
Abstract
BACKGROUND:
P-Element-induced wimpy testis (PIWI) proteins, when in combination with PIWI-interacting RNA (piRNA), are engaged in the epigenetic regulation of gene expression in germline cells. Different types of tumour cells have been found to exhibit abnormal expression of piRNA, PIWIL-mRNAs, and proteins. We aimed to determine the mRNA expression profiles of PIWIL1, PIWIL2, PIWIL3, & PIWIL4, in hepatocellular carcinoma patients, and to associate their expression patterns with clinicopathological features.
METHODS:
The expression patterns of PIWIL1, PIWIL2, PIWIL3, PIWIL4 mRNA, was assessed via real-time quantitative polymerase chain reaction (RT-QPCR), on tissue and serum samples from HCC patients, their impact for diagnosis was evaluated by ROC curves, prognostic utility was determined, and In Silico analysis was conducted for predicted variant detection, association with HCC microRNAs and Network Analysis.
RESULTS:
Expression levels were significantly higher in both HCC tissue and serum samples than in their respective controls (
CONCLUSION:
PIWIL mRNAs are overexpressed in HCC tissue and serum samples, the expression patterns could be valuable molecular markers for HCC, due to their association with age, tumour grade and pattern. To the best of our knowledge, our study is the first to report the expression levels of all PIWIL mRNA and to suggest their remarkable values as diagnostic and prognostic biomarkers, in addition to their correlation to HCC development. Additionally, a therapeutic opportunity might be also suggested through in silico miRNA prediction for HCC and PIWIL genes through DDX4 and miR-124-3p.
Keywords
Introduction
Liver cancer continues to be a global health problem, with global rates increasing [1, 2]. By 2025, the disease is predicted to affect over 1 million people [3]. Hepatocellular Carcinoma (HCC) is the most common type of primary liver cancer, accounting for around 90% of cases. HCC is seen as a problematic health problem in Egypt for example, with the number of patients doubling over the last decade [4]. Several risk factors are recognised for the development and progression of HCC, but the most prominent ones are viral infections, i.e., Hepatitis B virus (HBV) and hepatitis C virus (HCV). Cirrhosis is also considered at a risk factor developing HCC [3]. HCC aetiology is additionally correlated with mutational changes ascribed from exposure to tobacco and aristolochic acid (AA), and non-alcoholic steatohepatitis (NASH), all of which were determined as probable pathogenetic cofactors in HCC [5]. There is a widely accepted protocol for diagnosing chronic liver disorders (CLD), which involves evaluating liver function using a series of serum-level enzyme assays and a significant tumour marker,
Recent studies have shown that P-element-induced wimpy testis (PIWI) proteins could be used as markers for their diagnostic and prognostic values [7, 8]. Early Carcinogenesis has been linked to multiple epigenetic abnormal events such as; global hypomethylation of DNA, post-transcriptional changes of histones, dysregulation of noncoding RNAs (ncRNAs), and reactivation of transposable elements (TE) [9, 10, 11, 12, 13, 14]. PiRNAs (P-element induced wimpy testis (PIWI)-interacting RNAs) are short single-stranded ncRNAs, typically 25–33 nucleotides, and interact with PIWI proteins of the Argonaute family. PIWI proteins are involved in the synthesis of piRNAs and assemble ribonucleoproteins known as PiRNA-induced silencing complexes (pi-RISCs) in the cytoplasmic perinuclear foci or ‘nuage’, the mechanism operates at the transcriptional and post-transcriptional stages, and is based on complementarity with short RNA strands (piRNAs, miRNAs, and siRNAs). PIWI proteins and piRNAs were first thought to be implicated in germline and stem cells, with involvements in development, gametogenesis, proliferation differentiation, and maintenance of its integrity and stability via the inhibition of transposable elements’ (TEs) activation [15, 16, 17, 18, 19]. Their roles were also identified for self-renewal, fertilisation, organogenesis and epigenetic activation, expression of genes and proteins, maturation and plasticity of the brain, pancreas functions, and even fat metabolism [20, 21]. Emerging evidence has found their role in carcinogenesis and are related with prominent cancer hallmarks [5]. In humans, the PIWI protein family consists of four proteins: PIWIL1/HIWI, PIWIL2/HILI, PIWIL3, and PIWIL4/HIWI2 [22], belong to the class of cancer/testis antigens (CTAs), and their dysregulation is associated with cancer cell maintenance of proliferative signalling, apoptosis, stemness, genomic integrity, activating invasion, metastasis, mediating genomic instability, and boosting cell growth, to mention a few [23]. The abnormal expression of PIWIs in cancer were first discovered in 2011 [24] and the molecular mechanisms underlying PIWI’s oncogenic actions are controversial. Studies have also supported that PIWI proteins can be utilized cancer prognosis, and in combination with piRNAs they could also be employed for diagnosis [25]. Overexpression of PIWIL1/HIWI gene is seen in various cancers, including seminoma cell hyperplasia [25], oesophageal squamous cell carcinoma, gastric cancer [26] and pancreatic adenocarcinoma [27]. PIWIL2 gene variants transcribed by intragenic promoters, and shorter mRNAs were implicated in various cancers due to their carcinogenic characteristics. Thus, altered PIWI proteins and their variations seen in somatic malignant tumours may serve as diagnostic and prognostic biomarkers and therapeutic targets [28, 29]. Due to inconsistent findings in the literature, we examined the expression levels of the four human members of the PIWI family, both RNA levels by quantitative RT-PCR, in HCC patients (
