
Abstract
Aim:
We describe a cohort of five patients with limb-girdle muscular dystrophy (LGMD) 2G/LGMD-R7 in a South-east Asian cohort.
Background:
LGMD2G/LGMD-R7-telethonin-related is caused by mutations in the TCAP gene that encodes for telethonin.
Methods:
We identified consecutive patients with LGMD2G/LGMD-R7-telethonin-related, diagnosed at the National Neuroscience Institute (NNI) and National University Hospital (NUH) between January 2000 and June 2021.
Results:
At onset, three patients presented with proximal lower limb weakness, one patient presented with Achilles tendon contractures, and one patient presented with delayed gross motor milestones. At last follow up, three patients had a limb girdle pattern of muscle weakness and two had a facioscapular humeral pattern of weakness. Whole body muscle MRI performed for one patient with a facioscapular-humeral pattern of weakness showed a pattern of muscle atrophy similar to facioscapular-humeral dystrophy. One patient had histological features consistent with myofibrillar myopathy; electron microscopy confirmed the disruption of myofibrillar architecture. One patients also had reduced staining to telethonin antibody on immunohistochemistry.
Conclusion:
We report the unique clinical and histological features of a Southeast Asian cohort of five patients with LGMD2G/LGMD-R7-telethonin-related muscular dystrophy and further expand its clinical and histopathological spectrum.
ABBREVIATIONS
American College of Medical Genetics Creatine kinase Cytochrome oxidase Deoxyribonucleic acid Facioscapulohumeral dystrophy Modified Gömöri trichrome Hematoxylin and eosin Limb-girdle muscular dystrophy Medical research council Magnetic resonance imaging Nicotinamide adenine Dinucleotide-tetrazolium reductase Succinic dehydrogenase
INTRODUCTION
Limb-girdle muscular dystrophy (LGMD)2G/LGMD-R7-telethonin-related is an autosomal recessive muscle disease, caused by mutations of the TCAP gene. Telethonin is a protein localized to the Z-disc of adult striated and cardiac muscles, where it interacts with titin and other sarcomeric proteins.
LGMD2G/LGMD-R7 was first recognized amongst Brazilian patients of Italian origin [1]. While cases have since been described globally, it remains a rare condition with an estimated prevalence of 1.2 per million amongst east Asians, and less than 0.05 per million within other populations [2]. Mutations of the TCAP gene have also been reported to cause congenital muscular dystrophy [3], hypertrophic and dilated cardiomyopathy [4–6]. The clinical and histopathological spectrum of this condition continues to expand. Understanding the full disease spectrum of this condition is essential.
We report the unique clinical and histological features of a cohort of five patients with LGMD2G/LGMD-R7-telethonin-related seen in the tertiary neuromuscular referral centers at NNI and NUH in Southeast Asia.
METHODS
Patients were identified retrospectively from the institutional neuromuscular database, which is a prospective and retrospective registry of patients with various neuromuscular disorders, diagnosed by neuromuscular specialists, at the National Neuroscience Institute, Singapore and National University Hospital, Singapore [between January 2000 and June 2021].
We identified patients with possible LGMD2G/LGMD-R7-telethonin-related through two means: Patients seen for evaluation of muscular dystrophy who underwent genetic testing. For patients with muscular dystrophy where genetic testing was not previously performed, muscle biopsy specimens were reviewed for the absence or reduced staining for telethonin, on muscle immunohistochemistry. For such patients, subsequent sequencing of the TCAP gene was performed on DNA extracted from the specimen, where possible.
The diagnosis of LGMD2G/LGMD-R7-telethonin-related was based on meeting of either one of the following criteria: identification of biallelic pathogenic mutations of the TCAP gene absent staining for telethonin on muscle immunohistochemistry
Clinical records of identified patients were retrospectively reviewed, and following clinical data were extracted: Demographic data: age at onset of weakness, gender Clinical symptoms and signs: symptoms at onset, pattern of muscle weakness at last follow up (based on the established classification [7, 8]), muscle strength based on the medical research council (MRC) scale, contractures, hyperlordosis, age at which ambulatory aid was initiated, other associated non-neuromuscular symptoms Serum creatine kinase (CK) levels [last available value] Neuro-electrophysiological data Respiratory function, as evaluated by spirometry. Forced vital capacity (FVC) and forced expiratory volume (FEV1) were extracted, and severity of lung restriction was classified according to FVC [FVC>60% of predicted-mild, FVC 51-59% of predicted –moderate, FVC≤50% of predicted –severe] [9]. Cardiac investigations: electrocardiogram, 24-hour holter, echocardiogram [last available]. Muscle histopathology, electron microscopy, and immunohistochemistry
Muscle magnetic resonance imaging (MRI)
Whole body muscle MRI was performed using unenhanced axial T1 and T2 short tau inversion recovery (STIR) sequences from the neck to the ankles, on a 1.5T MR scanner (Ingenia: Philips Medical Systems, The Netherlands). Severity of muscle atrophy and T1 hyperintensity was staged based on the Mercuri criteria into the following categories: Stage 0: normal, stage 1: mild atrophy, stage 2: moderate atrophy, stages 3 and 4: severe atrophy [10].
Muscle biopsy
Muscle samples were obtained from the vastus lateralis (patients 1, 2, 4 and 5) for diagnostic purposes after obtaining informed consent. The samples were immediately frozen in liquid nitrogen and stored at –80 °C. Routine histological and histochemical procedures, including hematoxylin and eosin (HE) staining, modified Gömöri trichrome (m-GT) staining, and nicotinamide adenine dinucleotide-tetrazolium reductase (NADH-TR) staining, succinic dehydrogenase (SDH), cytochrome oxidase (COX) were performed. Electron microscopy was performed on the muscle biopsy specimens of patients 1 and 4. Biopsied muscle specimens were fixed in 2.5% glutaraldehyde and post-fixed with 2% osmium tetroxide. Semithin sections stained with toluidine blue were examined by light microscopy. Ultrastructural analysis was carried out on longitudinal and transverse ultrathin sections after staining with uranyl acetate and lead citrate, using a transmission electron microscope.
