
Abstract
In order to produce an improved strain of the entomopathogen Beauveria bassiana, fungus transformation system was developed based on resistance to the herbicide glufosinate ammonium, conferred by the bar gene. B. bassiana strain Bb 9112, characterized by its resistance to UV light, was transformed with the plasmid pBarGPE1, previously cloned with the marker gene coding for green fluorescent protein (GFP). Transformed colonies were selected in a minimal medium containing 25 µg/ml of the herbicide glucosinate ammonium. The expression of the protein GFP in the transformed protoplasts and the mycelium regenerated from those protoplasts was confirmed by UV light microscopy. Pathogenicity tests indicated no significant differences in percent pathogenicity between the transformed and non-transformed strains. In order to increase the pathogenicity of Bb9112 against the coffee berry borer, it was transformed with the plasmid pBarGPE1-pr1A containing a subtilisin - protease gene (pr1A) isolated from Metarhizium anisopliae. In the transgenic strain the presence of the pr1A gene was identified by PCR. The expression of the protein was confirmed by isoelectrofocus and enzymatic activity.
Introducción
El hongo entomopatógeno Beauveria bassiana ha sido ampliamente usado como controlador biológico de insectos (Ferron 1978). Sin embargo, los experimentos de control biológico empleando hongos entomopatógenos han originado frecuentemente resultados inconsistentes y la tardanza en producción de mortalidad en comparación con insecticidas químicos ha detenido el desarrollo comercial de estos productos. Debido a esto, las consideraciones de la sostenibilidad de un hongo entomopatógeno con propósitos comerciales inevitablemente conlleva a mejorar su desempeño como biocontrolador.
En el caso del ecosistema cafetero colombiano, B. bassiana juega un papel importante en el control de Hypothenemus hampei; este hongo se encuentra infectando naturalmente la broca del café en casi todas las regiones de Colombia y experimentos llevados a cabo en Cenicafé, han demostrado que el control del insecto en campo es posible empleando dosis de 1x108-12 esporas por árbol, las cuales causan 80-90% de mortalidad en los insectos (Posada 1998). Sin embargo, el uso de esta concentración de esporas es costoso y una de las formas de reducir el costo de estas dosis altas es aumentar la virulencia y patogenicidad de las cepas de B. bassiana. De esta manera, sería posible reducir la dosis de esporas requerida para controlar el insecto. Por otra parte, la mortalidad del insecto se obtendría más rápidamente, disminuyendo así el daño que causa el insecto a los frutos.
El primer paso para la producción de un hongo mejorado, a través de transformación genética, es el desarrollo de métodos eficientes de transformación y la manera más apropiada de evaluar los métodos de transformación genética de un organismo es el uso de genes marcadores y genes de selección. Los genes marcadores codifican enzimas que no están presentes en las cepas no transgénicas; su expresión puede ser visualizada fácilmente sin el requerimiento de ensayos costosos o complicados enzima- sustrato. Las mediciones cuantitativas de los niveles enzimáticos son posibles y fáciles. La actividad específica de las proteínas no compite con otras enzimas presentes en las células y no interfiere con el metabolismo normal del organismo. Los genes de selección confieren resistencia a sustancias tóxicas, codifican do productos que permiten la supervivencia de los organismos transformantes en medios tóxicos, que generalmente contienen antibióticos o herbicidas. Sólo los organismos transformantes tienen la habilidad de sobrevivir en estos medios.
La transformación de B. bassiana con genes de selección y marcadores no sólo permitirá la identificación eficiente de las cepas transformadas, sino que también facilitará el seguimiento de los procesos de infección y patogenicidad de estas cepas en condiciones de laboratorio y campo. En laboratorio, un requerimiento clave en el estudio de la interacción entomopatógeno-hospedero es la habilidad de detectar el hongo dentro del tejido del insecto. Cepas transformadas conteniendo genes de selección o marcadores proveerán una nueva herramienta de detección y los medios para monitorear el desarrollo del hongo y la interacción con el hospedero. En el campo, estos genes permitirán la recuperación e identificación de cepas del hongo asperjadas en el campo, lo cual conllevará al entendimiento de los procesos de transmisión, infectividad y persistencia del hongo en el medio ambiente.
Como genes de selección, el gen bar se ha usado en sistemas de transformación de hongos entomopatógenos. El gen aislado de Streptomyces hygroscopicus confiere resistencia a los herbicidas bialafós y glufosinato de amonio. Los herbicidas en cuestión, contienen como ingrediente activo phosphinothricin, un análogo de ácido glutámico, el cual inhibe la actividad de la enzima glutamina sintasa, la cual causa muerte de las células por acumulación de amonio. El gen bar codifica la enzima phosphinothricin acetiltransferasa, la cual inactiva phosphinothricin al acetilarlo (Avalos et al. 1989).