Methods
Patients and sampling
The present study was conducted at Theodor Bilharz Research Institute (TBRI), Egypt. Patients who were diagnosed with HCC by multi-slice triphasic CT and increased alpha fetoprotein levels were selected. Institutional Approval was acquired from the Research Institute Board office (IRB) (NHTMRI-IRB) (Serial: 2-2019), and Theodor Bilharz Research Institute (TBRI-IRB). The research was conducted according to the declaration of Helsinki for human subject research guidelines (2013). Prior to enrolment, all patients and volunteers signed an informed consent form. Participants’ information was collected in strict confidence. 50 patients undergoing liver resection were sampled for tumour and tumour-adjacent samples, the surgery for liver resection was conducted within the department of surgery NHTMRI Hospital, Egypt, matching blood samples were also collected, and blood samples from 25 healthy volunteers were used as controls. Individuals suffering from other liver diseases (e.g., Autoimmune hepatitis, Hemochromatosis, Schistosoma), or diseases such as HIV, and ischemic heart diseases were excluded. Patients with HCV who were taking immunomodulatory interferon therapy were also excluded.
Sample processing
Blood samples were allowed to clot, centrifuged at 500 xg for 10 minutes, serum was collected, centrifuged, aliquoted, and stored at
Biochemical parameters
Laboratory tests including alanine aminotransferase (ALT), aspartate aminotransferase (AST), Bilirubin, albumin (ALB) and alpha-fetoprotein (AFP) were performed for all subjects as routine tests upon admission, AFP was also measured by ELISA using a commercially available kit (ABCAM, AB79801, Cambridge, UK).
RNA extraction and cDNA synthesis
Total RNA was extracted using the miRNeasy extraction kit (Qiagen, Valencia, CA) was used to according to the manufacturer’s instructions for both tissue and serum samples. Samples were extracted in duplicates, then the quality and concentration of the samples were measured using a NanoDrop-1000c spectrophotometer (Thermo-Fisher Scientific, Cinisello Balsamo, Italy). For transcription of the mRNA samples into cDNA The QuantiTect Reverse Transcription Kit (Qiagen, Valencia, CA), was used according the manufacturer’s instructions, with 1
Real-time quantitative polymerase chain reaction (RT-QPCR)
The selected primers included the four isoforms PIWIL1/HIWI; PIWIL2/HILI; PIWIL3, PIWIL4 and all assays were acquired from Qiagen, and were performed according to the manufacturer’s instructors. Quantitative values were respective to the Cycle number (Ct Value), where fluorescence was directly proportional to growth of PCR products, this was performed by QuantiTect SYBR Green PCR Kits (Qiagen, Valencia, CA). Glyceraldehyde 3-phosphate dehydrogenase, (GAPDH), was also used, as an endogenous control because of its transcripts’ prevalence, to normalize each sample. All reactions were run in duplicates. Finally, the
In Silico variant detection for PIWIL genes and Network Analysis
Hepatocellular Carcinoma primary cancer database (
Statistical
All statistical analyses were performed using statistical software SPSS (Statistical Package for Social Science) statistical program version 21.0. A Power test, indicated that the standard deviation of control is 0.8 and the standard deviation for the regression errors will be 1.9. Regression was at 1.1, and that 50 study subjects and 25 normal controls will be an appropriate representation via regression with a probability of 85%, and Type I error was 0.05. as adapted from HCC molecular marker research [33]. Normality tests determined continuous variables, described as mean
Demographics and Clinico-pathological characteristics for HCC patients
Demographics and Clinico-pathological characteristics for HCC patients
Alanine aminotransferase (ALT), aspartate aminotransferase (AST), albumin (Alb) and alpha-fetoprotein (AFP). No. of masses are represented as Mean and SD. But Alpha feto-protein, Tumour size, and Steatosis (Fatty degeneration of hepatocytes (% of cells)) are represented as Median and Interquartile Range IQR (25%–75%). While Sex, Grade, Pattern, Stage, HAI (Hepatitis Activity Index (grade of hepatitis), Hepatomegaly, Ascites, Splenomegaly, and oedema Lower Limbs are represented as Frequency and percent.
