
Abstract
Hepatocellular carcinoma (HCC) is a cancer with relatively high mortality, yet little attention has been devoted for related prognostic biomarkers. This study analyzed differential expression of m5C RNA methyltransferase-related genes in normal samples and tumors samples in TCGA-LIHC using Wilcoxon test. K-means consensus clustering analysis was implemented to subdivide samples. Independent prognostic factors were screened by univariate and multivariate Cox regression analyses. KEGG pathway enrichment analysis was performed on the screened independent prognostic factor using GSEA tools. qPCR was conducted to test mRNA expression of key m5C RNA methyltransferase-related genes in tissues and cells. There were 7 m5C RNA methyltransferase-related genes (NOP2, NSUN4, etc.) differentially expressed in HCC tumor tissues. HCC samples were classified into 3 subgroups through clustering analysis according to the expression mode of m5C RNA methyltransferase-related genes. It was also discovered that patients in different subgroups presented significant differences in survival rate and distribution of grade. Additionally, NOP2, NSUN4 and NSUN5 expression notable varied in different grades. Through regression analyses combined with various clinical pathological factors, it was displayed that NSUN4 could work as an independent prognostic factor. KEGG analysis showed that NSUN4 mainly enriched in signaling pathways involved in ADHERENS JUNCTION, RNA DEGRADATION, MTOR SIGNALING PATHWAY, COMPLEMENT and COAGULATION CASCADES. As examined by qPCR, NSUN4 was conspicuously upregulated in HCC patient’s tissues and cells. Altogether, our study preliminarily developed a novel biomarker that could be independently used in prognosis of HCC, which may provide a new direction for the study of related molecular mechanism or treatment regimen.
Introduction
Hepatocellular carcinoma (HCC) is a cancer with relatively high mortality, strong invasiveness and frequent recurrence. Identified clinical risk factors for HCC are liver cirrhosis, alcohol abuse, steatohepatitis, obesity, diabetes, intake of the aflatoxin B1, etc. [1]. A study unveiled that HCC morbidity is increasing in patients with chronic liver disease and liver cirrhosis [2]. Due to the high incidence of hepatitis B, more than half of HCC patients in the world are in China [3]. Metastatic HCC may achieve distant metastasis directly or indirectly via the circulatory system, arriving at lungs or lymph nodes in the chest through the lymph system [4]. Presently, clinical treatment for HCC includes surgery, chemotherapy, radiotherapy and immunotherapy, but the prognosis of these remains dissatisfied with the 5-year survival rate lower than 50% [5]. Differential expression of some molecules can be used as a biomarker for the prognosis of HCC. For instance, a study pointed out that the expression level of Enolase-1 enzyme can be an independent prognostic factor of HCC [6]. Despite this, few studies about biomarkers related to the prognosis of HCC are available. Finding new biomarkers can provide new entry points and thoughts for the study of HCC-related molecular mechanism.
An increasing number of evidences indicate that post-transcriptional modification of RNA involves in the progression of various cancers in different degrees. Highly expressed NSUN4, for example, is considered to be relevant to the processes of methylation and demethylation and HCC patient’s survival [7]. NSUN4 is a 5-methylcytosine (m5C) methyltransferase responsible for mitochondria rRNA methylation, and it can participate in the assembly of ribosomes in mitochondria and the translation of related molecules by forming a stable dimer complex with MTERF4 [8]. In addition, other members of the M5C methyltransferase family also play an important role in RNA post-transcriptional modification. For example, NSUN1 and NSUN5 modify cytoplasmic ribosomal RNA [9, 10]. Cytoplasmic transport RNA is methylated by NSUN2, NSUN6, and DNMT2 [11, 12, 13]. A study demonstrated that NSUN4 is essential in embryonic development, and knocking out NSUN4 can kill embryos and can sometimes induce myocarditis in the myocardium of mice [14]. Besides, a relevant report uncovered by bioinformatics analysis that m5C distribution and expression are significantly different in HCC tissues and para-carcinoma tissues, and the peak value of m5C methylation in HCC tissues is much higher than that in para-carcinoma tissues [15]. These suggest that m5C methyltransferase family may be involved in the progression of HCC. However, there are rare studies or reports about the relationship between NSUN4 and HCC.
In recent years, the mining of cancer prognostic signals and establishment of prognostic models have been the focus of research. A relevant study explored long non-coding RNAs (lncRNAs) in glioblastoma via constructing a competing endogenous RNA (ceRNA) network mediated by cancer-related lncRNAs [16]. Moreover, a study dug immune-related genes that can guide prognosis using immunological database and ImmPort database, and a prognostic model constructed by 30 immune-related genes was built which can predict patient’s overall survival (OS) [17]. Another study applied multivariate Cox regression analysis to indicate that the 5 studied lncRNAs are all independent clinical pathological factors of HCC [18]. Since previous studies disclosed that highly expressed NSUN4 is significantly correlated with the survival of HCC patients, this study focused on m5C methyltransferase-related genes and mainly analyzed expression differences of these genes. Consensus clustering analysis was performed to classify HCC subgroups. The survival status of patients and the expression mode of m5C methyltransferase-related genes in different subgroups were investigated. Univariate and multivariate Cox regression analyses were adopted to screen genes related to HCC survival and prognosis. The independence of these genes in prognosis was identified. Kyoto encyclopedia of genes and genomes (KEGG) pathway enrichment analysis was undertaken on the independent prognostic factor using gene set enrichment analysis (GSEA) tools.
