
Abstract
Tissue engineering of articular cartilage requires accurate imaging of the chondrocyte cytoskeleton. Past studies have applied various fixation and permeabilization protocols without optimization of parameters. In this study, we have examined procedures using glutaraldehyde and paraformaldehyde as fixatives and Triton X-100 and Octyl-POE as permeabilizing detergents. A four-color fluorescence confocal method was developed to simultaneously image actin, tubulin, vimentin, and the nucleus. We found optimal preservation and morphology of the chondrocyte cytoskeleton after simultaneous fixation and permeabilization with glutaraldehyde and Triton X-100. These images displayed less cellular shrinkage and higher-resolution filamentous structures than with paraformaldehyde or when permeabilization followed fixation.
C
Chondrocytes were enzymatically isolated from the humeral head cartilage of young bovine shoulders and encapsulated in 6-mm diameter X 2-mm-thick agarose disks as described previously (Tran-Khanh et al. 2005). On day 15 of culture, agarose disks were vertically cut into 600-μm-thick slices with a vibratome in standard Hank's balanced salt solution (sHBSS) [1.29 mM CaCl2·2H2O, 5.37 mM KCl, 0.44 mM KH2PO4, 0.49 mM MgCl2·6H2O, 0.41 mM MgSO4, 136.89 mM NaCl, 0.34 mM Na2HPO4·7H2O, 5.55 mM glucose, 4.2 mM NaHCO3, 15 mM HEPES (C8H18N2O4S), pH 7.4]. Good cell viability (>95%) was confirmed in these slices using Calcein AM and ethidium homodimer (Molecular Probes; Eugene, OR) as previously described (Dumont et al. 1999). The cytoskeletal stabilization medium subsequently used for fixation and permeabilization was a modified Hank's balanced salt solution (mHBSS): 136.9 mM NaCl, 5.36 mM KCl, 2 mM MgCl2, 0.336 mM Na2HPO4, 0.44 mM KH2PO4, 4 mM NaHCO3, 2 mM EGTA, 5.55 mM