Immunohistochemistry and Western-blot analysis for telethonin was performed using anti-telethonin antibodies (LS-C340495; LifeSpan BioSciences, USA).
For each sample, a normal muscle control specimen was used to allow comparison between the patient and control sections.
Molecular analysis
For patients who underwent clinical genetic testing during the evaluation of muscular dystrophy, deoxyribonucleic acid (DNA) was extracted from peripheral blood. Targeted next generation sequencing panel includes the following genes associated with LGMD2/LGMD-R: ANO5, CAPN3, DYSF, FKRP, SGCA, SGCB, SGCD, SGCG and TCAP was performed. Sequences were searched for using the National Center for Biotechnology Information (NCBI) protein database, and variants were described with reference to the following transcripts: ANO5 (NM_213599.2), CAPN3 (NM_000070.3), DYSF (NM_003494.3), FKRP (NM_001039885.2), SGCA (NM_000023.4), SGCB (NM_000232.4), SGCD (NM_000337.5), SGCG (NM_000231.2), TCAP (NM_003673.3). For patient 1, additional targeted next generation sequencing panel including 36 genes (ACTA1, BAG3, CFL2, CRYAB, DES, DNAJB6, EPG5, FHL1, FLNC, GNE, KBTBD13, KLHL40, LAMP2, LDB3, MATR3, MEGF10, MYH2, MYH7, MYOT, NEB, ORAI1, PABPN1, PLEC, RBCK1, SEPN1, SIL1, STIM1, TCAP, TIA1, TNNT1, TPM2, TPM3, TRIM32, TTN, VCP, VMA21) known to cause myofibrillar myopathy syndrome by next generation sequencer (IonPGMTM) was performed. Identified TCAP variants were referenced against public databases including ClinVar (www.ncbi.nlm.nih.gov/clinvar/) and Varsome (varsome.com), and their pathogenicity was classified based on the American College of Medical Genetics (ACMG) criteria [11]. These variants were annotated based on the nomenclature recommended by the Human Genome Variation Society [12].
Sanger sequencing of the TCAP gene was performed on DNA extracted from muscle biopsy specimens for patients who had not undergone prior genetic testing. DNA was first extracted from frozen muscle tissue using Reliaprep gDNA tissue Miniprep system (Promega) following manufacturer’s guidelines. The TCAP gene (NG 009490.1) exons 2 to 4 were amplified by polymerase chain reaction using exTEN 2X PCR Master Mix (1st BASE) and primers (see supplementary Table 1) at an annealing temperature of 60°C. After amplification, PCR products were treated with FastAP Alkaline Phosphatase (Thermo Scientific) and Exonuclease I (Thermo Scientific) and subjected to direct sequencing using BigDye Terminator Cycle Sequencing Kit (Applied Biosystems) on an ABI 3130xl Genetic Analyzer (Applied Biosystems).
This study was approved by the Singapore Health Services Centralized Institutional Review Board (CIRB 2018-2341). Written informed consent was obtained from all patients.
RESULTS
A total of five patients (from four unrelated families) with LGMD2G/LGMD-R7-telethonin-related mutations were identified [Fig. 1]. Patients 2–4 were previously briefly reported in an abstract by Yee et al. [13]. Muscle biopsy was performed on four patients. One patient (patient 3) was a symptomatic sibling of patient 2, and was identified through cascade genetic testing.
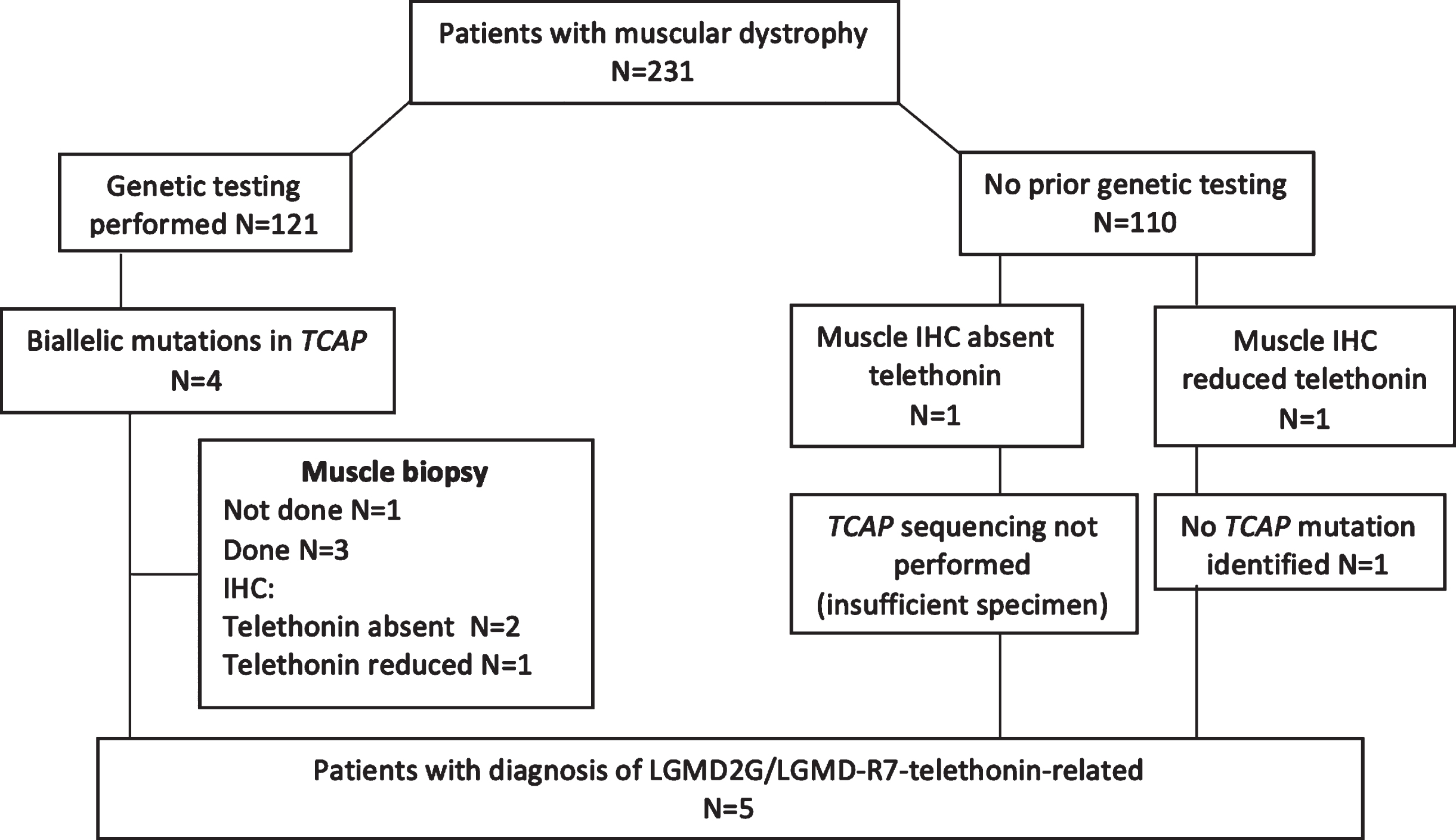
Flowchart describing identification of patients with LGMD 2G/LGMD-R7.