Como gen marcador, el cDNA que codifica la proteína verde fluorescente (GFP) (Chalfie et al. 1994), ha sido exitosamente expresada en un amplio número de organismos. Esta proteína aislada de Aequorea vistoria, la cual confiere bioluminiscencia a invertebrados acuáticos del orden Cnidaria, está compuesta de 238 aminoácidos. El uso amplio de esta proteína se debe a su fácil identificación: oxígeno y luz azul o ultravioleta son los únicos requerimientos para visualizar la proteína. GFP ha sido expresada en hongos basidiomicetes como es el caso de Ustilago maydis (Spelling et al. 1996), y hongos filamentos como Aureobasidium pollulans (Vanden Wymelenberg et al. 1997), Cochliobolus heterostrophus (Maor et al. 1998), y los entomopatógenos Paeciloтуces fumosoroseus (Cantone y Vandenberg 1999) y Metarhizium anisopliae (Inglis et al. 2000) y no se ha manifestado que tenga efectos negativos en el hongo o interfiera con sus procesos patogénicos.
La literatura menciona la transformación de B. bassiana con genes de selección como es el caso del gen de β tubulina resistente a (MBC) aislado de Neurospora crassa, el cual confiere resistencia al fungicida 1,2 benzimidazol carbamato (MBC) (Pfeifer, y Khachatourians 1992). De igual forma, Daboussi et al. (1989) demuestran la transformación del hongo con el gen gen nia D. Thorvilson y San francisco (2001) registran el uso del gen β glucoronidasa (GUS) como gen marcador en un sistema de transformación de B. bassiana. Sin embargo, hasta ahora no se ha indicado la transformación de este hongo con GFP, bar o genes involucrados en procesos de patogenicidad.
Luego del desarrollo de un sistema eficiente de transformación, el siguiente paso consiste en la producción de cepas con incremento en su patogenicidad. St. Leger y colaboradores (1996) han clonado varios genes que están involucrados en los procesos de infección y patogenicidad de Metarhizium anisopliae. Uno de los genes que se expresan cuando el insecto es atacado, codifica una proteasa tipo subtilisina denominada Pr1A, la cual solubiliza la cutícula proteinacea del insecto, permitiendo la penetración de las hifas del hongo en el insecto y el acceso a los nutrientes necesarios para su crecimiento.
La transformación de M. anisopliae con prlA (St. Leger et al. 1996, 1997) resultó en el desarrollo de un hongo entomopatógeno mejorado genéticamente. Cuando la cepa fue transformada y copias adicionales del gen codificando esta proteasa fueron insertadas en el genoma del hongo, la cepa resultante produjo la proteína durante el proceso de infección y colonización del insecto, y ésta fue también secretada por el hongo en la hemolinfa del insecto Manduca sexta. La presencia de esta proteína en la hemolinfa, activó en éste el sistema inmunológico de prefenoloxidasa. El efecto tóxico combinado de prlA junto con los productos de la reacción de fenoloxidasa causó, en los insectos expuestos al hongo transformado, una reducción del 25% en el tiempo de muerte y reducción de 40% en el consumo de alimento, comparado con los insectos infectados con el hongo no transgénico. Además, los insectos infectados con el hongo transformado mostraron una rápida mecanización y los cadáveres resultantes fueron un substrato pobre para la esporulación del hongo.
El objetivo de este estudio fue llevar a cabo la modificación genética de B. bassiana empleando vectores de transformación para hongos, que contienen como marcadores selectivos el gen bar y el gen marcador que codifica GFP y la proteasa prlA. Las cepas obtenidas serán usadas para realizar, en condiciones de laboratorio, el seguimiento de los procesos de infección y patogenicidad hongo vs. insecto y dilucidar los procesos de transmisión, infectividad y persistencia del hongo en condiciones de campo. Este estudio permitirá investigar el mantenimiento, estabilidad, proliferación y los efectos patogénicos de B. bassiana a través del uso de estos genes. Se cree que los genes marcadores selectivos no deben tener efecto alguno sobre la patogenicidad del hongo a menos que debido al sitio de inserción en el genoma, puedan interferir con algún gen involucrado en los procesos de patogenicidad. Este trabajo permitirá dilucidar esta afirmación.
Materiales y Métodos
Ingeniería de los vectores de transformación
El vector de transformación pBarGPE1 (Pall 1993) se obtuvo del Fungal Genetics Stock Center, Department of Microbiology, University of Kansas Medical Center. El plásmido se extrajo y purificó de cultivos de E. coli.