We tested three families of processing methods on these chondrocyte-laden slices: (a) sequential fixation/permeabilization by 0.1% glutaraldehyde and/or 1% to 4% paraformaldehyde for 30 min at 37C, followed by permeabilization with 5% to 10% Triton X-100 for 20 min at 37C; (b) smooth fixation/permeabilization by 0.125% glutaraldehyde in the presence of 1% to 2% Triton X-100 for 20 min at 37C, prior to postfixation with 1% glutaraldehyde or 4% paraformaldehyde for 30 min at 37C; (c) simultaneous fixation/permeabilization with 0.3% to 0.6% glutaraldehyde and 2% to 5% Triton X-100 for 30 min at 37C or simultaneous fixation/permeabilization with 0.1% glutaraldehyde and 1% to 4% paraformaldehyde and 5% to 10% Triton X-100 for 30 min at 37C. An autofluorescence block was performed on the same day or on the following day by incubating slices in 5 mg/ml NaBH4. Antibody penetration was then facilitated by digesting slices in 200 mU/ml chondroitinase ABC and 400 mU/ml keratanase as described previously (Langelier et al. 2000). Nonspecific antibody-binding sites were blocked by incubation in 10% goat serum (Sigma; St Louis, MO) and 0.01% Tween-20 in mHBSS for 2 hr. The buffer in the following steps was mHBSS containing 1% goat serum and 0.05% Tween-20 with agitation. Blocked slices were incubated with 10 μg/ml monoclonal anti-tubulin-β (Chemicon; Temecula, CA) for 16 hr at 4C, followed by three 20-min washes. Slices were then incubated in 10 μg/ml polyclonal goat anti-mouse coupled to Alexa Fluor 405 (Molecular Probes) for 4 hr and washed four times during 20 min. The buffer in the following steps was mHBSS containing 1% BSA and 0.05% Tween-20 with agitation. Tubulin-labeled slices were labeled for actin and vimentin with 0.2175 μg/ml Alexa Fluor 488-phalloidin (Molecular Probes) and 1.43 μg/ml monoclonal antibody anti-vim-Cy3 (Sigma) and washed three times during 20 min. Nuclear DNA was stained by first removing nonspecific RNA signal by 2-hr incubation with 1 mg/ml RnaseA (Qiagen; Mississauga, Canada) in PBS and 1% BSA