together.
Genetic subgroups of LGMD2A and Genotype- phenotype correlation
We categorized the 84 LGMDR1 patients into three genetic subgroups (Table 3). Patients with two nonsense/ out of frame/ splice-site mutations and two missense/ inframe mutations were categorized in group-1 (N = 13; 15.5%) and group-2 (N = 24; 28.5%), respectively. Group-3 (N = 47; 56%) consisted of patients with only one missense/in-frame mutation or one nonsense/out-of-frame/splice-site mutation, or both together.
Demographic features of genetic subgroups identified in 84 LGMDR1 patients
Among 84 genetically characterized LGMDR1 patients, LGMD phenotype was observed in 92.7% [76/82] cases. Advanced stage symptoms such as difficulty in climbing stairs, rising up from a chair or getting up from the floor/ fall while walking was noted in 21.4% (18/84) patients. Notably, only 10.7% patients (9/84) were confined to wheelchair and/or non ambulatory state at the time of presentation, attaining this state after 10–23 years after onset of first symptom. These 18 patients (1) were distributed in the mutant groups as follows: 3/13 (23.1%) in mutant group-1, 5/24 (20.8%) in mutant group-2 and 8/47 (17%) in mutant group-3 (p > 0.05). There was male preponderance (M:F ratio 2.3:1), lower age at onset in females (17.2±10.8 years vs 20.3±13.9 years), and higher CK levels in females (6453 IU vs 1544 IU) as compared to males. Clinical findings available for the patients are summarized in Table 1. Approximately 13% of patients (11/84) presented with atypical features i.e. facial weakness (3 patients), distal Miyoshi type weakness (4 patients) and metabolic myopathy (3 patients).
Cases with calpain-3 total loss versus partial loss showed lower mean age of diagnosis (24.4 years vs 27.2 years) and age at onset (18.6 years vs 20 years), higher mean CK level (5238 vs 3349 units), more frequent family history (20.9% vs 15.8%), and greater presence of lobulated fibers (11.6% vs 7.9%) (Table 1). However, these clinical, biochemical and histological differences were not statistically significant.
The difference in the mean age of onset of symptoms in mutant group-1 patients (17.1 years), group-2 (19.7 years) and group-3 (18.5 years) patients was statistically insignificant (p > 0.05). After excluding two patients with LOAF and one patient without immunoblot, total loss of calpain-3 protein was seen in 53.1% (43/81 cases), and partial loss in 46.9% (38/81 cases). Notably, a significantly higher number of mutant group-1 (p = 0.008) and mutant group-3 (p = 0.02) patients showed total loss of calpain-3, when compared to mutant group-2. A non-significant distribution of calpain-3 protein loss was observed between mutant group-1 and group-3 (p = 0.1) (Fig. 1F). However, mutant group-1 patients showed a significantly lower male: female ratio than mutant group-3 patients (p = 0.04). Similarly, mutant group-1 patients also showed a trend towards higher CK level than mutant group-3 patients (p = 0.06). However, no correlations could be found between the genetic subgroups and age at diagnosis, clinical features, family history, or presence of lobulated fibers.