Materials and methods
Acquisition of HCC clinical information and gene expression data for gene expression analysis
Patient’s clinical information (Table S1) and gene expression profile (FPKM format) in TCGA-LIHC were downloaded from TCGA database (
Consensus clustering analysis
To determine whether HCC could be subdivided by the expression mode of m5C RNA methyltransferase-related genes in HCC, “ConsensusClusterPlus” package was applied to undertake k-means consensus clustering analysis on samples [19]. The resampling method was adopted, with each sampled 80% and repeated 1,000 times. The cluster number K value was set from 2 to 10 in turn to evaluate the stability of the results and the optimal K value was chosen to subdivide the samples. To explore the difference in the survival rate in different subgroups divided by consensus clustering analysis, “survival” package was used to carry out Kaplan-Meier survival analysis on samples in each group [20]. The test method was log-rank test (
Screening of prognosis-related genes and construction of risk-model by univariate and multivariate Cox regression analyses
To screen prognosis-related m5C RNA methyltrans-ferase-related genes, “survival” package was first applied to perform univariate Cox regression analysis on 7 m5C RNA methyltransferase-related genes (
In the formula,
HCC samples were scored according to the risk model. Samples were divided into high- and low-risk groups by the median risk score to continue the following survival analysis. “survivalROC” package was used to draw receiver operating characteristic (ROC) curves to calculate corresponding area under the curve (AUC) values for 1-year OS and 3-year OS [21]. The accuracy of the prognosis predicted by the model was evaluated by the AUC value.
To explore the independence of the signature genes in prognostic assessment, whether the signature genes were related to prognosis was firstly determined by univariate Cox regression analysis (
KEGG pathway enrichment analysis on GSEA
Signature genes that had independent prognostic ability were obtained via univariate and multivariate Cox regression analyses. The threshold value was defined by the median expression of the feature genes, and samples were divided into high- and low-expression groups. KEGG pathway enrichment analysis was performed on the feature genes using GSEA tools to predict potential signaling pathways [23].
Clinical samples
Clinical samples used in this investigation were provided by HCC patients treated in Tangshan Gongren Hospital. Recruited patients all met the subsequential conditions: i. Patients received surgery for cancer therapy purpose; ii. The associated clinical information and medical histories of patients were accessible; iii. Patients didn’t receive preoperative treatments (radiotherapy and chemotherapy, etc.); iv. Patients didn’t have other tumors except for HCC. Clinical samples included 18 HCC patient’s tumor tissues and corresponding adjacent normal tissues. Fresh samples were preserved at
Cell culture
LO2, HepG2, Hep3B, SMMC-7721, MHCC97L cells were procured from BeNa Culture Collection and were routinely grown in Dulbecco’s Modified Eagle Medium (Gibico, BRL, USA) containing 10% fetal bovine serum (FBS).
qPCR detection
Total RNA isolation was performed using Trizol (Invitrogen, USA). RNA concentration was calculated using Nanodrop2000 (Thermo fish, USA). TransScript First-Strand (TransGen, China) was employed for RNA reverse transcription. Green qPCR SuperMix and 7500 sequence detection system were used for real-time quantitative amplification. NSUN4 primers were: forward: AGCGTTGCTTCAGACTGGCTGT; reverse: CCTGATCTCTTCAGGCACATAGC; GAPDH primers were: forward: GTCTCCTCTGACTTCAACAGCG; reverse: ACCACCCTGTTGCTGTAGCCAA. Calculation method for relative expression data was 2(̂-delta delta CT).
Results
Expression of m5C RNA methyltransferase-related genes in HCC samples
Wilcoxon test was used to analyze the differences in the expression of 7 m5C RNA methyltransferase-related genes (NOP2, NSUN2, NSUN3, NSUN4, NSUN5, NSUN6, NSUN7) in 50 normal samples and 374 HCC samples. The results showed that these 7 m5C RNA methyltransferase-related genes had highly significant differential expression in normal samples and HCC samples (Fig. 1A and B). Besides, the expression differences in the genes in different stages and grades of tumor were analyzed. It was displayed that NSUN4 showed different expression in varying stages and grades (Fig. 1C and D), and the expression level of NSUN4 increased as HCC progressed (Fig. 1E and F).