followed by DNA staining with 3 μM of TOTO3 (Molecular Probes) in PBS for 2 hr. These four-color-stained slices were immersed for 30 min in glucose oxidase/catalase (GOC) anti-fading reagent and mounted with Mowiol on slides with coverslips N° 1.5 as described previously (Langelier et al. 2000). Fluorescence imaging of chondrocytes in 600-μm agarose slices was performed using a LSM 510 META Axioplan 2 confocal laser scanning microscope with C-Aprochromat X40/1.2 water-immersion objective (Carl Zeiss; Jena, Germany). Laser lines used were 488-nm argon laser, 543- and 633-nm heliumneon lasers, and 810 nm- (two-photon) pulsed titanium sapphire laser (VerdiV10/Mira 900; Coherent, Santa Clara, CA) using dichroic HFT UV/488/543/633 for conventional laser lines and HFT KP650 for the pulsed laser. The meta function was used to select filter and dichroic mirror configurations that minimized overlap from the four different fluorochromes. Images were recorded at 810-nm excitation using a BP 390–465 IR band pass filter for Alexa Fluor 405, at 488-nm excitation using a BP 510–520 IR band pass filter for Alexa Fluor 488, at 543-nm excitation using a BP 565–615 IR pass filter for Cy3, and at 633-nm excitation and a BP 644–676 meta filter for TOTO-3. The pinhole size was adjusted to obtain the optimal spatial resolution and high-magnification images were recorded with a 0.45-μm z-step and 0.08 μm x/y pixel size. Calcein-loaded chondrocytes under the viability test were also imaged as described above for Alexa Fluor 488 but using a water- immersion objective IR-Achroplan X63/0.9 dipped directly in sHBSS. Image stack images were deconvolved with Huygens2 software and presented with Imaris 4.0 software (Bitplane AG; Zurich, Switzerland). Images are representative of at least three sets of individual chondrocytes.
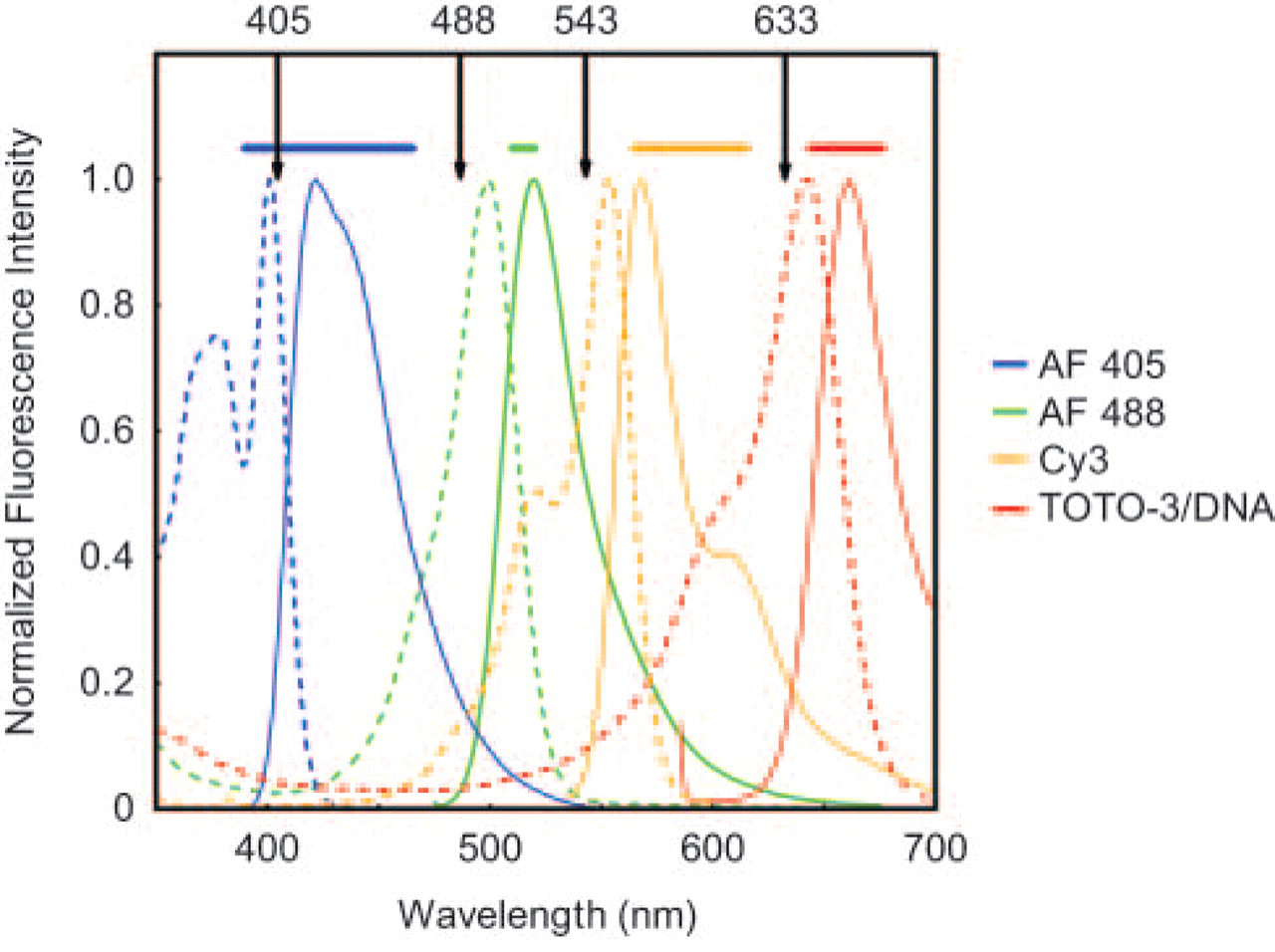