Individual patients’ clinical, histopathological and genotypic data are summarized in Table 1. Four patients were of Chinese ethnicity while one patient was Indonesian. Median age of onset of symptoms was 20.5 years (range 0–33 years); patients 1 to 4 had adult-onset disease while patient 5 presented in infancy. Median duration of follow up was 107 months (range 24–265 months). Three patients presented with proximal lower limb weakness at onset, one patient presented with Achilles tendon contractures, and one patient presented with delayed gross motor milestones (patient 5, crawling at 7 months old, cruising at 12 months old, walking with aid of knee-ankle foot orthosis at 24 months). At last follow up, three patients (patients 1,3 and 5) had a limb girdle pattern of weakness and two patients (patients 2 and 4) had a facioscapular-humeral pattern of muscle weakness. Median serum CK was 1418 U/l (range 104–2212 U/l). None of the patients had significant respiratory or cardiac involvement. Muscle biopsy was performed for patients 1, 2, 4 and 5, all of which showed myopathic histology. Muscle biopsies of patients 1, 2 and 4 showed the presence of rimmed vacuoles while patient 1 had additional findings indicative of myofibrillar myopathy. Three (patients 1, 4 and 5) showed absent staining for telethonin on immunohistochemistry, while one (patient 1) showed reduced staining for telethonin. Western-blot analysis was performed for patient 2, which showed complete absence of telethonin protein (supplementary figure 1). Electron microscopic examination (patients 1 and 4) showed ultrastructural features suggesting the disruption of myofibrillar architecture in patient 1, and subsarcolemmal inclusions in patient 4.
Clinical characteristics, histopathological and genotypic findings of patients
$in years; #at last follow-up; –: absent;+: present; CK: creatine kinase; D: distal; F: female; LL: lower limb; M: male; P: proximal; P>D: proximal weakness more severe than distal weakness; UL: upper limb; NA: not available; ND: not done. a,b,c,d: denote 4 unrelated families.
Clinical histories of patients 1 and 4 are discussed in further detail as illustrative cases.
Patient 1
Patient 1 was a Chinese male who presented, at age 32 years, with gradually progressive bilateral lower limb weakness. Developmental milestones were normal. The patient was physically active up to age 20, when he developed a tendency to fall while playing football. Since age 24, patient developed progressive difficulty climbing stairs and rising from a squatting position. There was no parental consanguinity. The patient had two older brothers (aged 37 and 35) and one younger sister (aged 28). He was married, and had a daughter. All family members were well.
Examination revealed hyperlordosis with asymmetric muscle atrophy over bilateral thighs and mild bilateral calf hypertrophy. There was no facial, bulbar and neck weakness. Upper limb power was normal, apart from mild weakness [MRC grade 5-/5] of bilateral finger abduction. There was slightly asymmetric bilateral proximal lower limb weakness (Hip flexion: right 2, left 3+; hip extension: right 4+, left 4+; knee flexion: right 4+, left 4+; knee extension: right 2, left 2; ankle dorsiflexion and plantarflexion: 5 bilaterally). Serum CK was 1036 U/L. Needle electromyography showed myopathic changes. Electrocardiogram showed normal sinus rhythm. He had no respiratory symptoms.