Expression of m5C RNA methyltransferase-related genes in samples. A: Heatmap of the expression of m5C RNA methyltransferase-related genes (NOP2, NSUN2, NSUN3, NSUN4, NSUN5, NSUN6, NSUN7) in normal samples and HCC samples; B: Violin plot of the expression of m5C RNA methyltransferase-related genes in normal samples and HCC samples, with blue plot representing the normal group and red plot representing the tumor group; C-D: Heatmaps of the expression of m5C RNA methyltransferase-related genes in different tumor stages and grades; E-F: Boxplots of the expression of NSUN4 in different tumor stages and grades; *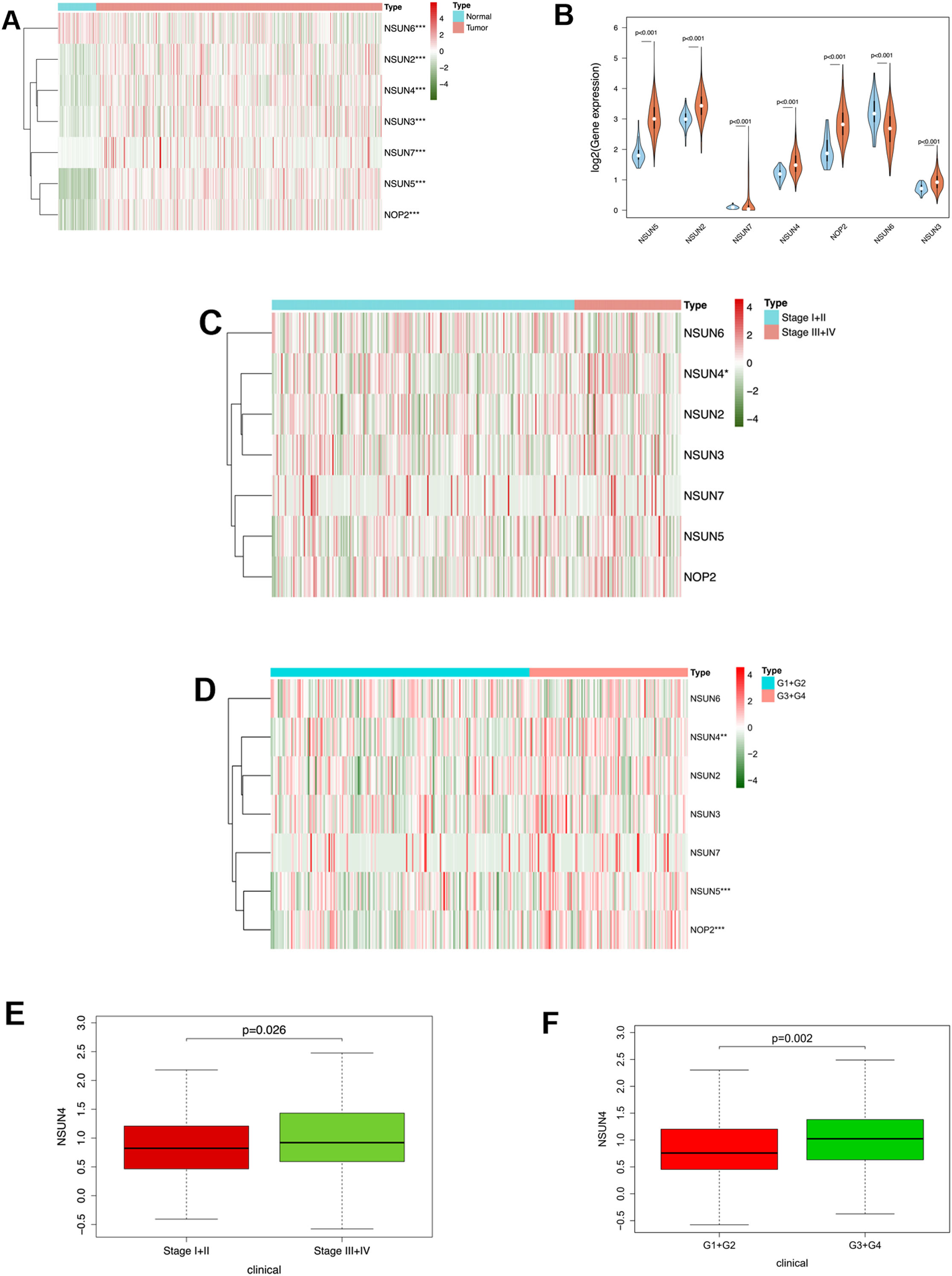
In order to explore whether HCC could be subdivided by the expression mode of m5C RNA methyltransfer-ase-related genes, unsupervised consensus clustering analysis was performed on the profile of these genes in 374 HCC samples in TCGA-LIHC dataset. The results showed that when k went from 2 to 3, the area difference under the CDF curve was the largest (Fig. 2A and B). Meanwhile, when
HCC subgroup analysis based on k-means consensus clustering analysis. A: Curves of cumulative distribution function (CDF) of consensus clustering as cluster number ranging from 2 to 10; B: Curve of CDF delta area as cluster number ranging from 2 to 10; C: Heatmap of sample consistency as cluster number K 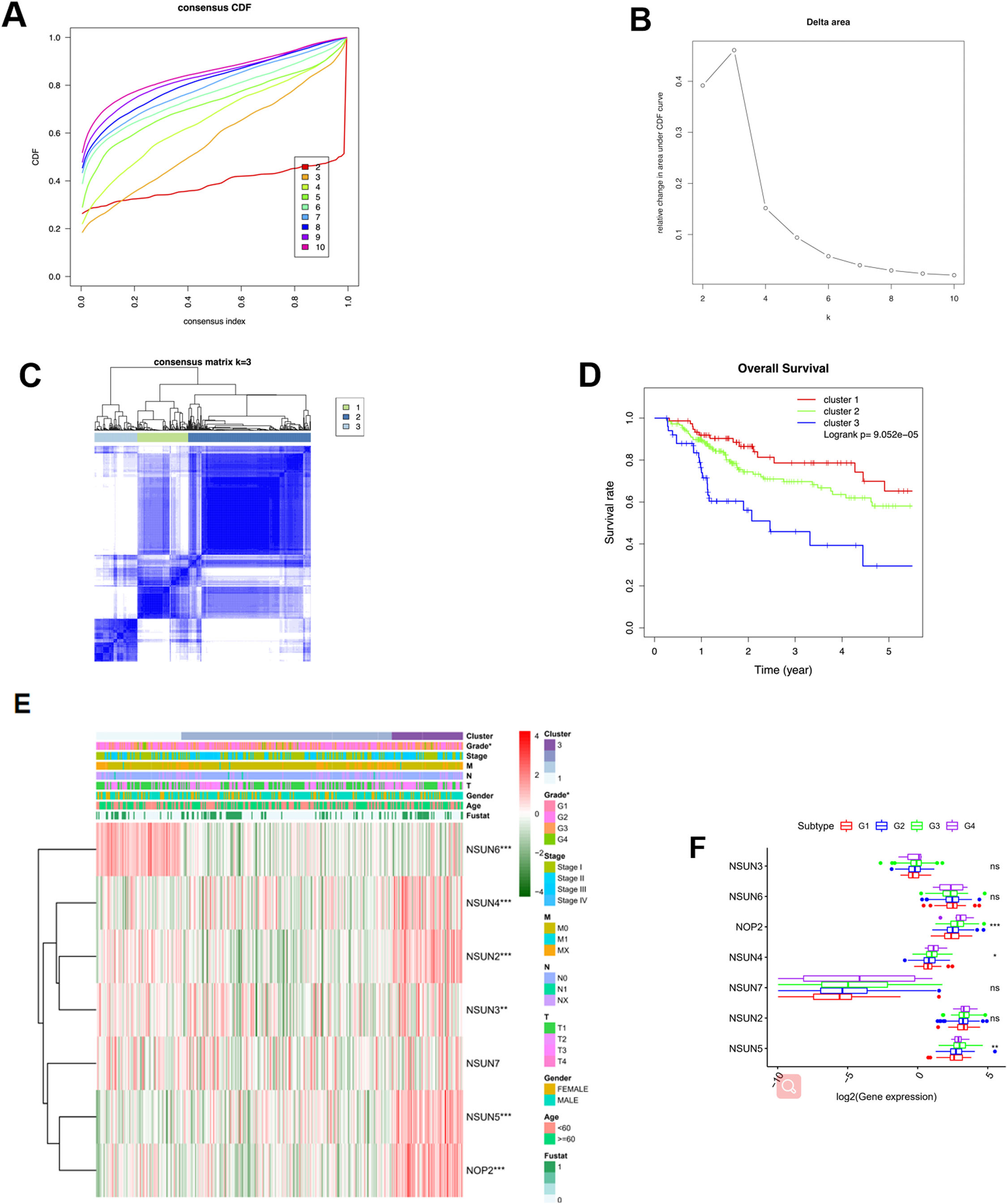
