
Abstract
The initial stages of nerve outgrowth carried out by growth cones occur in three fundamental cyclic steps. Each of these steps appears to require myosin II activity to variable degrees. The steps include the following: (a) exploration, involving extensions and retractions that are driven and controlled by the interaction of actin retrograde flow and polymerization; (b) adhesion of new extensions to the substrate, which has been shown to be mediated by complex interactions between extracellular matrix proteins, cell adhesion proteins, and the actin cytoskeleton; and (c) traction force generated during forward advance of the growth cone, resulting in the production of tension on the neurite.
N
Steps in Growth Cone-mediated Nerve Outgrowth
Step a: Exploration
Regulation of extension and retraction of filopodia through the influence of extracellular signals is important in determining the direction of neurite outgrowth (Gallo and Letourneau 1999; Isbister and O'Connor 2000). The formations of stable new filopodia extensions are an essential first step in the cycle of directed growth cone advance (Bentley and Toroian-Raymond 1986). Recently, Mallavarapu and Mitchison (1999) showed that the apposing processes of actin retrograde flow and actin polymerization underlie filopodial extensions and retractions in neuroblastoma cells. The sum of these two opposing processes dictates whether filopodia grow, retract, or remain stationary. Each process appears to be independently regulated, leading to a complex array of possible responses in terms of speed, direction, and duration. If F-actin turnover or polymerization is inhibited, then myosin-dependent contraction or flow causes retraction of growth cones (Gallo et al. 2002). Although the mechanics of actin polymerization are fairly well documented (Chen et al. 2000), the mechanisms that mediate actin retrograde flow require a closer look. The most likely candidate for the molecular motor that mediates the retrograde flow of actin within neuronal growth cones is one of the myosins. The inhibition of myosins by microinjecting NEM-inactivated myosin S1 fragments or by applying the general inhibitor of myosin ATPase, 2,3-butanedione-2-monoxime (BDM) onto Aplysia bag cell neurons resulted in a cell phenotype that exhibited a complete lack of actin retrograde flow (Lin et al. 1996). The myosin inhibitors that were used in these experiments were not completely target-specific and may have influenced other processes occurring within growth cones. However, these data remain the best evidence for implicating a role of myosins in actin retrograde flow. The exact type(s) of myosin driving retrograde flow remains unknown.
Work done by Wylie et al. (1998), using antisense oligodeoxyribonucleotides complementary to myosin IIB, resulted in an attenuation of the production of myosin IIB in neuroblastoma cells and produced a significant decrease in outgrowth. Likewise, an attenuation of filopodial extensions was also reported, although no quantitative data to support this assertion were presented. The effect of myosin IIB on the attenuation of retrograde actin flow was also not assayed. The reduction in filopodial extensions could be taken to imply that myosin IIB may be directly involved in extension as opposed to being a motor that influences or controls retrograde flow. A direct involvement of myosin IIB in filopodial extension has not been directly investigated, but recent data do not support such a role. Results using transgenic mice collected by Tullio et al. (2001) and Bridgman et al. (2001) confirmed myosin IIB's importance in neuronal outgrowth and normal growth cone behavior of primary neurons. Knockout (KO) of the myosin IIB heavy chain (B-/B-) turned out to be lethal starting around E14 up to P0, which coincides with the timing for up-regulation of myosin IIB (Bridgman and Elkin 2000). Neurons cultured from the superior cervical ganglia (SCG) of B-/B- mice between E18–P0 were shown to have decreased rates of outgrowth (Tullio et al. 2001). In addition, growth cones were smaller and had distinctly narrow morphology compared with controls (Bridgman et al. 2001; Tullio et al. 2001). Protrusion and retraction locations and dynamics were also altered. However, unlike the results from neuroblastoma cells, the frequencies of protrusions and retractions (filopodia + lamellipodia) were significantly increased (but average areas decreased) in the KO when normalized for the different growth cone size. Actin organization was also altered in the KO growth cones. Actin bundles throughout the cone were decreased, although they were still present in filopodia. These findings in myosin IIB KO mice substantiate myosin IIB's role in the exploration step of neurite outgrowth. Because both the extension and retraction frequencies of protrusive structures (mainly filopodia, as KO cones have very few lamellipodia) are altered in the myosin IIB KOs, it appears likely that myosin IIB plays a pivotal role in this important first step in axon outgrowth and pathfinding. These data also suggest that it is unlikely that myosin IIB activity drives filopodial protrusion. Recent data from Dan Jay's lab also support this conclusion (Diefenbach et al. 2002). MicroCali of myosin II did not affect filopodia protrusion or retraction rates. It appears more likely that myosin IIB may be acting to influence actin organization via effects (direct or indirect) on actin retrograde flow. Because myosin IIA and IIB have overlapping distributions (Rochlin et al. 1995), both may influence or directly affect retrograde flow. When myosin IIB is absent myosin IIA could influence the retrograde flow of actin, but with altered dynamics because of its potential ability to contract at greater speeds (Kelley et al. 1996). In any case, it is clear that any alteration in retrograde flow properties will alter actin organization and ultimately the stability of protrusions such as lamellipodia. To determine the exact influence of myosin IIA on retrograde actin flow, it will be necessary to knock out the function of both myosin II isoforms in growth cones.