El gen de la proteasa tipo subtilisina prlA de M. anisopliae var. acridum, identificada en la base de datos del GenBank del National Center for Biotechnology Information (USA) con el número de acceso AJ251925.1 se obtuvo del Dr. Ray St-Leger, Departamento de Entomología, University of Maryland, College Park. MD. USA. El gen había sido previamente clonado en el sitio múltiple de clonación del vector pBluescript SK (PBS) (Stratagene, La Jolla) entre los sitios de restricción EcoRI y Xho I. Εl plásmido se amplificó y el gen prlA se separó del plásmido PBS por digestión con las enzimas de restricción Sma I y BamH I. Luego fue ligado en el sitio múltiple de clonación del vector pBarGPE1 bajo el control del promotor de gpdA y la región terminadora del gen trpC (ambos provenientes de Aspergillus nidulans), generándose el vector de transformación pBarGPE1-pr1A. Células competentes de E. coli se transformaron con la solución de la ligación, las colonias bacterianas que contenían el plásmido recombinante se identificaron y el plásmido se amplificó y secuenció. Los resultados de la secuencia mostraron que el gen estaba correcto.
El gen que codifica la proteína verde fluorescente (GFP) se clonó en el vector pBarGPE1 de la misma manera descrita para la proteasa prla y fue obtenido del Dr. Ray St-Leger.
Cepa de Beauveria bassiana
La cepa de B. bassiana Bb9112 aislada en Caldas, Colombia, de un lepidóptero de la familia Geometridae, se escogió para desarrollar los trabajos de transformación debido a que mostraba una patogenicidad superior al 85% contra la broca del café y resistencia a luz U.V.
Producción de protoplastos
Esporas obtenidas a partir de un cultivo del hongo crecido en Agar Sabouraud Dextrosa (ASD) por 15 días a 27°C, se resuspendieron en 10 ml de Tween estéril al 0,02%. Con esta suspensión se inocularon 100 ml de Líquido Sabouraud Dextrosa (LSD) suplementado con 0,1% de extracto de levadura. El cultivo creció a 27°C y 120 rpm por 36 h. El micelio se recuperó por filtración a través de una capa de miracloth estéril y se lavó con agua desionizada estéril. El micelio se incubó por 3h, en agitación lenta en 20 ml de una solución 1,2 M sorbitol y 50 mM MES, que contenía 0,8% Novozyma 234 y 0,3% B-glucoronidasa. La presencia de protoplastos se verificó por observaciones al microscopio. La solución de micelio y protoplastos se filtró, a través de una capa de miracloth estéril y los protoplastos se precipitaron por centrifugación a 2.000 gpm y 4°C, posteriormente se lavaron 2 veces por resuspensión y centrifugación en 20 ml de solución osmótica STC (1,2 M Sorbitol, 10 mM Tris pH 7,5, 20 mM CaCl2). Finalmente, los protoplastos se resuspendieron a una concentración de 1x108 protoplastos/ml de STC, obteniéndose 1 a 5 ml de protoplastos en total.
Transformación de Beauveria bassiana con los vectores pBarGPE1-pr1A, pBarGPE1-GFP
La transformación del hongo con pBarGPE1-pr1A, y pBarGPE1-GFP se hizo usando una solución de 200 µl de protoplastos a 1 x 108 protoplastos/ml. A ésta se adicionaron 50 µg de DNA plasmídico en un volumen de 50-100 µl, se incubó en hielo por 30 min. Luego, se agregaron 50 µl de polietilenglicol 3000 (PEG) al 60%, en STC y se incubó en hielo por 10 min. Finalmente, se adicionó 1 ml de la solución de PEG y se incubó a temperatura ambiente por 10 minutos. La solución se resuspendió en 4 ml RM (1,2 M Sorbitol, 10 mM Tris pH 7,5, 0,1% K2HPO4 0,05% MgSO4 y 0,3% NaNO3), para obtener un volumen final de 5 ml de transformantes. Estos 5 ml de transformantes se dividieron en 2 tubos falcon de 50 ml conteniendo cada uno 2.5 ml; a cada tubo se le adicionaron 47,5 ml de RM líquido con Bacto-agar al 1%, a una temperatura de 35°C. 10 ml de esta solución se sirvieron en cajas de Petri. Estas cajas, con los protoplastos transformantes en medio mínimo, se incubaron por 18 a 24 h y luego se cubrieron con 10 ml de RM con Bacto-agar al 1% y glufosinato de amonio a una concentración final de 25 µg/ml [Finale (5% ingrediente activo) AgrEvo USA. Wilmington DE].
Evaluación de las colonias de B. bassiana transformadas
Los genes se amplificaron con reacciones de PCR en un volumen de 50 µl, conteniendo 500 ng de ADN fúngico, 1X PCR buffer, 25 pmoles de iniciadores, 0,2 mM de cada deoxinucleótido, 2,0 mM de MgCl2, 1U de Taq DNA polimerasa. Ciclo de PCR: 94°C x 5 min, 94°C x 1 min, 55°C x 1 min, 72°C x 1 min, (40 ciclos), 72°C x 5 min.