Relative expression representation for PIWIL mRNA in HCC (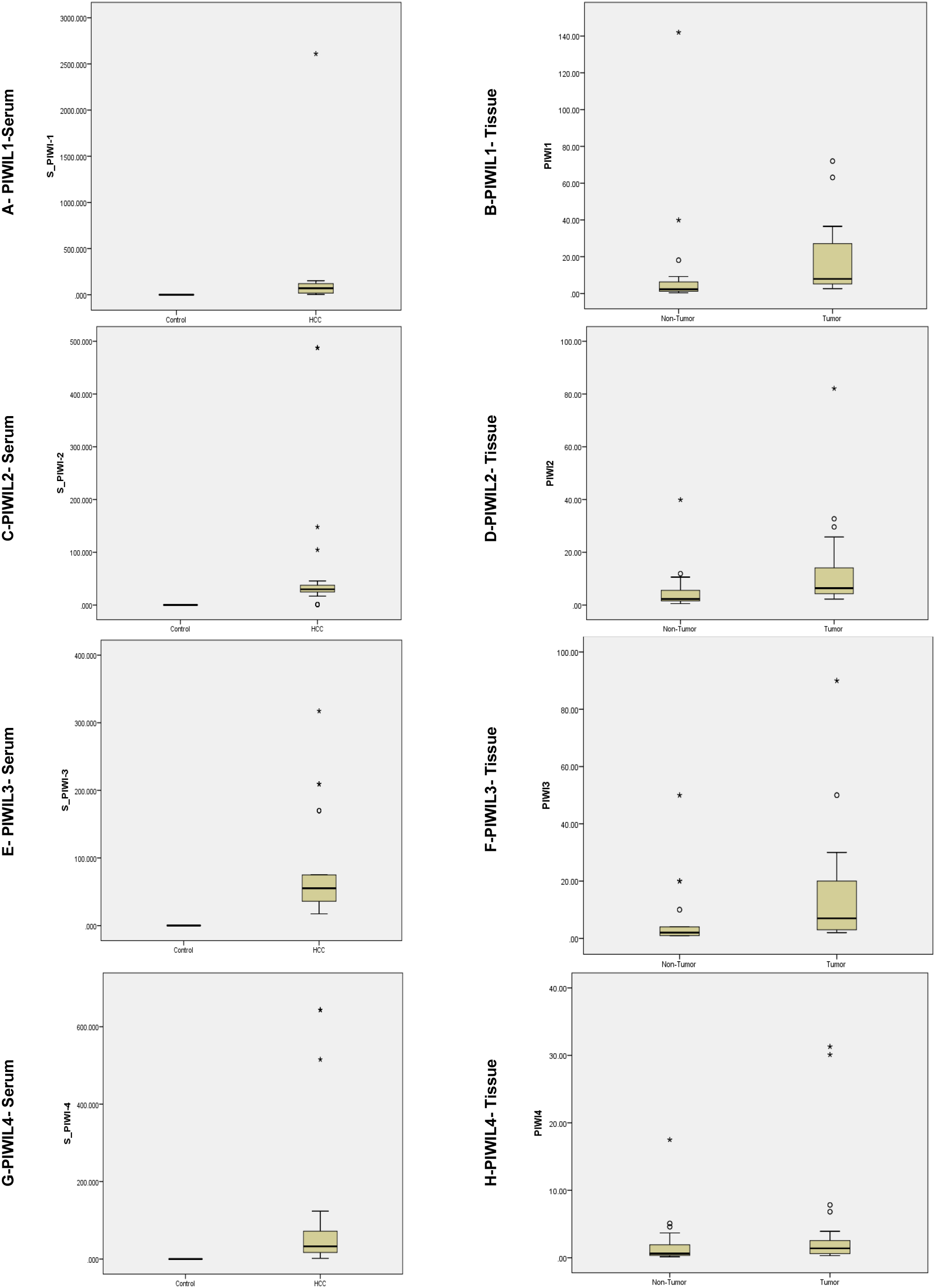
Patient characteristics
A total of 50 HCC participants were recruited in this study, the patients included 28 males (56%) and 22 females (44%), with a mean age of 57.2
Clinico-pathological characteristics for HCC patients and their association with PIWIL mRNA expression
Clinico-pathological characteristics for HCC patients and their association with PIWIL mRNA expression
The studied genes are represented as Median and Interquartile Range IQR (25%–75%); the data were analysed by Mann-Whitney U test. *p < 0.05, **p< 0.001. Grade, Pattern, Stage, Hepatomegaly, Ascites, Splenomegaly, and oedema Lower Limbs are represented as Frequency and percent.