Recently, the hypothesis that myosin II plays a role in retrograde flow has been challenged (Diefenbach et al. 2002). MicroCali of myosin 1c, but not myosin IIB, was shown to slow retrograde flow. The authors suggest that myosin 1c is the only motor that drives retrograde flow. However, if this were the case, then one must devise a mechanism through which myosin 1c is able to generate retrograde flow rates severalfold higher than its F-actin sliding rate (Zhu et al. 1996). This is in contrast to a myosin II-dependent mechanism that relies on local contractions orthogonal to the direction of flow to “pull” the actin meshwork rearward (Svitkina et al. 1997; see step c below). In addition, myosin 1c expression does not appear to be upregulated during development, is not enriched in growth cone particles, and either is not detectable or is expressed only at very low levels in growth cones of some neuron types (Ruppert et al. 1995; and our unpublished observations). Therefore, if myosin 1c does drive retrograde flow in some growth cones, it must do so by a yet to be defined cell specific mechanism.
Although it has long been known that the actin cytoskeleton and the protrusive structures that it supports are required for directed outgrowth of axons and growth cone turning (Marsh and Letourneau 1984; Bently and Toroian-Raymond 1986; O'Conner et al. 1990; Zheng et al. 1996), whether or not myosins play a role in turning has been less clear. Very recently it has been shown that the local application of a myosin II inhibitor (ML-7) to one side of a growth cone causes the growth cone to turn away from the drug source (Zhou et al. 2002). This appears to be because myosin II regulates actin bundling which, in turn, affects other cytoskeletal elements. Under certain adhesive conditions, retraction normally induced by ML-7 can be prevented and the application results in the loss of actin bundles including those that form the core of filopodia. However, the F-actin meshwork is only minimally affected. The selective loss of actin bundles results in decreased local penetration of microtubule ends into the peripheral domain and turning for the following reason. In the absence of actin bundles (and contacts?) microtubules ends are continually swept rearward by the retrograde flow of the actin meshwork found in lamellipodia, and therefore do not advance. In the presence of actin bundles, especially the prominent bundles found in filopodia, polymerizing microtubules are stabilized and guided by the bundle, and the rate of microtubule extension is the sum of the microtubule polymerization rate and F-actin retrograde flow rates (Schaefer et al. 2002). Therefore, stabilization of microtubule ends by actin bundles in the peripheral domain appears to be a necessary prerequisite for microtubule advance and the continued regional extension and maturation of the growing neurite. When actin bundles are selectively lost from one side of the growth cone turning results. In the Zhou et al. (2002) study, inhibition of retrograde flow by a general myosin inhibitor (BDM) caused microtubules to penetrate the peripheral region even in the absence of actin bundles. Therefore, retrograde flow, rather than the actin meshwork itself, inhibits peripheral microtubule advance. Penetration of microtubules into actin-rich peripheral regions in non-neuronal cells appears to play a role in maturation of focal contacts (Kaverina et al. 2002). Microtubule ends may also influence the site of new actin polymerization (Rochlin et al. 1999; Dent and Kalil 2001). Taken together, these results support the idea that myosin II/actin interactions can influence other cytoskeletal components and are important for stabilizing the extensions that lead to both advance and growth cone turning.