Las secuencias de los iniciadores que se usaron para la amplificación fueron:
Gen bar: 5′-3′ ATGCGCCCAGAACGACGCCC. 3′-5′ GCAGGACCGGACGGGGG
Gen pr1A: 5′-3′ TCTTCTCACTCTTCTCCCA. 3′-5′ TTAGGCACCGTTGTAGGCA
GFP: 5′-3′ ATGGTGAGCAAGGGCGAGGAGC 3′-5′ TTACTTGTACAGCTCGTCCATGCCG
La reacciones de PCR, se corrieron en geles de agarosa al 1% y tras observación bajo luz UV se determinó la presencia de las bandas con un tamaño molecular correspondientes al esperado para cada gen.
Identificación de las proteínas pr1A en las cepas transformadas
Identificación de proteasas por isoelectroenfoque (IEF)
La presencia de las proteínas y su actividad proteolítica también se determinó a través de separación de las proteínas por electroforesis de isoelectroenfoque (IEF) y degradación de gelatina (Bidochka y Khachatourians 1994). Para esto, las cepas de B. bassiana transformadas y no transformadas crecieron en medio mínimo líquido, suplementado con 1% (p/v) de quitina de caparazón de cangrejo o quitina al 1% (p/v) más N- acetilglucosamina al 1% (p/v) durante 48 horas. 25 ml del medio de cultivo se concentraron por ultracentrifugación usando unidades de ultrafiltración Centricon con membranas de exclusión de 10kDa (Amicon). Se preparó un gel de poliacrilamida al 5% más anfolitos 3/10 Biolyt e al 2%. 2 ml de las muestras concentradas se aplicaron sobre el gel y se separaron por isoelectroenfoque usando una cámara mini-IEF-cell modelo 111 (Bio-Rad). La separación se llevó a cabo a 100 V por 15 min, seguido de 200 V por 15 min y 450 V por 1 h. La presencia de las proteasas se visualizó al poner en contacto el gel con una película para radiografía, la cual está recubierta de una capa de gelatina.
Pruebas de patogenicidad cepas transformadas con la proteína verde fluorescente
Se llevaron a cabo pruebas de patogenicidad, con las cepas transformadas con la proteína verde fluorescente. Las cepas transformadas crecidas en medio ASD más glufosinato de amonio a una concentración de 25 µg/ml y las cepas control no transformadas se sometieron a control de calidad, de tal manera que se evaluaron las siguientes características: 1) germinación superior al 85% en un tiempo de incubación de 24 h, 2) porcentaje de pureza mayor del 90%. La germinación, la pureza y viabilidad, así como las pruebas de patogenicidad (15 individuos por tratamiento con 4 repeticiones) se determinaron siguiendo el método descrito por Vélez et al. (1997).
Resultados y Discusión
Cepas transformadas con la proteína verde fluorescente
Luego de la transformación, se observó la solución de protoplastos putativamente transformados bajo el microscopio y se detectó la presencia de protoplasto verde fluorescente (Fig. 1). De 5-10 días luego de la transformación, las colonias putativamente transformadas empezaron a desarrollarse en el medio selectivo (Fig. 2). Inicialmente, dos colonias crecidas en medio mínimo con 25 µg/ml del glufosinato de amonio, que mostraron fluorescencia verde bajo el microscopio de luz- UV, fueron subcultivadas en SDA. El crecimiento de las colonias se siguió diariamente. Cuan do se observó la esporulación de los cultivos, las esporas se subcultivaron en medio fresco. Los subcultivos se hicieron por 3 generaciones. En la tercera generación, se aisló el DNA genómico del micelio y una amplificación por PCR de los genes bar y GFP se realizó usando iniciadores específicos para cada uno de éstos. Ambos genes se amplificaron por PCR en las cepas transgénicas pero no en el tipo silvestre Bb9112 (Fig. 3). En las cuatro cepas transformadas, identificadas como T1-1, T1-2, T2-2 y T2-3, el gen GFP se amplificó por PCR (Fig. 4). En el micelio de las 4 cepas, la expresión de la proteína GFP se confirmó por observaciones al microscopio de luz UV. Sin embargo, el grado de expresión de la proteína verde fluorescente no es el mismo en todo el micelio y en algunos casos se observan partes de micelio y esporas que no expresan la proteína; esto se puede deber a vejez del micelio, o a que los cultivos no son puros. Por esta razón, se ha iniciado un proceso de producción de cultivos monoespóricos a partir de estas cepas transformantes, con el propósito de producir cultivos puros que expresen la proteína de una manera uniforme. Estos cultivos están siendo sometidos a pruebas de control de calidad y patogenicidad sobre la broca del café.
Expresión de GFP en protoplastos transformados de Beauveria bassiana Bb 9112 con el plásmido pBarGPE-GFP. Microscopía con luz UV (40x).