Muscle biopsy of the left vastus lateralis showed changes suggestive of myofibrillar myopathy [Fig. 2]. HE stain and m-GT stain showed several fibers with uneven dark staining and several fibers with rimmed vacuoles. Rare eosinophilic cytoplasmic bodies were also observed. The NADH-TR stain showed many lobulated fibers. SDH and COX stains showed many fibers showing “wiped-out” areas devoid of oxidative enzyme activity. The Congo red stain did not show any obvious congophilic staining. Myosin ATPase stains showed a random fiber type distribution with atrophy of both type 1 and type 2 fibers. Immunostain for telethonin showed reduced staining while Immunostain for desmin showed increased staining pattern in several fibers.
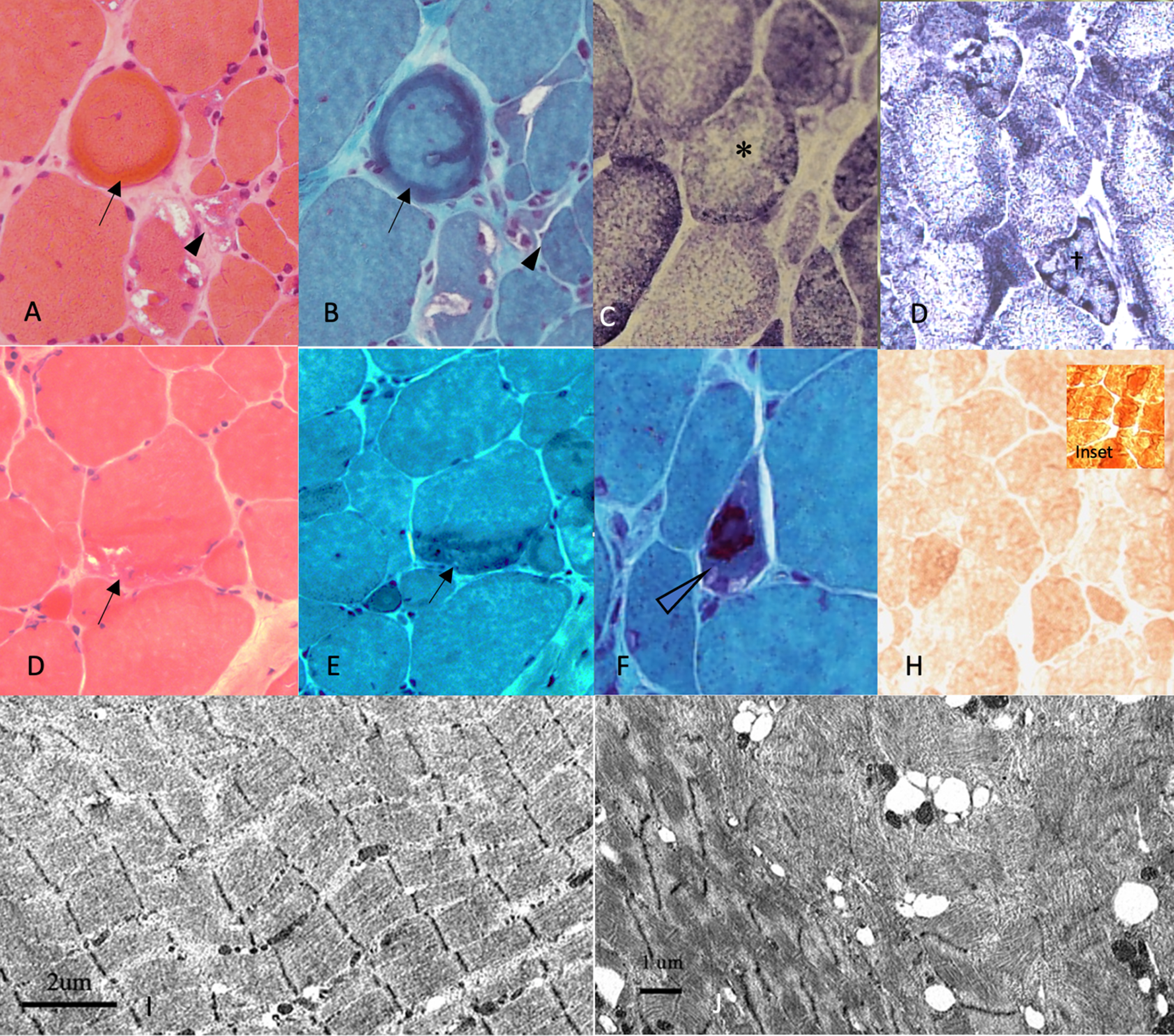
Histopathological findings in Patient 1 suggestive of myofibrillar myopathy. (A, B, E, F) sarcoplasmic, subsarcolemmal protein aggregates (arrows), irregularly shaped vacuoles (arrowheads), associated with (C) focal loss of NADH staining The same fibres show loss of NADH staining (*). (D) Many lobulated fibres (†) are seen. (G) cytoplasmic bodies (hollow arrowhead) are visible. (H) Immunostain for telethonin shows patchy reduced staining compared to normal control (inset). Electron microscopy reveal (I) sporadic areas of Z-disk disarray, producing streaming, undulation and smudging of the Z-disks. (J) Areas of subsarcomeric vacuoles containing the filamentous remnants. (A, D: Hematoxylin and eosin (HE) stain, original magnification×20; B,E: Gomori-trichrome (GT) stain, original magnification×20; F: Gomori-trichrome (GT) stain, original magnification×40; C,D: NADH-TR stain, original magnification×20; H: telethonin immunostain, original magnification x 20, I: scale bar 2μm, J: scale bar 1μm.).
Ultrastructural examination revealed sporadic areas where the Z-disks of the sarcomeric unit was disarrayed, producing streaming, undulation and smudging of the Z-disks. In more severely affected areas, there was a generalized effacement of the sarcomeric architecture. These abnormal areas had been sealed off in subsarcomeric vacuoles containing the filamentous remnants of the disrupted sarcomere.