Following univariate Cox regression analysis, 4 m5C RNA methyltransferase-related genes of survival significance were screened: NOP2, NSUN2, NSUN4, NSUN5 (Fig. 3A). Afterwards, multivariate Cox regression analysis was carried out, and 2 high-risk factors, NOP2 and NSUN4 (HR
Screening of prognosis-related genes, construction and assessment of risk model. A: Screening of prognosis-related genes based on univariate Cox regression analysis; B: Screening of signature genes based on multivariate Cox regression analysis; C: Survival curve of high- and low-risk groups (Risk value of each sample was calculated based on prognostic prediction model. The median of risk value was used as the node to divide the samples into high and low risk groups); D: The predictive performance of the prognostic prediction model was evaluated by drawing the ROC curve based on the survival time of patients; E: Heat map of NSUN4 and NOP2 expression in high and low risk groups based on common clinical features; F: Boxplots of the risk score in different clinical stages and grades; *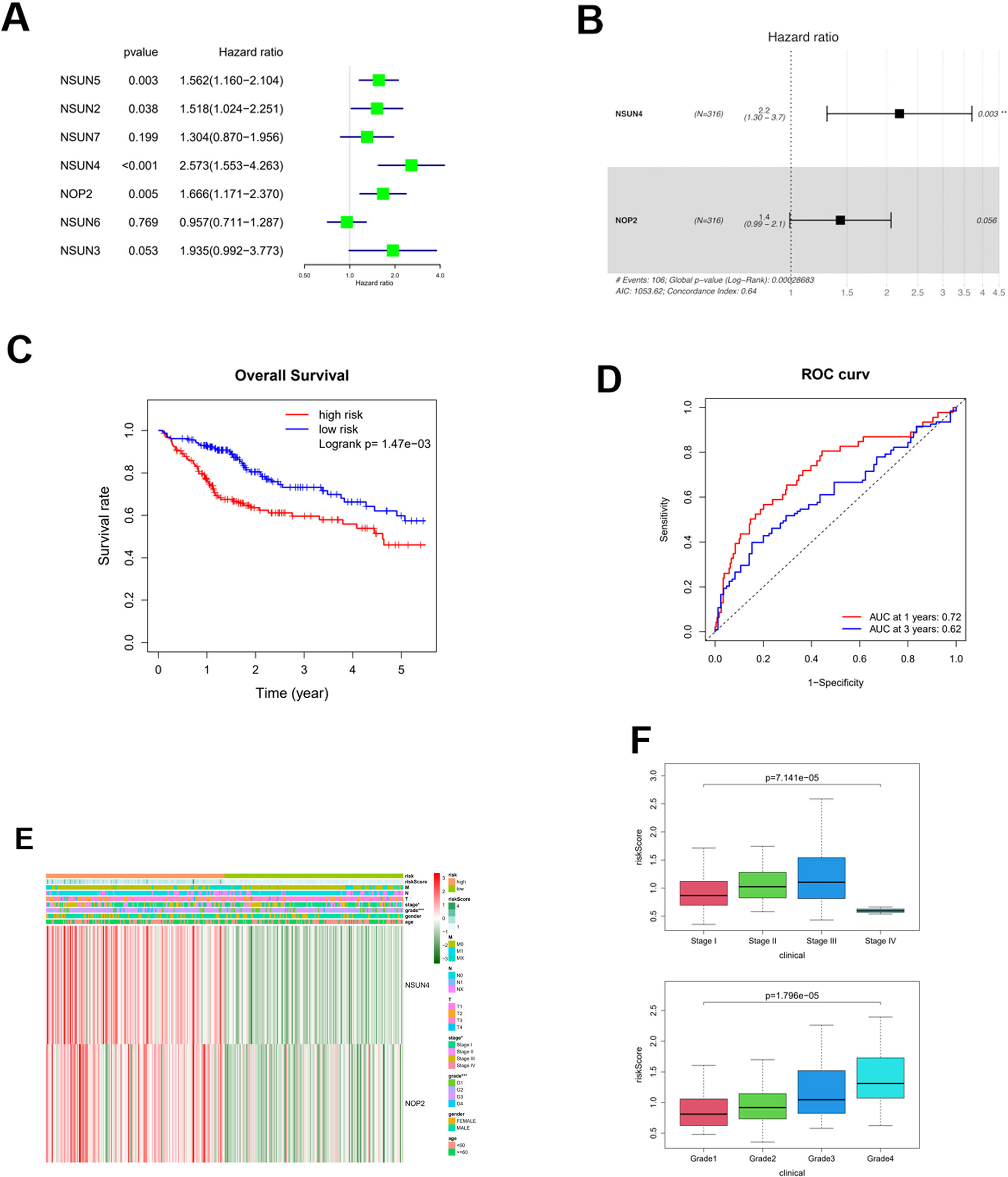
To investigate whether the screened prognosis-related genes (NOP2, NSUN4) could work as an independent prognostic factor to assess the prognosis of HCC, firstly, univariate and multivariate Cox regression analyses were performed on the 2 genes with clinical pathological factors. Results of univariate Cox regression analysis displayed that NOP2 and NSUN4 were notably associated with prognosis in addition to clinical stage and T stage (Fig. 4A). Multivariate Cox regression analysis was undertaken on NOP2 and NSUN4 combined with clinical information, and it was illuminated that only NSUN4 could be an independent factor to predict the prognosis of HCC (Fig. 4B). Afterwards, TIMER website was used to analyze survival status of HCC patients with high or low NOP2 and NSUN4 expressions. Similarly, the prognosis of HCC patients with high NSUN4 expression was poor (Fig. S1). Later, samples were divided into NSUN4 highly and lowly expressed groups with the median NSUN4 expression as the threshold. GSEA was employed for KEGG pathway enrichment analysis (Fig. 4C). The results manifested that NSUN4 mainly enriched in signaling pathways like ADHERENS JUNCTION, RNA DEGRADATION, MTOR SIGNALING PATHWAY, COMPLEMENT and COAGULATION CASCADES.