Expresión de GFP en una colonia transformada de Beauveria bassiana Bb 9112 con el plásmido pBarGPE-GFP. Microscopía con luz UV (2,5x).
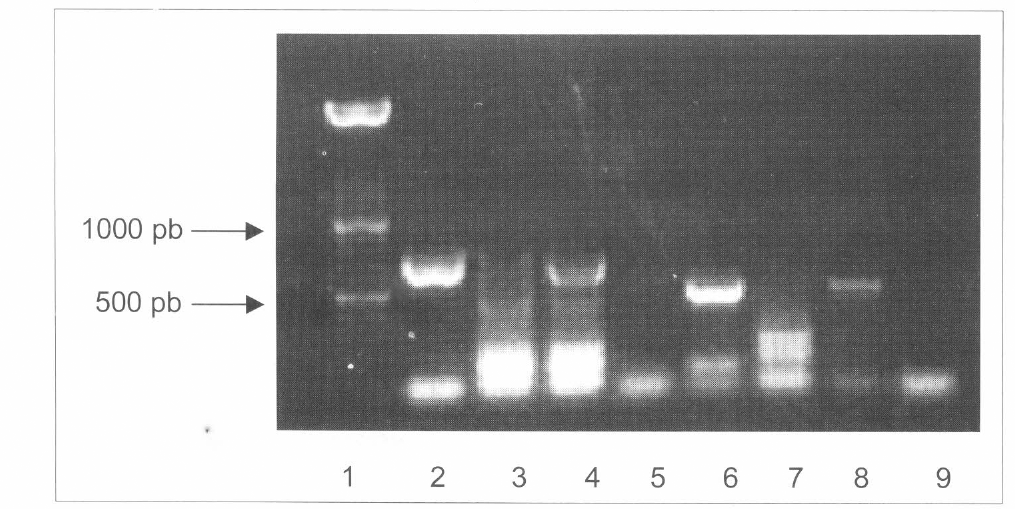
Separación en agarosa de los productos de PCR de los genes GFP y bar amplificados en cepas de B. bassiana transformadas con pBarGPE1-GFP. Una banda de 742 pb indica la presencia del gen GFP. Una banda de 537 pb indica la presencia del gen bar. Líneas 2 a 5 contienen las muestras amplificadas con los iniciadores para GFP. Línea 1 fue cargada con 500 ng de DNA marcador de peso molecular XIV (Boehringer). Línea 2 contiene la reacción del plásmido pBarGPE1-GFP. Línea 3 fue cargada con la reacción de una cepa no transformada. Línea 4 fue cargada con la reacción de una cepa transgénica. Línea 5 fue cargada con la solución maestra sin DNA. Líneas 6 a 9 contienen muestras amplificadas con iniciadores del gen bar. Línea 6 fue cargada con la reacción de pBarGPE1-GFP. Línea 7 fue cargada con la reacción de una cepa no transformada. Línea 8 fue cargada con la reacción de una cepa transgénica. Línea 9 fue cargada con solución maestra sin DNA.
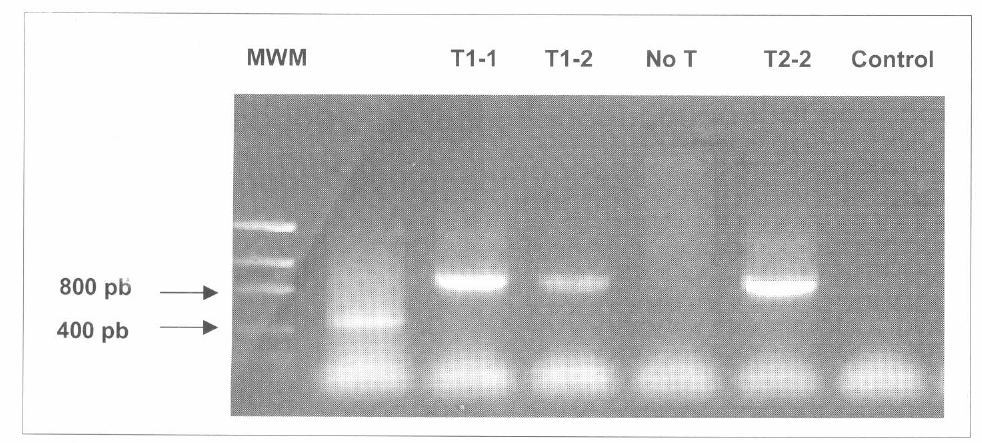
Separación en agarosa de los productos de PCR de los genes GFP en cepas de B. bassiana transformadas con pBarGPE1-GFP. Una banda de 742 pb indica la presencia del gen GFP. Línea MWM fue cargada con 500 ng de DNA marcador de peso molecular (Low DNA Mass ladder) (Gibco). Línea T1-1, T1-2 y T2-2 contiene la amplificación de cepas transformadas. Línea NT fue cargada con la reacción de una cepa no transformada. Línea Control fue cargada con la solución maestra sin DNA.