DISCUSSION
LGMDR1 is the most common LGMD the world over, including in India, but its genetic landscape is not studied in the Indian population [1]. In this cohort, we studied 95 patients for CAPN3 mutations, of which 88.4% cases showed pathogenic or potentially pathogenic genetic alterations. The majority of the patients (94.2%) had at least two mutations, similar to the observation of Fanin M et al. [5] but had more frequent identification of two mutant alleles than in studies by Saenz et al. [7] and Groen et al. [8]. We found 71 novel potentially pathogenic and 32 previously reported pathogenic mutations, like other first-time reports from different populations [7, 13–19]. Detection of the large number of novel mutations in the present study could be due to the different population spectrum and ethnic background of the Indian population. About 67% of CAPN3 variations identified were clustered in 9 exons and their border intronic regions, nearly similar to studies from France [18], Italy [20] and Brazil [27] where approximately 80–90% of mutations were clustered in 6–8 exons. Overall, the different types of mutations identified in the present cohort were comparable with genetic changes reported in CAPN3 Leiden database, where substitutions, deletions, insertion, delins and duplication constituted about 66%, 22%, 0.2%, 9% and 1.8%, respectively, hence supporting the validity of our findings. Recently, Ankala A et al. and Khadilkar SV et al. [25] reported two CAPN3 founder mutations (c.2338G>C and c.2099-1G>T) in two different haplotype backgrounds in patients from unrelated families of Agarwal community of India. In a recent US based study [30] utilizing next-generation sequencing gene-panel that also encompasses patient samples from India, reported mutation c.2051-1G>T, c.380-1G>A, c.1838delA, c.1774C>T, c.1377G>A, c.2051-1G>T, c.351C>T that were also identified in our patients. Although the study did not mention number of patients from India but these variations are also not reported in any other population except our, as per CAPN3 mutation database http://www.dmd.nl. This further supports the novel population specific mutations identified in our cohort. Overall, 5/84 (6%) patients showed homozygous CAPN3 mutation, similar to reported in a large cohort of 4656 LGMD patients [30]. We also found 6 patients harboring c.2338G>C variations. Two patients with LOAF [Pt. 24 and Pt. 31] showed variations in two and one allele, respectively. These variations were not found in autocatalytic domain of CAPN3 but such changes outside autocatalytic domain with LOAF have been reported before [5].
Clinicopathological correlations
Approximately 86% of our genetically characterized LGMDR1 cases showed mean age at onset about 15 years which is similar to most earlier studies [7, 32]. We observed an association of early age at onset, increased CK levels, relatively more frequent presence of lobulated fibers, and family history in patients with total loss of calpain-3 as compared to partial loss; similar correlation was described by Fanin et al. [20]. The degree of dystrophic features in muscle biopsy did not have any significant association with calpain-3 protein status.
The clinical symptoms of our LGMDR1 patients were mostly similar to earlier defined LGMDR1 phenotype [1, 33]. Advanced features of the disease were present in about one fifth of patients, but overall, the disease progression and severity in this cohort were slow when compared to reports from UK, Brazil, Italy and few other European populations [1, 33]. Paula FD et al. reported that African–Brazilian calpainopathy patients are more severely affected than Caucasians [4]. Thus, the disease course may vary in different ethnic populations.
Genotype–phenotype correlation
It is already known that most of the patients with total loss of calpain-3 protein show mostly null mutations, [5, 12] while missense type mutations are associated with unpredictability of their effect at the protein level and are usually associated with a milder phenotype [7, 35]. We also demonstrated that mutant group-1 and group-3 patients showed significant correlation with calpain-3 total loss, as compared to group-2. The lack of statistically significant differences in the mean age at onset between the mutant groups could be attributed to decreasing number of nonsense/out-of-frame/splice-site mutations associated with older age at onset, as reported in earlier studies [4, 5]. However, these findings are discordant with some other reports [7, 8]. The mean CK level in our patients of mutant group-1 (mean CK = 4688.2) and group-3 (mean CK = 2787.8) was lower than group-2 (mean CK = 5048.6). Zatz M et al. [31] and de Paula [4] have showed that the mean serum CK activity did not differ significantly with the type of the mutation/ mutant subgroups, as CK is known to be extremely variable during the disease course. In addition, the frequency of presence of lobulated fibres and family history in our mutant group-2 was lower than group-1 and group-3, and this aligns with the severity of clinical course seen in our patients with regards to mutant groups. These observations are in concordance with the previous findings that patients with two nonsense/splice-site/out-of-frame mutations have relatively severe clinical presentation and reduced protein levels than patients with at least one missense/in-frame mutation [5, 37].