Screening of independent prognostic factors and signaling pathway analysis. A: Forest map of univariate Cox regression analysis on the expression of NOP2 and NSUN4 combined with clinical information; B: Forest map of multivariate Cox regression analysis on the expression of NOP2 and NSUN4 combined with clinical information; C: GSEA enrichment analysis of high- and low-expression groups of NSUN4 based on KEGG database. FDR 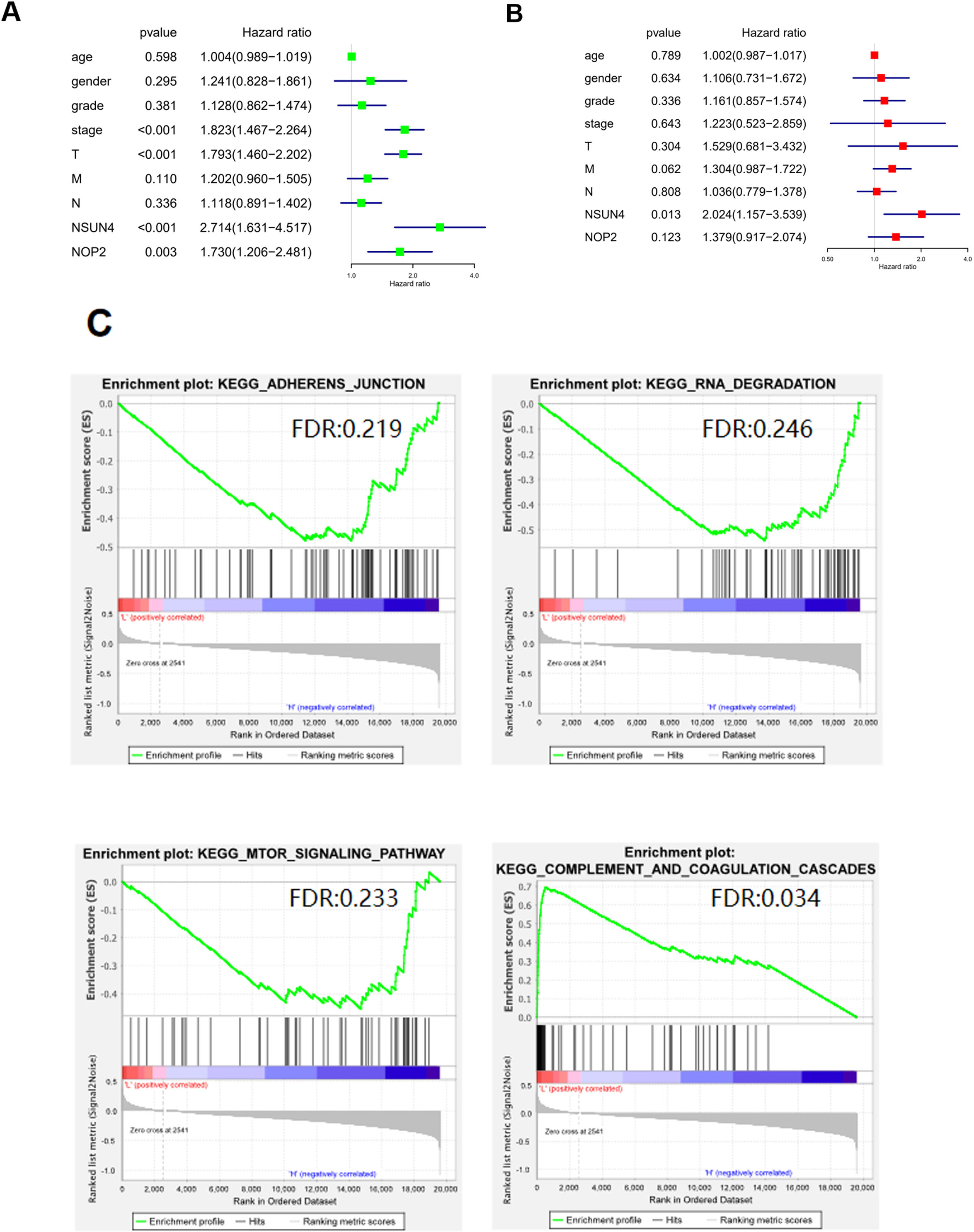
Patient’s tumor tissues and corresponding normal tissues were gathered for validation of bioinformatics prediction (Table S3). NSUN4 expression was also measured in normal liver cell line (LO2) and HCC cell lines (Hep3B, Huh-7, HepG2, SMCC7721, MHCC97L) Then, NSUN4 expression was found to be conspicuously upregulated in tumor tissues and HCC cells (Fig. 5A and B).