Patogenicidad de las cepas que expresan la proteína verde fluorescente
Se realizaron pruebas de patogenicidad por duplicado en diferentes épocas del año, evaluando las diferencias estadísticas entre las cepas Bb9112 no transgénicas y dos cepas transformadas con la proteína GFP, cepas T1-1 y T2-2. Los resultados se muestran en las tablas 1, 2 y 3. El análisis de varianza con un nivel de significancia del 5%, y la prueba de Tukey, mostró que no existen diferencias significativas con respecto al porcentaje de patogenicidad sobre la broca del café causado por B. bassiana Bb9112 y las cepas transformadas. Tampoco se observaron diferencias con respecto al tiempo de mortalidad ni el comportamiento de las cepas. Esto indica que la inserción de los genes se produjo en lugares en el genoma que no interfieren con genes involucrados en los procesos de patogenicidad del hongo. En el caso de estudios preliminares, Thorvilson y San Francisco (2001) muestran que las cepas transformadas con el gen GUS fueron menos efectivas en matar a los insectos en condiciones de laboratorio; los autores aseguran que esto se puede deber al sitio de inserción del gen GUS en el genoma del hongo, el cual puede interferir con algún gen involucrado en la patogenicidad. En el presente estudio, el proceso de transformación con los genes marcadores no afectó los procesos patogénicos de las cepas.
Las cepas obtenidas en este estudio, transformadas con GFP pueden usarse en laboratorio para estudios de la interacción entomopatógeno-hospedero, ya que sería fácil la detección del hongo dentro del tejido del insecto. Por otra parte, se demuestra que estos genes no interfieren con los procesos patogénicos del hongo. Al nivel de campo, estas cepas marcadas permitirían monitorear el desarrollo del hongo y la interacción con el hospedero. En las pruebas de campo, la identificación de estos genes permitirá la recuperación de cepas asperjadas, lo cual conllevará al entendimiento de los procesos de transmisión, infectividad y persistencia del hongo en el medio ambiente.
Cepas transformadas con la proteasa pr1A
Después de obtener transformantes expresando GFP, se realizó la transformación con el plásmido pBarGPE-PrIA. Los transformantes se seleccionaron por resistencia a 25 µg/ml de glufosinato de amonio. De 5 a 10 días luego de la transformación, diez colonias se aislaron de los medios selectivos y tres de estas colonias (pr1A-1, pr1A-2 y pr1A-3) se seleccionaron para la identificación de la proteína PrlA.
La actividad enzimática de las muestras se observa en la figura 5. Todas las cepas incluyendo el control (Bb9112 no transformado) produjeron una proteasa similar a la PrlA de M. anisopliae durante el crecimiento en el medio con quitina, la cual induce la producción de Pr1A. Sin embargo, solamente las cepas transformadas produjeron proteasas cuando se cultivaron en el medio que contenía quitina más NAcetil-glucosamida. En este medio, la NAcetil-glucosamida reprime la síntesis de proteasa tipo PrIA en cepas no modificadas. El resultado del experimento, implica que el prlA se incorporó en el genoma de los transformantes y la proteína se sintetizó y secretó constitutivamente. Cuando se analizó por isoelectroenfoque y se identificó la actividad proteolítica, los medios de cultivo suplementados con 1% de quitina o 1% de la quitina más 1% de NAcetil-glucosamina, donde habían crecido el hongo control y el transformado (pr1A-2), se encontró que la mayor cantidad de proteasas la produjo la cepa Pr1A-2 creciendo en medio con quitina y N-Acetil glucosamina. Este resultado confirma la transformación de la cepa y la producción constitutiva de la proteasa (Fig. 6).
Resultados de patogenicidad de B. bassiana Bb9112 y Cepa T1-1 sobre H. hampei
Tratamientos identificados con la misma letra no difieren estadísticamente.
Resultados de patogenicidad de B. bassiana Bb9112 у Сера Т1-1 y T2-2 sobre H. hampei
Tratamientos identificados con la misma letra no difieren estadísticamente.
Resultados de patogenicidad de B. bassiana Bb9112 у Сера Т2-2 sobre H. hampei
Tratamientos identificados con la misma letra no difieren estadísticamente.
La cepa Pr1A-1 se subcultivó y luego de dos generaciones, el ADN genómico se aisló del micelio. Iniciadores específicos se usaron para la amplificación de los genes bar (no se muestra los datos) y prlA (Fig. 7). Ambos genes se amplificaron en la сера transgénica pero no en el tipo silvestre 9112. La presencia del gen prla amplificado por PCR también se observa en la cepas Pr1A-2 y Pr1A-3 (Fig. 8). Se tienen 3 cepas transformadas con este gen.