Clinico-pathological characteristics for HCC patients and their correlation with PIWIL mRNA expression
Alanine aminotransferase (ALT), aspartate aminotransferase (AST), albumin (Alb) and alpha-fetoprotein (AFP). No. of masses are represented as Mean and SD. But Alpha feto-protein, Tumour size, and Steatosis (Fatty degeneration of hepatocytes (% of cells)) are represented as Median and Interquartile Range IQR (25%–75%). Sex, represented as Frequency and percent
15 (30%) patients were rated as AI, 35 (70%) were A2 while none of the patients were A3. Abdominal ultrasounds detected hepatomegaly in 9 (18%) patients, ascites in 20 (40%) patients and splenomegaly in 29 (58%) patients and finally 13 (26%) out of the 50 HCC patients were detected with oedema lower limbs (Table 1).
ROC curves of the studied PIWIL mRNA in (a) serum and (b) tissue samples. (C & D) are combined curves for serum and tissue, respectively. (E) ROC curve for AFP measured by ELISA in serum samples of HCC patients. Graphs depict the diagnostic performance of all PIWIL mRNAs in terms of specificity & sensitivity.
ROC curve analysis for diagnostic performance for each of the studied PIWIL mRNAs in serum and tissue samples
Sn: Sensitivity, Sp: Specificity, AUC Area under curve and C.I: 95% Confidence Interval. * p< 0.05, ** p< 0.001.
RT-QPCR results showed mRNA levels were upregulated for PIWIL1, PIWIL2, PIWIL3 and PIWIL4 in serum & tissue samples at (
Association of clinicopathological features with the expression PIWIL mRNA transcripts
Tumour grades of patients were associated with expression of PIWIL1-4. No association was found for serum, but for tissue samples, a significant association for PIWIL1 (
HCC risk results of PIWIL mRNA in HCC patients
HCC risk results of PIWIL mRNA in HCC patients
OR: Odds Ratio; C.I: Confidence Interval
Receiver Operating characteristic (ROC) analysis of PIWIL1, PIWIL2, PIWIL3 and PIWIL4 in both serum, and tissue samples of HCC patients. They showed a sensitivity of 100%, specificity of 100%, area under curve (AUC) of 1.00 (
Logistic regression analysis of PIWIL’s
To assess the relative risk of HCC presented from PIWIL mRNA, logistic regression analysis model was performed (Table 5). It revealed that PIWIL1-4 is significantly associated with increased risk for HCC in serum (
In Silico analysis for PIWIL genes
In Fig. 3B the role of PIWIL genes in HCC progression is indicated, where the highest implication being for PIWIL2 at 4% structural variant detection with an association for HCC; Fig. 3B shows the frequency of PIWI mutations according to HCC types; miRNet prediction was also employed for HCC association with PIWIL genes (Fig. 3A) and a direct association was detected between PIWIL2, PIWIL4 and MiR-124-3p.1, a known potential tumour suppressor associated with diverse processes including proliferation, apoptosis, and metastasis. The Frequency of PIWIL mutations were caried out through TCGA; via BioPortal (Fig. 3C), the results indicated that the highest frequency for reported mutations were for PIWIL2 at 4%, and the type of reported alterations were deep deleted deletions, remaining PIWIL variants had 1.4% which also had missense mutations as alterations. We identified the top 24 neighbouring genes with the highest frequency association with differential expressed PIWIL targets. The functions of these related were predicted using Metascape. The top 29 GO enrichment were described in (Fig. 4), which mainly included gene silencing by RNA, lncRNA, siRNA biogenesis and RISC complex assembly; Pathway enrichment analysis represented pathways a strong associated between PIWIL2, PIWIL4 and DDX4, DEAD-box helicase 4 (DDX4) and DDX39 was previously found to be upregulated in HCC, and that knocking down the DDX4 significantly decreased tumour formation in vivo and in vitro, as well as reduces tumour metastasis in vivo [34, 35] (Fig. 4F); indicating a relationship with HCC progression. The PPI network and MCODE components are shown in (Fig. 4C–F).