Genetic testing revealed two heterozygous variants in the TCAP gene (NM_003673: c.[23_24insCGAGGTGT(;)110 + 5 G>A];p.[(Glu12Argfs)(;) (?)]. Genetic testing for genes associated with myofibrillar myopathy was negative.
Patient 4
Patient 4 was a Chinese male who presented with gradually progressive bilateral upper and lower limb weakness. Early developmental milestones were normal. He was physically active until the age of 14 years-old when he developed calf pain and difficulty standing after running in school. This was associated with toe walking, eventually requiring bilateral Achilles tendon release when he was 19 years-old. From age 20, he complained of progressive proximal limb weakness which progressed to involve the upper limbs at age 30. There was no parental consanguinity. The patient had two older brothers and one younger sister. He was married, and had one daughter. All family members were well. He was first evaluated at age 32. At that time, he was noted to have asymmetric scapular winging, left humeral wasting, wasted sternal head of pectoralis major, associated with proximal and distal lower limb atrophy and weakness, without significant facial weakness. Muscle biopsy performed at age 32 demonstrate a chronic, dystrophic myopathic process, associated with rimmed vacuoles and lobulated fibers. Immunostain for telethonin showed absent staining. Ultrastructural examination revealed abnormal subsarcolemmal myofibrillar and mitochondrial inclusions.
He was lost to follow up for a few years, and was re-evaluated at age 44, by which time he had developed facial weakness and progressive limb weakness, with requirement of walking stick for ambulation.
Examination at ages 44 and 54 [Fig. 3] revealed hyperlordosis with symmetric muscle atrophy over bilateral arm and bilateral proximal and distal lower limb muscles. Contractures were noted of bilateral knee and ankle joints. There was facial and eye closure weakness without ptosis or bulbar weakness and neck weakness. Upper limb examination revealed predominant proximal weakness with disproportionate asymmetric weakness of the elbow flexor and extensor. Scapular winging with an over-riding was noted on shoulder abduction. There was slightly asymmetric bilateral proximal lower limb weakness (Hip flexion: right MRC grade 2, left 3+; hip extension: right 4+, left 4+; knee flexion: right 4+, left 4+; knee extension: right 2, left 2; ankle dorsiflexion and plantarflexion: 5 bilaterally). There was abdominal flexion weakness. Serum CK was elevated at 576 U/L). Needle electromyography showed myopathic changes. Electrocardiogram showed normal sinus rhythm.
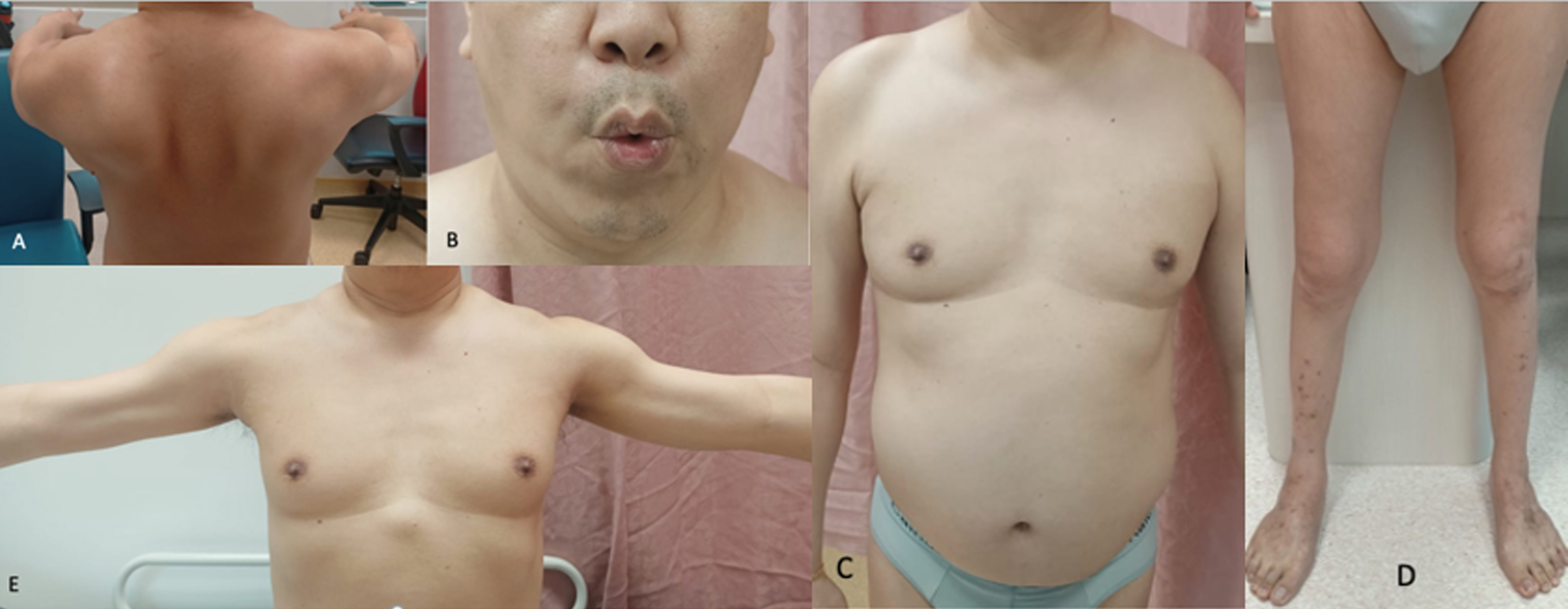
Patient 4: A: Posterior winging of the scapula on shoulder flexion; B: Facial weakness with inability to fully pucker mouth; C: Chest wall showing asymmetric wasting of pectoral muscles with prominent axillary fold, protruberant abdomen; D: Wasting of bilateral distal thigh and bilateral legs; E: High-riding scapula when attempting to abduct the arms caused by elevation of the scapula. Wasting of the bilateral biceps.