Atypical clinical and pathological features:
Atypical clinical features have been noted in approximately 12% of genetically characterized LGMDR1 patients [33]. LGMDR1 phenotype with the Erb presentation may be misdiagnosed as FSHMD [38, 39]. Patients with clinical presentation of LGMD have been found to have a dystrophinopathy [40], and vice a versa [41]. We encountered 9 cases which, on histopathological examination, had been suspected to be SMA, desminopathy, neurogenic atrophy, polymyositis, FSHMD and other myopathies. Presentations including neurogenic pattern with muscular dystrophy, SMA, proximal type SMA [32, 43], desminopathy [8], dystrophinopathy [41], hyperCykemia [44], pseudometabolic myopathies, eosinophilic myositis and polymyositis [17, 44] have been described in the literature, suggesting that clinical and pathological spectrum of LGMDR1 is wide, and mutation analysis is necessary for accurate diagnosis.
Using a combined calpain-3 immunoblotting and autolytic assay-based diagnostic strategy, we obtained a diagnostic accuracy of 88.3% for LGMDR1. Our diagnostic sensitivity is relatively high to other reports and is comparable to 80–83% reported by Fanin M et al. [5]. Moreover, using Bayesian multiple analysis with dependence, Groen et al. [8] reported that if the phenotype and the western blot suggested a calpainopathy, the probability of identification of CAPN3 mutation was as high as 90.8%, which is similar to our observations.
We assume that the 5 patients with mutations in only one allele may either be true heterozygotes for a pathogenic CAPN3 allele, or may harbor a yet unrecognized hypomorphic allele variation; similar findings have been reported in about 20–30% of LGMDR1 patients [4, 45]. The second mutations may either be localized in noncoding regions such as introns or gene promoter, or may happen to be large genomic rearrangements that may have escaped detection by the mutation analysis methods used, and could possibly be identified by cDNA sequencing [45, 46] or high throughput genome wide screening.
Now it is well accepted that CAPN3 in-frame c.643_663del21, c.598_612del15deletions and missense variant, c.1333G>A constitutes a dominantly inherited form of calpainopathy [47, 48] and few of our patients with single mutation may belong to this LGMD type. Further, in 11 patients, no mutations were identified, or we likely missed possibly because some mutations may be localized in regions with deletions or duplication of one or more exons, or deletion of the entire CAPN3 gene [49, 51]. Although calpain-3 protein is stable in muscle tissue, the calpain-3 protein amount can be partially reduced by the artificial degradation that occurs due to partial thawing of muscle tissue or moisture when muscle tissue is handled or stored under conditions that promote rapid calpain-3 autolysis. This could lead to false protein loss on immunoblotting, seen in 10–20% of cases [4, 52], resulting in degradation of calpain-3 which has a very short half-life. Further, secondary calpain-3 loss has been described in LGMD2I/R9 or titinopathy, which could lead to an erroneous diagnosis of LGMDR1 and subsequent failure to detect CAPN3 mutations [7, 54].
To conclude, the present study is the first systematically designed, large cohort of patients with mapping of CAPN3 alterations in LGMDR1 Indian patients, which found 71 novel potentially pathogenic and 32 known pathogenic mutations, including four recurrent mutations, in a large proportion of cases, with significant genotype- protein phenotype correlation. The combined molecular diagnostic approach adopted by us had a high probability of obtaining calpainopathy diagnosis in patients with protein deficiency or functional inactivity. Interestingly, nearly two third of CAPN3 variations were present in 9 exons and their border intronic regions. Thus, we propose that, in Indian patients, the 9 CAPN3 exons enriched with mutations could be analyzed initially for the genetic diagnosis of LGMDR1, particularly in resource-limited settings, decreasing the turnaround time and cost involved in more extensive genetic analysis.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest to report.
Footnotes
ACKNOWLEDGMENTS
We thank all the LGMD patients and their families. The original work by the authors was supported by CN Centre, Department of Pathology, AIIMS, and ICMR, New-Delhi (Project 3/1/2/1/Neuro/2008-NCD-I). The authors thank Dr. Mitali Mukerji (CSIR-IGIB) for providing sequencing facility and Mrs. Suman Singh and Mr. Varun Surolia (CSIR-IGIB), Delhi, India for their assistance in sequencing analysis. We acknowledge Mr. Anil Bisht and Mr. Rajeshwar Khadia for doing IHC/EHC, and Dr. Aanchal Kakkar for critical review of the manuscript.
The authors declare that they have no conflict of interest to report.