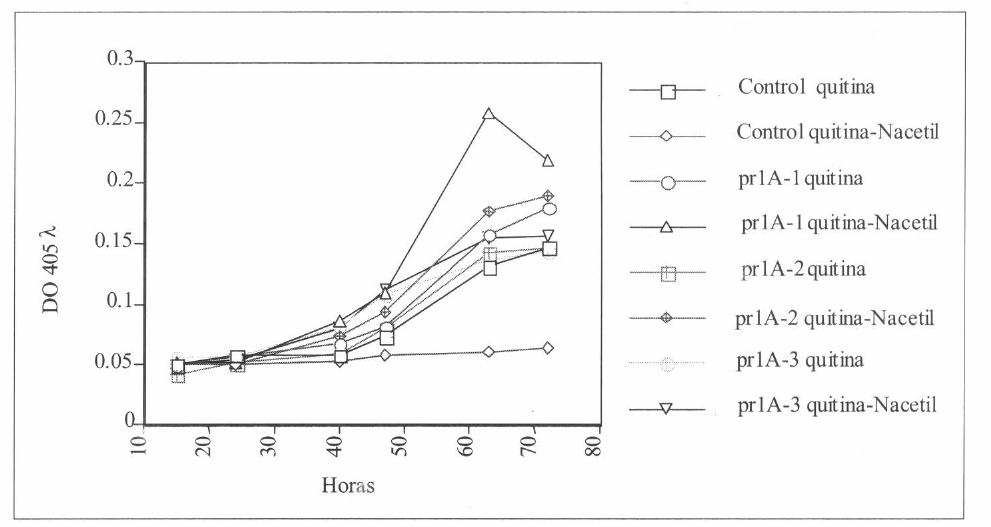
Actividad enzimática de pr1A a diferentes tiempos en la cepa Bb 9112 (control) y 3 cepas transformadas con pBarGPE1-prla en medio mínimo suplementado con quitina o quitina más N-acetil glucosamide.
La transformación de B. bassiana con los genes bar- GFP y bar-pr1A demuestra que se cuenta con un método de transformación confiable y la selección con el gen bar es adecuada. Hasta el momento este es el primer trabajo que muestra el uso de estos genes para la transformación de B. bassiana.
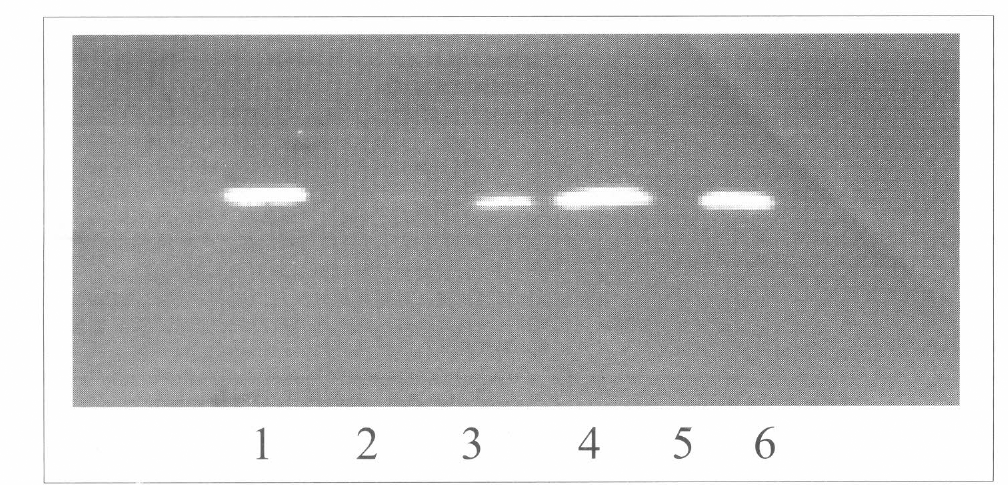
Actividad proteolítica del filtrado del cultivo de B. bassiana (Las proteasas presentes en el gel de isoelectroenfoque degradan la gelatina de la película, y se observa un halo transparente que indica actividad proteolítica específica de proteasa). Cepa Bb 9112 en medio básico suplementado con quitina (Línea 1 y 5) y medio básico suplementado con quitina más N-Acetil Glucosamida (Línea 2 y 6). Cepa pr1A-2 transformada con pBarGPE1-prla en medio básico suplementado con quitina (Línea 3) y medio básico suplementado con quitina más N-Acetil Glucosamida (Línea 4).