Whole body muscle MRI performed at age 54 revealed the following findings [full details in Fig. 4]. severe atrophy of the bilateral platysma and trapezius in the neck region. Severe atrophy of the bilateral serratus anterior, latissimus dorsi and pectoralis major, with sparing of the bilateral deltoid in the shoulder girdle. Severe asymmetric atrophy of the right brachialis and left biceps with severe atrophy of the bilateral triceps long head in the arm. Forearm muscles were largely spared. Severe atrophy of the obliques was noted in the abdominal region. There was diffuse lower limb involvement disproportionately involving the proximal lower limb muscles.
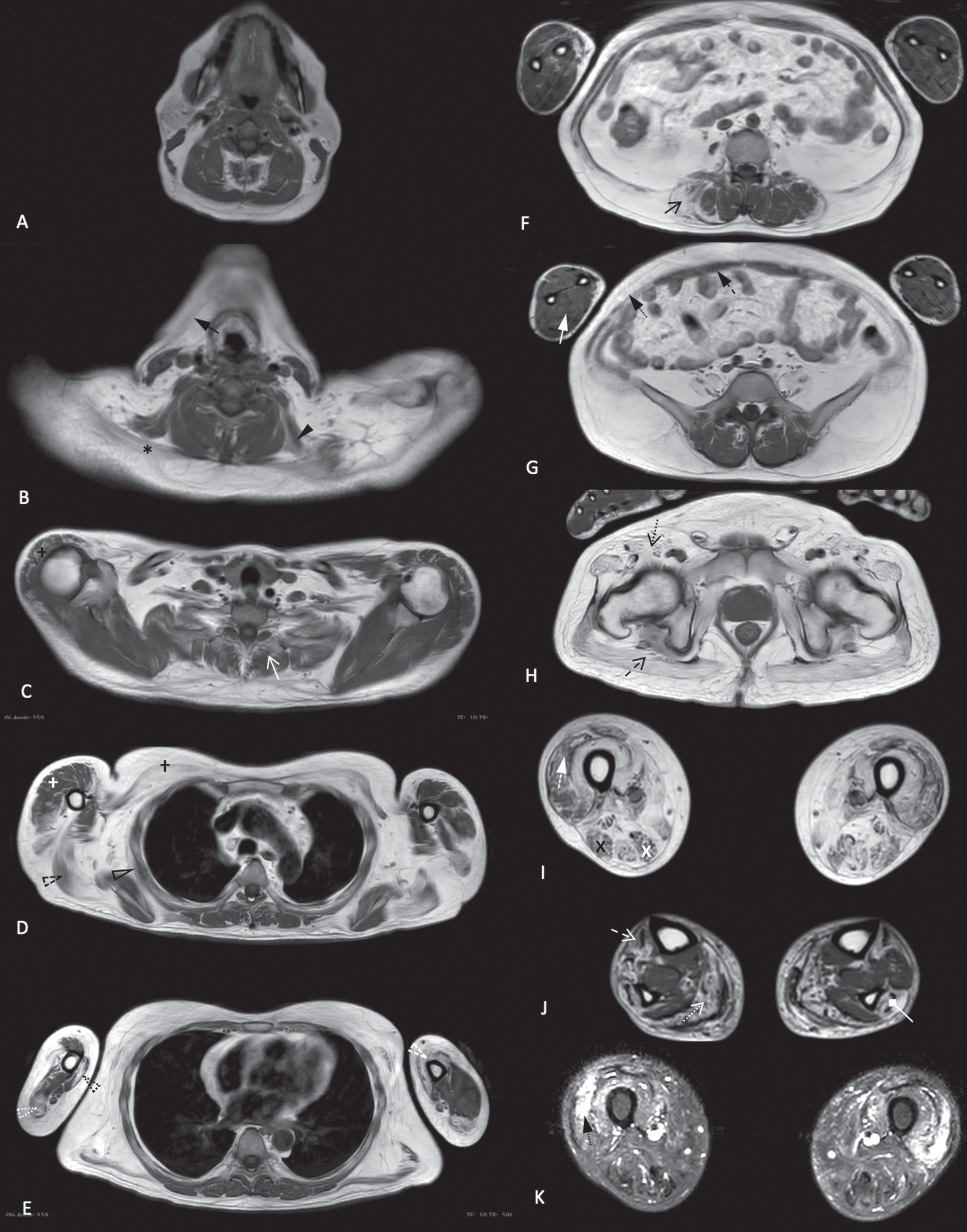
Whole body muscle MRI images of patient 4 performed at age 54 shows: Neck and axial region (A-F): severe atrophy of the bilateral platysma (black arrow) and trapezius (black *), moderate atrophy of the left levator scapulae (black arrowhead), mild atrophy of the thoracic paraspinal (white open arrow) muscles and focal asymmetric atrophy of the abdominal paraspinal muscles (black open arrow). Shoulder girdle (C-D): severe atrophy of the bilateral serratus anterior (black hollow arrowhead), bilateral latissimus dorsi (black dashed hollow arrowhead), bilateral pectoralis major (black †), with mild atrophy of the bilateral deltoid (white †). Arm (D-E): severe atrophy of the right brachialis (black dotted hollow arrowhead) and left biceps (white dashed hollow arrowhead) with moderate atrophy of the right medial biceps (anterior compartment). Severe atrophy of the bilateral triceps long head (white dotted hollow arrowhead), mild atrophy of the right triceps medial and lateral head (posterior compartment). Abdominal region and forearm muscles (E): severe atrophy of the obliques (black arrow with dotted tail) and mild to moderate atrophy of the rectus abdominis (black arrow with dashed tail). Forearm muscles (white arrow) are largely spared. Pelvic region (F): severe diffuse muscle atrophy of posterior (open black arrow with dashed tail) and anterior (open black arrow with dotted tail) compartments. Thigh region (G, I): severe atrophy apart from moderate atrophy of the bilateral vastus lateralis, bilateral rectus femoris (white arrow with dashed tail) (anterior compartment) and moderate atrophy of the right semimembranosus (black X) and right biceps femoris (white X) (posterior compartment). This is associated with edema of bilateral vastus lateralis (I, black arrow with dotted tail). Leg (H): moderate atrophy of the bilateral soleus, gastrocnemius (white open arrow with dotted tail) (posterior compartment), bilateral left tibialis anterior, right extensor digitorum longus (white open arrow with dashed tail) (anterior compartment) and right peroneal (white arrow with diamond head) (lateral compartment). (A-H) Axial T1, (I) Axial STIR.