Expression of NSUN4 in tissues and cells. A: qPCR detection of NSU4N expression in HCC tumor tissues and normal tissues; B: qPCR detection of NSU4N expression in LO2, Hep3B, Huh-7, HepG2, SMCC-7721, MHCC97L cells. *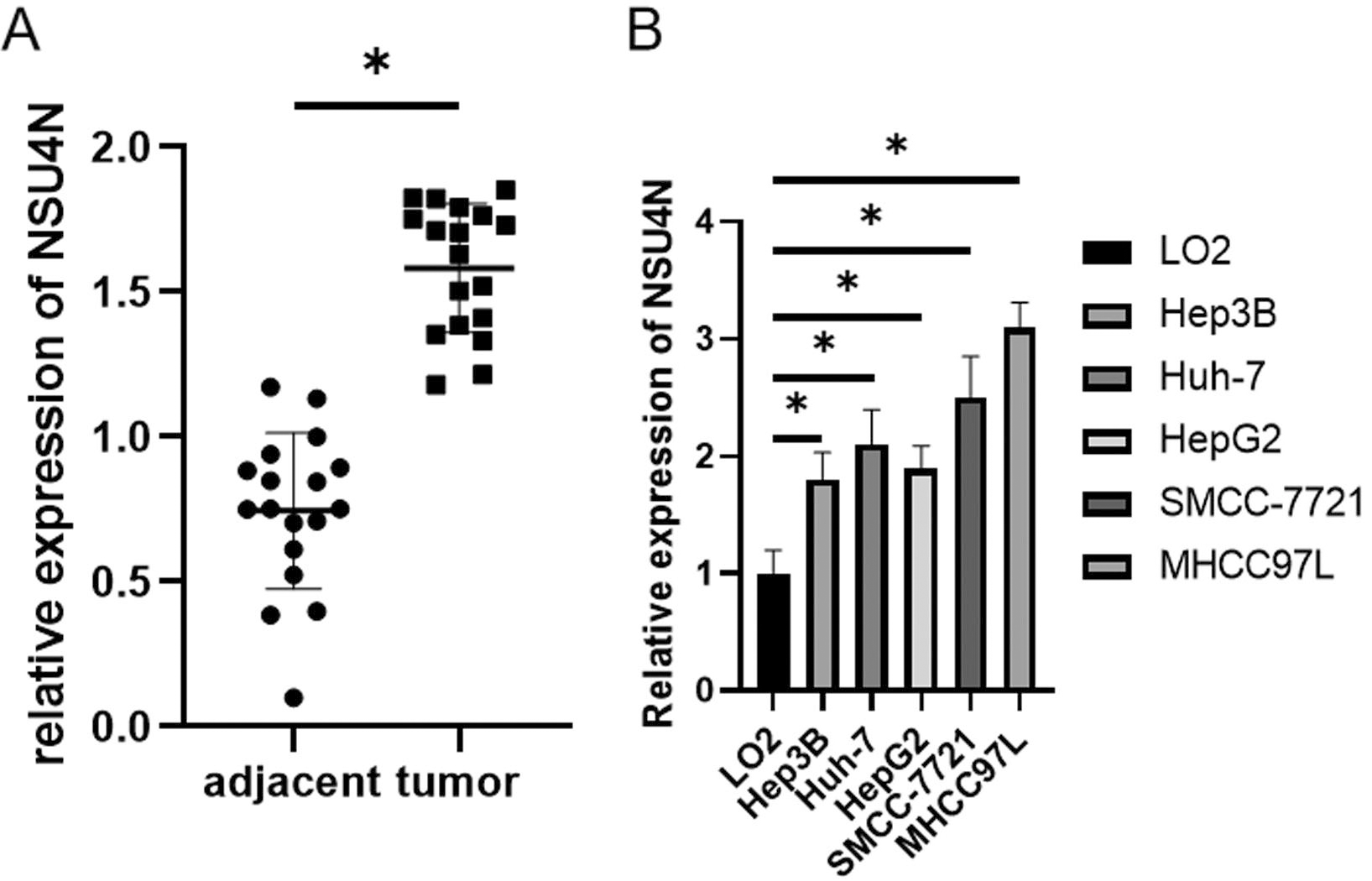
RNA epigenetic modification is an important biological process. In recent years, more and more evidences indicate that the aberrant expression of RNA epigenetic modification-related molecules is closely related to the progression of cancers. Relevant studies unmasked that the abnormal expression of NSUN family relates to a variety of diseases including cancers. For instance, NSUN2 mutation can result in autosomal recessive intelligence disorder [24], and knocking out NSUN2 inhibits the migration of glioma cells [25]. Furthermore, studies found that NSUN4 and MTERF4 participate in mitochondrial ribosomes together to modulate the expression of mitochondrial DNA, which is relevant to the regulation of mammalian oxidative phosphorylation [26]. However, abnormal oxidative phosphorylation is closely associated with the progression of cancers [27, 28, 29]. These suggest that m5C RNA methyltransferase-related genes may be involved in progression of cancers. This study mainly discussed the expression of m5C RNA methyltransferase-related genes in HCC and corresponding effect on prognostic prediction. It was disclosed that the expression of m5C RNA methyltransferase-related genes in normal samples and HCC samples was different. The expression levels of genes like NOP2, NSUN2 and NSUN4 in HCC were higher than that in normal tissues. A relevant study represented that high NOP2 expression can stimulate the migratory ability of prostate cancer. [30]. A study revealed that stable NOP2 helps to promote the proliferative ability of HCC [31]. However, direct evidence about the effect of NSUN4 in HCC is rare. Even so, a study clarified that the expression level of NSUN4 elevates with HCC progression [7], which is in good consistent with our research.