Discussion
PIWIL1, PIWIL2, PIWIL3 and PIWIL4, are designated as catalytic elements of the pi-RISCs complexes, the have implications in piRNAs’ biogenesis on the basis of complementarity [35, 36, 37]. Their roles vary at transcriptional and post-transcriptional epigenetic regulation. PIWIL1-2-3-4 RNAs were strongly expressed in HCC tissue, and circulating sera compared to the controls. The unexpected role of the PIWI-piRNA pathway has led to distinct functions of human PIWI proteins and mRNA in various cancer types [38, 39, 40]. Their involvement in multiple cancer hallmarks, has led to a possible representation as diagnostic and prognostic biomarkers [41, 42]. There are conflicting statements on the expression patterns of PIWIL1/PIWIL2/PIWIL3/PIWIL4, their prognostic, and predictive values, in addition to a complete absence for hepatocellular carcinoma in terms of mRNA transcript accumulation, and associated piRNAs, with recent studies on identifying novel piRNAs [43, 44, 45].
In terms of demographic data for the patients of the current study, the mean age was 57.2
(A) miRNet prediction of PIWIL and HCC. (B) Reported cases with mutated PWIL genes in Liver cancer (C) Frequency of mutated PIWIL genes and their predicted variants’ implication for HCC through TCGA; via BioPortal for Cancer Genomics (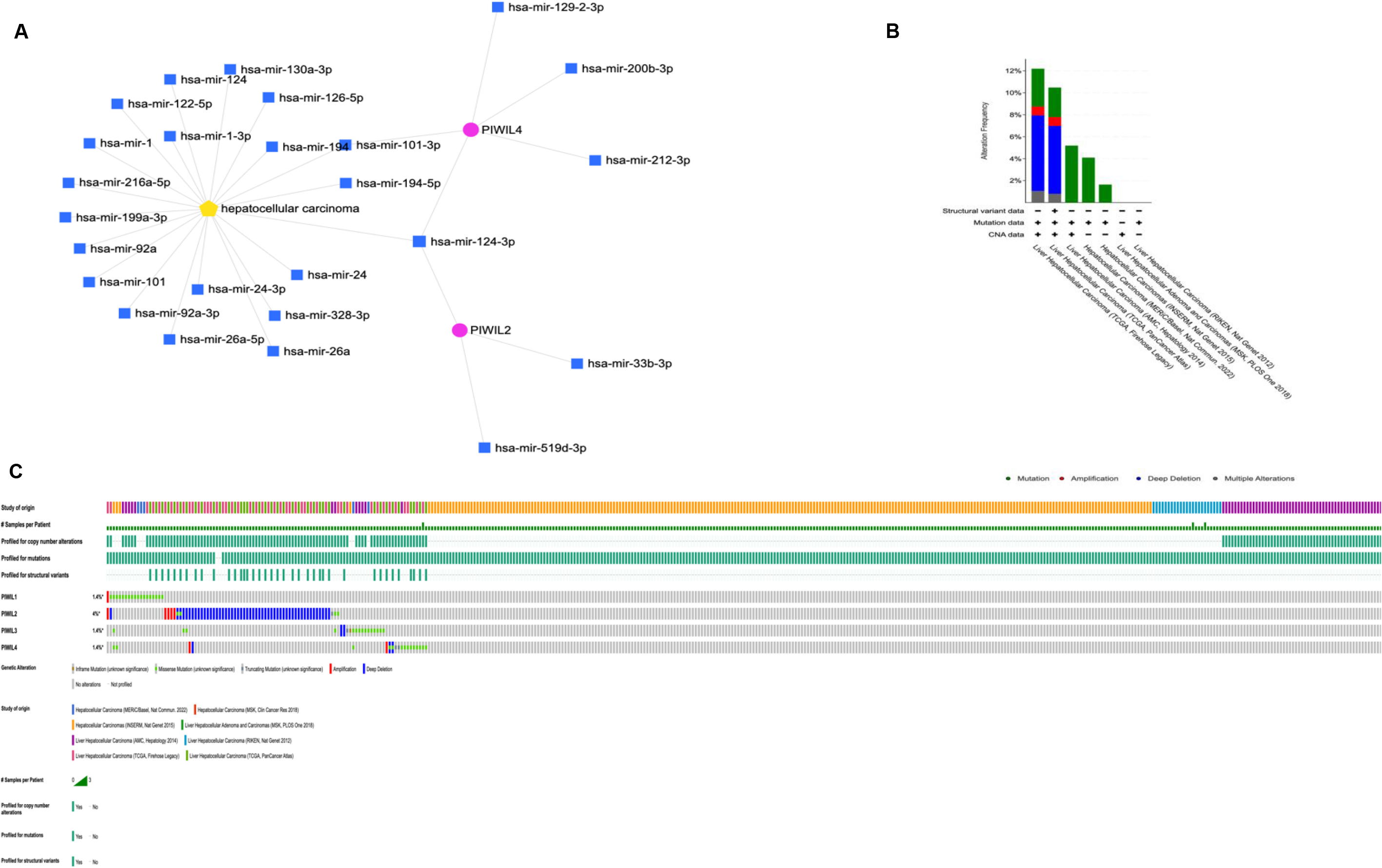
(A) Genemania generated Network for PIWIL1, PIWIL2, PIWIL3, PIWIL4 and function predictions (B) Metascape results for Enriched ontology clusters. (C) PIWIL Subset network with force-directed clustering (D) Enriched network according to statistical significance, the darker the color, the more statistically significant the node is. (E) protein-protein interactions’ network from PIWIL genes. (F) Results for MCODE algorithm to identify neighborhoods where proteins are densely connected.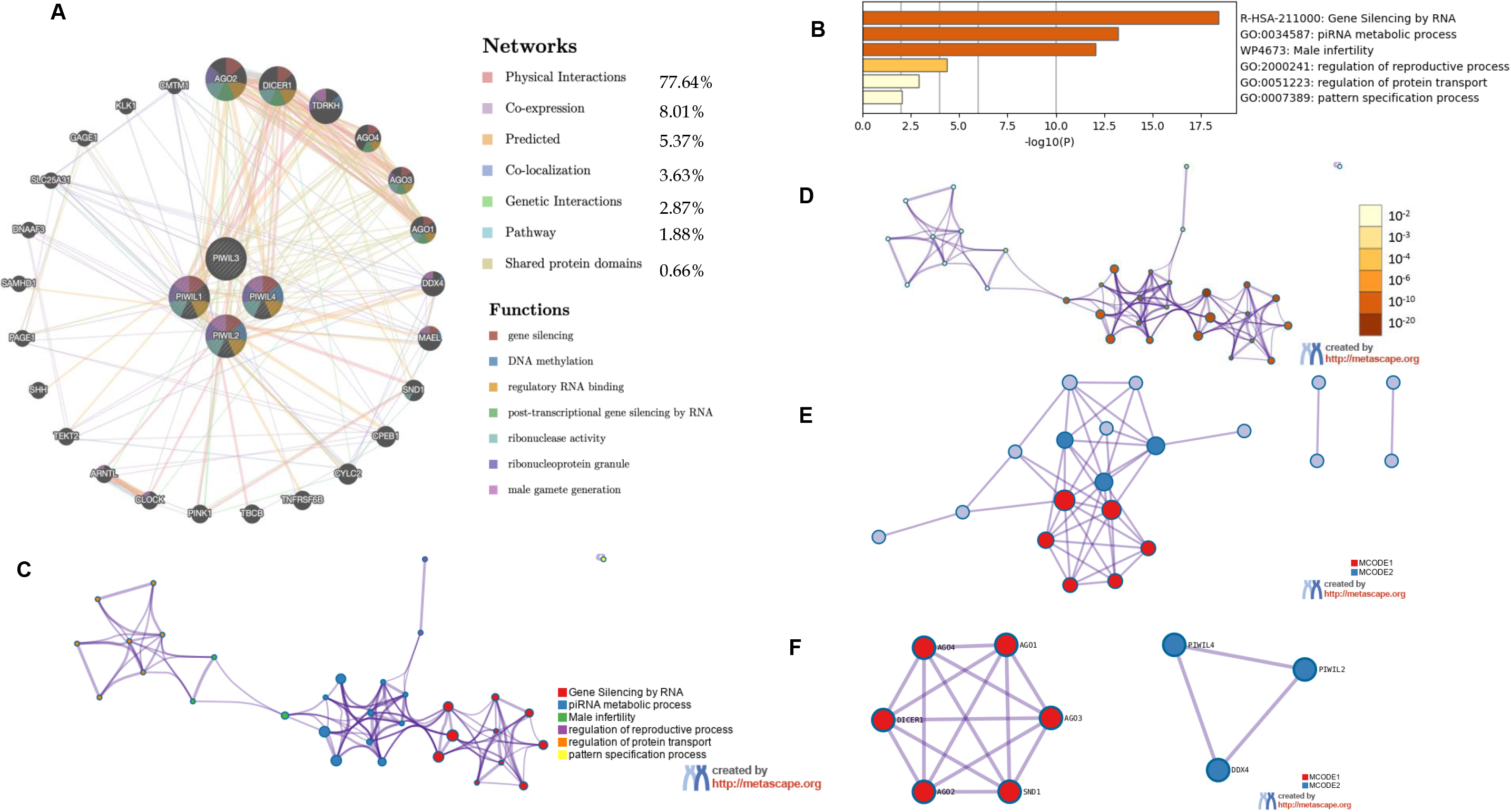
& PIWIL4 had significant difference with tissue expression, which was confirmed in a study by Litwin et al. for PIWIL1, & PIWIL2 in breast cancer [49].