Genetic testing revealed a homozygous variant in the TCAP gene: c.23_24insCGAGGTGT; p.Glu12Argfs, which is classified as pathogenic based on the ACMG criteria.
DISCUSSION
We present the clinical, histological, and genetic profile of a Southeast Asian cohort of patients with LGMD2G/LGMD-R7-telethonin-related. Our cohort includes the first description of myofibrillar myopathy as a pathological feature of LGMD2G/LGMD-R7, and presentations of LGMD2G/LGMD-R7 with facioscapular-humeral pattern of weakness [two patients] and congenital/infantile onset muscle dystrophy [1 patient]. We expand the full-body muscle MRI reports in LGMD2G/LGMD-R7, which have previously only been reported in five patients [3, 15]. Our cohort further expands the clinico-pathological phenotype associated with LGMD2G/LGMD-R7.
LGMD2G/LGMD-R7 has been reported to be associated with several phenotypic patterns: limb girdle distribution of weakness [1], congenital muscular dystrophy [3] as well as distal myopathy [16]. Previously reported cases are summarized in Table 2. We report two patients (patients 2 and 4) with a facioscapular-humeral pattern of weakness mimicking facioscapular-humeral muscle dystrophy (FSHD). Whole body muscle MRI (of patient 4) also demonstrated a pattern of muscle involvement similar to that of FSHD, with the disproportionate involvement of the trapezius, latissimus dorsi, pectoralis major, serratus anterior and arm muscles with significant asymmetry [17, 18]. This clinical and radiological pattern of muscle weakness has not been previously reported in patients with LGMD2G/LGMD-R7. While scapular winging [3, 19–22], a positive Beevor sign [23] as well as facial weakness [22, 25] have been reported in LGMG2G/LGMD-R7, they are reported in isolation and tend to occur later in the disease course.
Clinical characteristics, histopathological and genotypic findings of patients reviewed on available literature
aa, amino acid; Ab, antibody; Absent, absent staining; AM, adductor magnus; asym, asymmetrical; BF, biceps femoris; D, distal; EDL, extensor digitorum longus; EHL, extensor hallucis longus; FDL, flexor digitorum longus; FHL, flexor hallucis longus, GL, lateral gastrocnemius; GM, gluteus maximus; GRA, gracilis; IP, iliopsoas; LL, lower limb; MG, medial gastrocnemius; NA, detail not given; ND, not done; P, proximal; PF, piriformis; PL, peroneus longus Q, quadriceps; SAR, sartorius; SG, shoulder girdle; shBF, short head of biceps femoris; SM, semimbranosus; SO, soleus; ST, semitendinosus; Sym, symmetrical; TA, tibialis anterior; TP, tibialis posterior; UL, upper limb; VM, vastus medialis; WB, whole body; +, positive; –, negative.
It is therefore important to consider the diagnosis of LGMD2G/LGMD-R7 in a patient with a facioscapular-humeral pattern of muscle involvement on clinical and radiological examination, especially for a patient who presents late in the course of his/her disease. Useful clinical features to differentiate LGMD2G/LGMD-R7 from FSHD include initial presentation of lower limb involvement and prominence of joint contracture in LGMD2G/LGMD-R7. Based on the review of available literature (Table 2), the majority of reported cases of LGMD2G/LGMD-R7 presented with either lower limb weakness or Achilles tendon contracture as the first symptom.
We noted the finding of myofibrillar myopathy in patient 1, on muscle histopathology as well as on ultrastructural examination. Myofibrillar myopathy is characterized by the presence of the following histopathological features: presence of amorphous, granular or hyaline deposits varying in shape and size in trichrome-stained sections; focal areas with either reduction/loss of oxidative enzyme activity in many abnormal fiber regions, and especially in the hyaline structures; the presence of both rimmed and non-rimmed vacuoles; congophilic deposits associated with hyaline structure; an abnormal and ectopic expression of multiple protein such as desmin. Electron microscopy shows myofibrillar disorganization starting from the Z-disk and focal accumulations of compacted degraded myofibrillar elements distributed among the myofilaments [26].