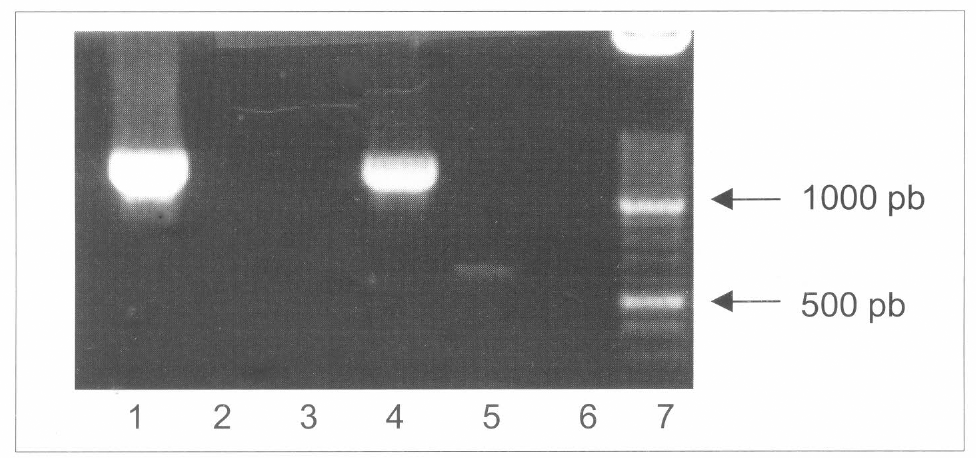
Separación en agarosa del gen pr1A amplificado por PCR. Una banda de 1189 bp indica la presencia del gen. Línea 1 contiene la reacción de PCR del plásmido pBarGPE1-pr1A. Línea 2 fue cargada con la reacción del plásmido pBarGPE-GFP. Línea 3 fue cargada con la reacción de la cepa no transformada 9112. Línea 4 contiene la reacción de la cepa pr1A-1. Línea 5 contiene la reacción de una cepa transformada con pBarGPE-GFP. Línea 6 contiene solución maestra sin DNA. Línea 7 contiene 500 ng de ADN marcador de peso molecular XIV (Boehringer).
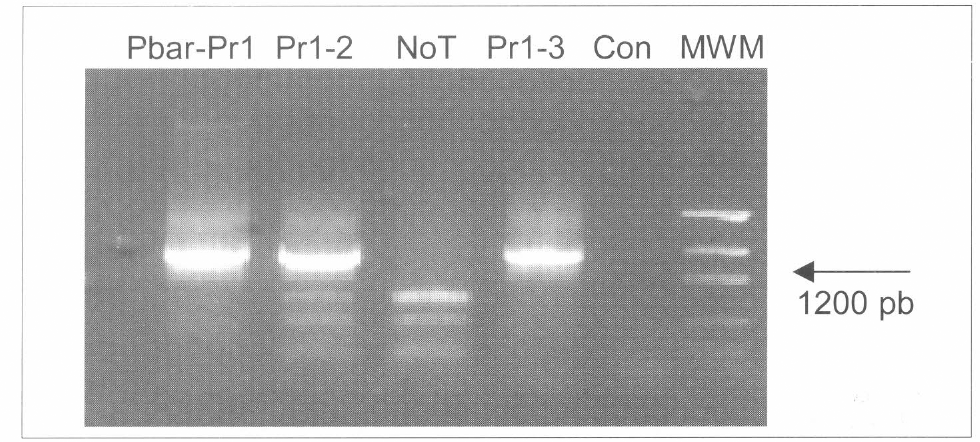
Separación en agarosa del gen pr1A amplificado por PCR. En cepas de B. bassiana. Una banda de 1189 pb indica la presencia del gen. Línea Pbar-Pr1 contiene la reacción del plásmido pBarGPE-GFP. Líneas pr1-2 y pr1-3 contiene la reacción de las cepas transformadas pr1A-2 y pr1A-3 respectivamente. Línea NoT fue cargada con la reacción de una cepa no transformada. Línea Con fue cargada con la solución maestra sin DNA. Línea MWM fue cargada con 500 ng de DNA marcador de peso molecular (Low DNA Mass ladder) (Gibco).
Conclusiones
Se logró llevar a cabo la modificación genética del hongo. Se cuenta con un método de transformación eficiente para Beauveria bassiana.
El gen de selección (bar) y el gen marcadores GFP no afectan el grado de patogenicidad ni el comportamiento de las cepas de B. bassiana contra la broca del café.
Estas cepas transgénicas se pueden usar para el seguimiento de los procesos de infección y patogenicidad sobre la broca del café en condiciones de laboratorio y campo. El siguiente paso es hacer el control de calidad de estas cepas y las pruebas de patogenicidad sobre la broca del café.
Footnotes
Agradecimientos
La autora agradece al Dr. Ray St. Leger por permitir el uso de los genes y a la Federación Nacional de Cafeteros de Colombia y Colciencias por el apoyo financiero dado a este trabajo.