Recently, several reports have indicated that aberrant expression of PIWI at the mRNA and protein levels occurs in various types of tumours [50, 51]. PIWIL1/HIWI was previously linked with several types of cancers, with a pattern of overexpression, moreover, it was correlated with tumour grading and staging [51, 52]. Our findings confirmed the apparent role of PIWIL mRNAs for HCC, with a pattern observed of overexpression for all PIWIL isoforms. Previously, both PIWIL1 & PIWIL2 was identified as overly expressed in colorectal, prostate, breast, cervical, gastric and bladder cancer [51], PIWIL1, 2 & 4 was observed as downregulated in renal cell carcinoma [50]. Meseure et al. conducted an investigation on the bio-pathological significance of the PIWI-PiRNA pathway through PIWIL1-2-3-4 mRNA expression levels in a panel of normal tissues and corresponding malignant tumours, and detected variable levels of expression across malignancies [53]. In addition, the relative expression of PIWIL2 mRNA was previously found to be higher in HCC tissues compared with adjacent normal liver tissues. A positive correlation was found between PIWIL2 expression and piR-Hep1 level according to Pearson’s correlation analysis [37, 38]. PIWIL2 acts as an oncogene by activating the STAT3/Bcl-xl cell signalling pathway through endogenous RNAi mechanism, hence inhibiting cell apoptosis and promoting cell proliferation. To the best of our knowledge, our study is the first to report the expression levels of all PIWIL mRNAs in serum and tissue, and to suggest their possible roles as diagnostic and prognostic biomarkers, in addition to their correlation to HCC development.
The involvement of PIWI proteins was linked with multiple hallmarks of cancer including invasion, apoptosis evasion, metastasis and cell proliferation, as such they possess prospective diagnostic factors and biomarkers for cancer prognosis [25, 54]. Significant increased level of PIWIL1 was reported for colon, bladder, and hepatocellular carcinoma. Expression of the four members of the PIWI proteins was viewed as distinct in tumour tissue when compared with the adjacent non-tumorous tissue [22, 55]. PIWIL3 and PIWIL1 were assessed for relative expression levels by [56] in colorectal cancer, and had non-significant expression statistically. Among all PIWIL genes, those assessed for expression and correlated with overall survival and recurrence-free survival were PIWIL3 & PIWIL4, in invasive urothelial bladder cancer [49]. Erber et al. reported a limitation of their study was not assessing the expression of PIWIL1 and PIWIL2 at the mRNA level, and depending on Immunohistochemistry, but they reported higher expression levels [57]. In the current study, expression of all PIWIL was assessed using real-time PCR in HCC patients. The levels of mRNA transcripts expression are reported for the first time for PIWIL1, PIWIL2, PIWIL3, PIWIL4 in tumour and nontumorous adjacent tissue, and matching serum samples HCC patients and healthy controls, the expression was correlated with clinical data. When compared to adjacent non-cancerous tissues, we found a significantly elevated expression for PIWIL1, PIWIL2, PIWIL3, &, PIWIL4 in HCC samples (
These findings are consistent with prior findings in colorectal cancer, in which PIWIL1 mRNA levels in non-cancerous tissue were low or undetectable, but were dramatically raised in malignant tissue [58]. Additionally, PIWIL1/HIWI was reported to have marked expression levels in HCC tissue, for patients who had undergone curative resection [42]. In breast cancer, PIWIL1 and PIWIL3 gene expressions were reported to be upregulated, whereas PIWIL2 and PIWIL4 were downregulated compared with normal breast tissue [53]. It is noteworthy to state that our research detected significant association between PIWIL1 & PIWIL4 expression with increasing tumour grade (
In terms of diagnostic performance, our findings showed that serum had an overall sensitivity and specificity of 100%, and an AUC of 1.0, in comparison to normal serum, which shows an indication of disease prevalence but should be adapted for clinical settings i.e., patients with chronic liver diseases. Tissue samples exhibited sensitivity of 70%, specificity of 68%, and AUC of 0.733. Both of which were significant