Histopathological features of previously reported cases of LGMD2G/LGMD-R7 include myopathic findings associated with rimmed vacuoles and lobulated fibers. While histological findings consistent with myofibrillar myopathy have not been previously reported, there have been reports whereby scattered myofibrillar disruption at the Z-disc, seen in individual sarcomeres, [22] and focal sarcomere architectural disturbance [16] was observed on electron microscopy. Additionally, analysis of zebrafish depleted of telethonin showed altered organization of the myofibrils rather than abnormal sarcomere assembly [27]. The disruption of myofibrillar architecture leading to myofibrillar myopathy in LGMD2G/LGMD-R7 may be under-reported due to the paucity of electron microscopic studies of patients with LGMD2G/LGMD-R7 in literature. This histological findings may have been previously missed as well due to the focal nature of abnormalities associated with myofibrillar myopathy [28].
It is uncertain at this point how mutations in the TCAP gene can lead to the downstream effect of myofibrillar disruption. It had been shown that telethonin plays a role in the assembly of titin and its anchoring to the Z-disk [29] and that depletion in telethonin protein leads to disruption of T-tubular development through the disruption the interaction between sarcomere and T-tubules rather than through disruption of sarcomeric assembly [27]. This mechanistic explanation is likely not complete as it is unable to explain all the histological abnormalities seen in patients with LGMD2G/LGMD-R7.
Given the histological findings seen in patient 1, one may postulate that mutations of telethonin may lead to pathophysiological phenomena that result in myofibrillar disruption. This implicates other processes that have been previously postulated to be associated with myofibrillar myopathy, and includes the effect of the altered protein on the formation and maintenance of the extra-sarcomeric intermediate filament network through direct and indirect protein interactions, cell signalling cascades [30–34], mitochondrial dysfunctions [28], as well as impairment of the proteolytic function of the ubiquitin–proteasome system [35].
Immunohistochemistry and Western blot findings, in reported literature, is summarised in Table 2. The absence of telethonin staining on immunohistochemistry in a muscle biopsy has been demonstrated to be a highly specific diagnostic feature for LGMD2G/LGMD-R7. Based on this information, the absence of telethonin staining may be used as a surrogate for the diagnosis LGMD2G/LGMD-R7 if genetic diagnosis were not available. On the other hand, reduced telethonin staining on immunohistochemistry appears to be a finding of low specificity. In our study, one patient with reduced staining did not have pathogenic variants identified in the TCAP gene, while one genetically confirmed patient had reduced staining for telethonin on immunohistochemistry.
Reduced staining for telethonin was also reported on one other occasion by Barresi et al., who reported a patient with a homozygous c.244 C>T; p.Gln82X variant. Immunohistochemistry showed absence of staining to an antibody acting on the telethonin C-terminus and while showing reduced staining to an antibody acting on full-length telethonin. The c.244 C>T variant was predicted to generate a truncated protein of 81 amino acids [22]. It was postulated that alterations in staining pattern may be secondary to changes in the 3-dimensional conformation of the mutant protein or nonsense mediated mRNA decay [22].
Genetic sequencing of patient 1 revealed two heterozygous variants in the TCAP gene (NM_003673: c.[23_24insCGAGGTGT(;)110 + 5 G>A]; p.[(Glu12 Argfs)(;) (?)]. The c.110 + 5 G>A variant consists of a guanine-to-adenine nucleotide change in a noncanonical splicing site previously reported by Chen et al [36], and is classified as likely pathogenic based on the ACMG guideline (supplementary text 1). RT-PCR analysis by Chen et al. showed a mis-splicing pattern of TCAP mRNA, while western blot analysis revealed a significant reduction of TCAP protein level. We postulate that reduced telethonin staining in patient 1 may have resulted from the c.110 + 5 G>A variant producing an altered protein product with reduced affinity to the telethonin antibody.
Echoing Barresi et al., we would still recommend that patients with muscle biopsy specimens showing reduced staining to telethonin undergo genetic sequencing for the TCAP gene. Additional functional testing in the form of western blotting and/or RT-PCR analysis should be considered for further functional corroboration in the event a reduction in telethonin staining is observed.
There are several limitations to our study. Firstly, this was a retrospective study with inherent biases of a retrospective chart-review. Clinical and laboratory evaluations were clinician-determined, and therefore not standardized. Muscle imaging had only been performed on one patient within our cohort. Genetic confirmation could not be made in the patient with LGMD2G/LGMD-R7 who presented with congenital muscular dystrophy. Nonetheless, our study provides useful insights into the phenotypic characteristics and genotypic features of a South-East Asian cohort diagnosed with LGMD2G/LGMD-R7.
In conclusion, we report the unique clinical and histological features of a Southeast Asian cohort of five patients with LGMD2G/LGMD-R7-telethonin-related muscular dystrophy and further expand its clinical, radiological and histopathological spectrum. LGMD2G/LGMD-R7 may result in a clinical as well as radiological facioscapular-humeral pattern of muscle weakness. LGMD2G/LGMD-R7 may present with myofibrillar myopathy on histological examination. Although it is a non-specific finding, patients with reduced staining of telethonin on immunohistochemistry should undergo genetic testing for the TCAP gene.
Footnotes
ACKNOWLEDGMENTS
The authors thank our patients and their families for their valuable contribution to the study. This study was funded by Singapore’s National Medical Research Council (ASLN by the Clinician-Scientist Transition Award (MOH-TA18may-0003) and by Singapore Health Services (ZC by the SingHealth Precision Medicine Institute (PRISM) grant (AM/PRM009/2020).
DISCLOSURE OF CONFLICTS OF INTEREST
The other authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
Drafting or revising the manuscript for intellectual content: ZC, JSK, MS, KP. Study design: ZC, JSK, MS, PSN Acquisition of data: ZC, KSST, SHK, ML, JYHC, ASLN, AT, KBT, SKHT, JMMT, JYHC. Analysis and interpretation of the data: ZC, JSK, MS, KP, JYHC. All authors approved the final version of the manuscript.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding authors, Z Chen upon reasonable request.