Using k-means consensus clustering analysis, HCC was classified into 3 subgroups according to the expression mode of m5C RNA methyltransferase-related genes. Previously, a study divided HCC into 4 subgroups according to the expression mode of hypoxia-related genes [32]. After subdividing HCC, this study analyzed OS of samples in each subgroup and explored whether the survival status of samples in different subgroups is different. Besides, samples in each subgroup were further subdivided, and it was discovered that the expression of m5C RNA methyltransferase-related genes was different in varying clinical grades. The results may provide new references and evidences for the clinical grading of HCC in the future.
A related study classified cancer samples into high- and low-risk groups by constructing a predictive model based on DNA methyltransferase-related gene signature, and then assessed the predictive accuracy of the model via AUC of ROC curve [33]. In this study, firstly, univariate and multivariate Cox regression analyses were applied to construct a risk model of HCC which contained 2 high-risk factors related to prognosis (NOP2, NSUN4). Afterwards, the accuracy of the model was evaluated through AUC of ROC curve. AUC values for 1-year OS and 3-year OS were 0.72 and 0.62, respectively, indicating that the model was relatively accurate in predicting the survival of HCC patients. Most studies adopted ROC curves to evaluate the accuracy of models in predicting survival [34, 35]. However, in addition to ROC curve, this study analyzed the difference in the risk score based on the model in different clinical stages and grades. According to the results, the risk score elevated with HCC progression, which further testified the accuracy of the model.
Combining with clinical information of samples, univariate and multivariate Cox regression analyses revealed that only NSUN4 could be an independent prognostic factor for HCC. A study established a 3-miRNA based predictive model, and testified these 3-miRNA signatures as independent predictors of vascular invasion in HCC patients by multivariate Cox regression analysis [36]. In the context, NSUN4 is potential to be a novel factor to guide the prognostic management of HCC patients. Besides, it was uncovered by KEGG pathway enrichment analysis that NSUN4 was mainly enriched in signaling pathways including ADHERENS JUNCTION, RNA DEGRADATION, MTOR SIGNALING PATHWAY, COMPLEMENT and COAGULATION CASCADES. Aberrant ADHERENS JUNCTION directly affects the invasive and migratory abilities of cancer cells. For example, prostate cancer can release exosomes under hypoxic conditions to regulate the expression of related molecules in the pathways involved in ADHERENS JUNCTION, thereby enhancing the invasive ability of the cancer [37]. Nevertheless, no study can support that NSUN4 can modulate the expression of ADHERENS JUNCTION-related molecules at present, which may provide a new idea for future research directions. So far, biological functions of NSUN4 in cells have not been fully dug. NSUN2 is the most well-known member of NSUN family so far, and its effect has been clarified in various cancers. For instance, NSUN2 shortage can cause skin cancer progression [38]. Another study about the expression of NSUN family in mouse embryos demonstrated that NSUN family may have overlaps in biological functions, indicating that NSUN4 may play an unknown biological function in cancers.
Viewed in toto, this study mainly analyzed the expression of m5C methyltransferase-related genes in HCC. HCC was firstly divided into 3 subgroups by k-means consensus clustering analysis. A risk model was then established based on m5C methyltransferase-related genes by univariate and multivariate Cox regression analyses. The accuracy of the model was also evaluated. Combining patient’s clinical information, it was found that NSUN4 could be an independent prognostic factor of HCC. However, there are some shortages in this study. For instance, this study didn’t use HCC datasets in other databases in validating the accuracy of the risk model, thus the accuracy of the risk model should be further examined.
Ethical approval
Ethical approval is not applicable for this article. This article does not contain any studies with human or animal subjects.
Competing interest
The authors declare no conflicts of interest.
Availability of data and materials
The data used to support the findings of this study are included within the article. The data and materials in the current study are available from the corresponding author on reasonable request.
Funding statement
Not applicable.
Authors’ contributions
Conception: Dr. Mingxin Cui and Dr. Fengzhi Qu.
Interpretation and Analysis of Data: Dr. Libing Wang and Dr. Xiaogang Liu.
Preparation of the manuscript: Dr. Fengzhi Qu and Dr. Zhaoyuan Tang.
Revision for Important Intellectual Content: Dr. Mingxin Cui and Dr. Daming Cheng.
Supervision: Dr. Jingkun Yu.
Supplementary data
The supplementary files are available to download from http://dx.doi.org/10.3233/CBM-210154.
sj-pdf-1-cbm-10.3233_CBM-210154.pdf - Supplemental material
Supplemental material, sj-pdf-1-cbm-10.3233_CBM-210154.pdf
sj-xlsx-2-cbm-10.3233_CBM-210154.xlsx - Supplemental material
Supplemental material, sj-xlsx-2-cbm-10.3233_CBM-210154.xlsx
sj-xlsx-3-cbm-10.3233_CBM-210154.xlsx - Supplemental material
Supplemental material, sj-xlsx-3-cbm-10.3233_CBM-210154.xlsx
sj-xlsx-1-cbm-10.3233_CBM-210154.xlsx - Supplemental material
Supplemental material, sj-xlsx-1-cbm-10.3233_CBM-210154.xlsx
Footnotes
Acknowledgments
Not applicable.