Excitation and emission spectra of the four dyes used in this study. Blue, green, orange, and red represent spectra from Alexa Fluor 405, Alexa Fluor 488, Cy3, and TOTO3 dyes, respectively. Vertical lines at 405 nm, 488 nm, 543 nm, and 633 nm represent excitation laser line used, whereas horizontal bars represent signal captured by emission filters.
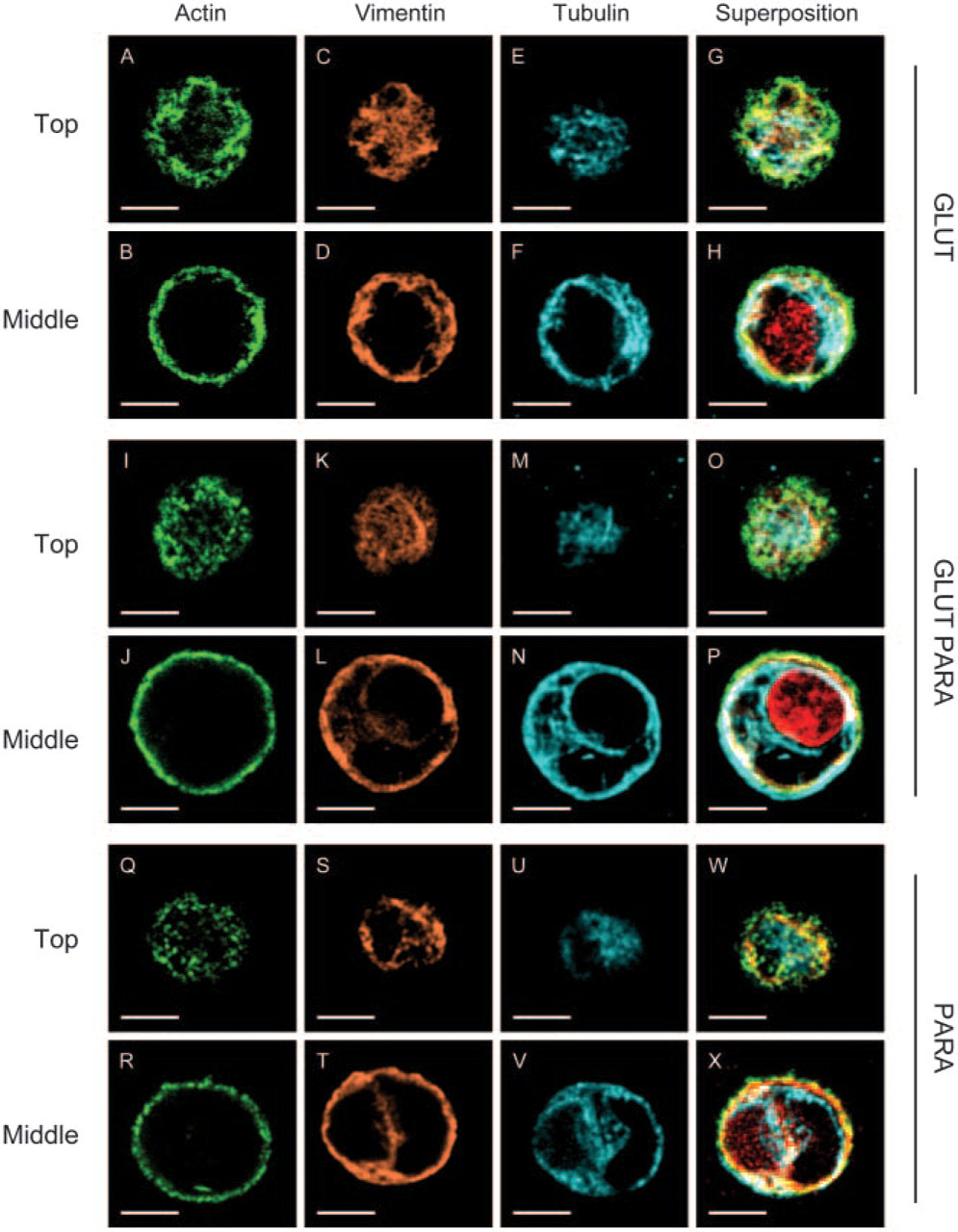
Comparison of processing methods for cytoskeletal preservation and labeling of chondrocytes in agarose. Chondrocytes were stained for actin (green), vimentin (orange), tubulin (blue), and nucleus (red) shown as individual channels and in superposition. Representative image planes from the top and from the middle of a chondrocyte from the same stack are shown. All samples were processed with simultaneous fixation/permeabilization using one of the following three variants: 0.6% glutaraldehyde and 5% Triton X-100 (
Because multiple stains have a tendency to overlap spectrally and spatially, especially when four stains are used, and cellular components of interest as microtubule and vimentin network are known to colocalize, we chose the Alexa Fluor 405, Alexa Fluor 488, Cy3, and TOTO-3 dyes to minimize overlap. A rigorous lambda mode analysis using the META function provided excitation and emission spectra of each dye to optimize filter configuration (Dickinson et al. 2001) and ensure a distinct separation of signals in the quadruple-stained specimens (Figure 1). Confocal z-series through representative chondrocytes grown in agarose showed a well-organized distribution of MT, MF, and vimentin IF (Figure 2) comparable to monostains previously described in situ and in agarose (Idowu et al. 2000). Because several previous studies have shown that precipitating fixatives such as acetone or alcohols produce cellular shrinkage and disruption with poorly preserved cytoskeletal organization (Bacallao et al. 1995), these precipitating fixatives were not examined in our study.