PIWIL genes have several transcripts, some of which appear to be transcribed by putative intragenic promoters rather than a canonical promoter, which was associated with tumorigenesis [65]. PIWIL expression was revealed to have a direct predicted influence on HCC progression, through PIWIL2, and PIWIL4, this was found through two novel associations DDX4 (Fig. 4E), and miR-124-3p.1 (Fig. 3A), Recent studies have shown that downregulation of miR-124-3p.1 was associated with poor survival, early recurrence and sorafenib sensitivity in HCC patients [42], and our findings have demonstrated a novel direct interplay between PIWIL2, PIWIL4 and miR-124-3p.1, the pathway additionally indicates a several target miRNAs which could be used as possible therapeutic targets for PIWIL2 miR-33b-3p and miR-519d-3p were identified, while for PIWIL4, miR-129-2-3p, miR-200b-3p, and miR-212-3p were found, all of which are related to miR-124-3p.1, a microRNA which when upregulated, negative affects proliferation and migration in hepatocellular carcinoma via a phosphoinositide 3-kinase catalytic subunit alpha (PIK3CA) pathway [68]. and miR-101-3p was additionally detected for PIWIL4, and was reported to negatively influence HCC proliferation and metastasis through the HGF/c-Met pathway [70]. Thus, aberrant expression of PIWIL2 and PIWIL4 is a possible mechanism of tumour suppressor inactivation Fig. 3A, and the associated miRNAs could represent attractive therapeutic targets for combined therapies where a specific antibody for PIWIL2 or the PIWI/miRNA RISC complex could be targeted [71]. PIWIL expression is also shown have a direct predicted influence on HCC progression, through PIWIL2, and PIWIL4, through association with DDX4 (Fig. 4). Studies have shown that elevated levels of DDX4, indicates its ability to promote the stemness of breast cancer stem cells by regulating the expression of proteins such as Oct3/4 and Sox-2 and promoting disease progression [72]. In this regard, its upregulation or overexpression promotes proliferation, suggesting an oncogenic role, its association through the PPI network (Fig. 4), shows a novel interaction with PIWIL2 and PIWIL4 and further analysis into PIWIL2 and PIWIL4 expression and silencing is recommended.
Finally, PIWI mRNA, PIWI proteins, and piRNAs were identified as germline markers. DNA methylation, histone methylation, histone acetylation, and histone ubiquitination not only play significant transcriptional regulatory roles, but PIWI family proteins can break mRNA under the supervision of piRNA, suggesting a post-transcriptional regulatory function. However, the method of control of PIWI/piRNAs in cancer appears to be unique. Most studies have established that PIWI mRNA, proteins, and piRNA appear to govern tumours as two distinct entities rather than as a unified entity. As a result, it is critical to investigate how the PIWI protein regulates tumours independently of piRNA. The specific molecular biological mechanism behind the effect of PIWIL on the occurrence, progression, and prognosis of HCC is currently unknown and deserves additional investigation. Our findings provide a unique viewpoint on the activities of PIWIL at the mRNA level in HCC development, as well as a different pattern of overexpression that provides potential candidates for HCC disease progression and risk assessment. PIWIL mRNAs are overexpressed in HCC tissue and serum samples, the expression patterns could be valuable molecular markers for HCC, due to their association with age, tumour grade and pattern. To the best of our knowledge, our study is the first to report the expression levels of all PIWIL mRNA and to suggest their remarkable values as diagnostic and prognostic biomarkers, in addition to their correlation to HCC development. Additionally, a therapeutic opportunity might be also suggested through in silico miRNA prediction for HCC and PIWIL genes through DDX4 and miR-124-3p. The epigenetic regulation of the identified changes in PIWIL1, PIWIL2, PIWIL3, and PIWIL4 at the transcriptional and protein levels warrants additional investigation, which could have significant clinical relevance, and should be examined for therapeutic roles. A large sample size investigation will aid in analysing the link between PIWIL expression/co-expression and HCC prognosis. This can serve as the preliminary foundation for PIWIL as molecular markers of early-stage diagnostic and prognostic evaluation, as well as targeted cancer therapies.
Footnotes
Supplementary data
The supplementary files are available to download from http://dx.doi.org/10.3233/CBM-230134.