MT, vimentin IF, and MF were all well preserved with intense staining and high signal-to-noise when chondrocytes were simultaneously fixed in glutaraldehyde and permeabilized with Triton X-100 (Figures 2A–2H). Overall, simultaneous fixation with 0.6% glutaraldehyde and 5% Triton X-100 or, equivalently, prefixation by 0.125% glutaraldehyde with 2% Triton X-100 followed by 1% glutaraldehyde provided the most reproducible, intense, and morphologically clear images (Figure 2). An acceptable although not optimal preservation of the three-cytoskeletal networks with lower signal-to-noise and less clarity was found when cells were fixed with 1% to 4% paraformaldehyde and 5% Triton X-100 in presence of 0.1% glutaraldehyde (Figures 2I–2P). The microtubular network was found to be fragmented and vimentin staining diffuse when chondrocytes were simultaneous fixed by 4% parformaldehyde and 5% Triton X-100 (Figures 2Q–2X). Thus, adding 0.1% glutaraldehyde to paraformaldehyde fixative appeared to be required to preserve the microtubular network when chondrocytes are simultaneously permeabilized with Triton X-100. This need for glutaraldehyde is possibly due to its bifunctional aldehyde for rapid and stable fixation, leading to optimal conservation of cellular morphology and preservation of microtubule and vimentin structure where antigenic sites were preserved and accessible. Finally, chondrocytes that were first fixed by 1% to 4% paraformaldehyde and then permeabilized with Triton were irreproducibly preserved and stained for tubulin and vimentin (data not shown). In all cases, a lower concentration of fixative and/or Triton X-100 than described above resulted in diffuse staining (data not shown). Shrinkage and distortion of cytoskeleton under axial projection was also observed with paraformaldehyde as fixative when Triton X-100 was simultaneously employed, with or without glutraldehyde, compared with live chondrocytes (Figure 3A compared with Figures 3C and 3D). Similar shrinkage and distortion were obtained when paraformaldehyde fixation sequentially preceded Triton X-100 permeabilization (data not shown). In contrast, a normal expanded chondrocyte morphology was better maintained with glutaraldehyde and Triton X-100 (Figure 3A compared with Figure 3B). Finally, when Triton X-100 was replaced by Octyl Poe (P-1140; Bachem Bioscience, King of Prussia, PA) in the prefixation methods, results were less reproducible in terms of obtaining high signal-to-noise and highly resolved images for tubulin and vimentin.
In summary, we have identified a reproducible and easily performed procedure to preserve and label all three filamentous structures of the chondrocyte cytoskeleton as well as the nucleus. Simultaneous fixation and permeabilization followed by autofluorescence blocking and specific labeling with four fluorochromes resulted in clearly defined and intensely labeled filamentous structures. These methods can be applied to examine changes in chondrocyte structure in tissue-engineered constructs or in states of disease and degeneration in arthritis, as well as in studies aimed at elucidating the biological consequences of mechanical stimulation of chondrocytes and cartilage
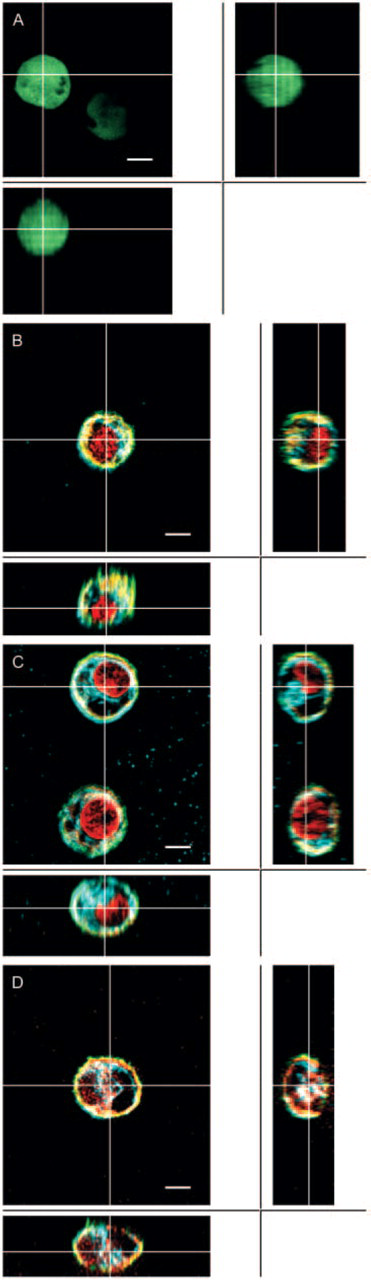
Comparison of different processing methods on morphological preservation vs shrinkage of chondrocytes in agarose. Central images (
Footnotes
Acknowledgements
Funding was provided by the Canadian Institutes of Health Research.