Step b: Adhesion
Wylie and Chantler (2000) have shown that focal contacts are formed at the leading edge of growth cones of neuroblastoma cells. As these mature into focal adhesions they are then stabilized by myosin IIA, which helps in the recruitment of paxillin and vinculin to the adhesion site. These findings were demonstrated by creating a functional KO of myosin IIA using anti-sense RNA application to neuroblastoma cells. The cells showing diminished myosin IIA levels expressed a significantly decreased capacity for adhesion, although it is important to note that the cells that did manage to adhere to the substrate had no reduction in outgrowth. Myosin IIA's role in recruiting paxillin and vinculin to adhesion sites was demonstrated by immunofluorescence. In the absence of myosin IIA, paxillin and vinculin were diffusely distributed throughout the cytoplasm. The work done on myosin IIA's role in adhesion was conducted in neuroblastoma cells where adhesion sites are much more static and well defined than in growth cones of a primary neurons undergoing rapid growth (Gomez et al. 1996; Renaudin et al. 1999). The adhesion complexes that form in growth cones of rapidly growing primary neurons do not mature to the extent observed in neuroblastoma cells, possibly because of the increased rate of growth and associated greater motility. Although previous work has shown that myosin IIA is most concentrated in the central region of the growth cone, consistent with a possible role in adhesion within this region (Rochlin et al. 1995), more peripheral staining was also seen. This peripheral staining often co-localizes with staining for β1-integrin, suggesting that there is a close relationship with developing focal complexes (our unpublished observations). It is therefore encouraging that myosin IIA's distribution in primary neuron growth cones is consistent with its proposed role in stabilizing adhesion sites. Recent work from Steketee and Tosney (2002) suggests that the location of adhesive contacts on filopodia is important for regulating veil (lamellipodia) advance that often occurs between adjacent filopodia. Adhesions along the filopodial shaft inhibited veil advance, whereas adhesions at the filopodial tips stimulated veil advance. Adhesions at the base of filopodia did not influence veil advance but were necessary for filopodia formation. It will be interesting to determine the signals that arise from these different adhesive sites and how they influence myosin II activity. The site of adhesions relative to the location of activated myosin might explain how traction force can be exerted in a localized region of the growth cone while retrograde flow continues in adjacent areas. Both myosin IIA and B isoforms may also be instrumental in mediating a molecular clutch function similar to that describe in fibroblasts by Smilenov et al. (1999), where the adhesion complexes remain mobile until they receive the proper signal to adhere tightly to the substrate. The adherence then allows the cell to migrate. Work done by Suter et al. (1998) on growth cones showed that beads coated with ApCAM move with retrograde flow, but if the beads' movement was restrained by a glass needle, the beads would stop and actin flow would continue unaffected. However, after a latency period the beads would couple tightly. The restrained beads halted retrograde flow locally, bending the needle. Apparently the molecular clutch was engaged, generating tension on the needle. This clutch mechanism is similar to a myosin-actin interaction, i.e., it results in a transient generation of force resulting in displacement. Recently, it was shown that the Src family of tyrosine kinases regulates the ApCam-cytoskeletal linkage (Suter and Forscher 2001).
Step c: Traction Force Generation
Once a growth cone has committed to a direction for continued extension and established adhesion sites to stabilize the leading edge, tension and contractile forces must be generated to pull the rest of the cytoplasm along to sustain neurite outgrowth. Although tension forces were first observed in growing neurons in 1979 (Bray 1979), and it has been directly shown that growth cone pull (Lamoureux et al. 1989), the question of how and where these forces are generated has remained elusive. Because actin is the predominant cytoskeletal component in the growth cone periphery and filopodia have already been implicated in force generation through contraction (Heidemann et al. 1990), it stands to reason that an actin-myosin interaction provides the mechanism for traction force and the resulting tension on the growing neurite. Although it has been known that there is an important and intimate inverse relationship between myosin-dependent retrograde flow and forward advance of the growth cone (Lin et al. 1996; Lin and Forscher 1995), a model that explains the site of force generation or molecular details of the actin-myosin interaction has been lacking. For example, the actin filament sliding rates of all myosins known to be present in neurons are slower than the maximal rates of retrograde flow that are observed. An explanation of how actin-myosin interaction could be translated into the intracellular movements and forces that drive growth cone motility comes from the unlikely source of the fish keratocyte. The explanation derived from studies on keratocytes is called the dynamic network contraction model (Svitkina et al. 1997). In this model, an actin-myosin II network undergoes continuous assembly in the lamellipodia, contraction and formation of bundles roughly parallel to the leading edge occurs in the transition zone, and disassembly of the bundles occurs in the cell body (Figure 1). One interesting aspect of this dynamic contraction model is that the velocity of contraction is twice the speed of filament sliding, and this velocity could potentially be translated and even increased by the movement of actin filaments orthogonal to the direction of contraction. Therefore, with an appropriately interconnected network of peripheral actin, the rate of retrograde flow could potentially exceed the rate of filament sliding by a single myosin head. Whereas neurite outgrowth and fish keratocyte motility may at first appear to be unrelated processes, on closer examination it appears that growth cones possess all the necessary characteristics required of this model. In the dynamic network contraction model, there must be a graded mixture of actin filament polarity from the leading edge to the transition zone. This is because myosin II bipolar filaments located in the transition zone must interact with oppositely oriented actin filaments to generate a contraction roughly orthogonal to the direction of retrograde flow. Negative stain and freeze-etch EM work on growth cones by Lewis and Bridgman (1992) confirmed early work on non-neuronal cells that actin filaments in the leading edge region of growth cones are predominantly oriented with their barbed end pointing towards the leading edge. However, a second population of actin filaments was also observed which were more randomly oriented with respect to the leading edge. The mixed polarity orientation of this population increased with increasing distance from the leading edge. Therefore, in the transition zone there are sufficient numbers of oppositely oriented actin filaments to allow myosin II-dependent contraction. In addition, the transition zone also contains more actin bundles than the periphery, which may arise as a consequence of a myosin-dependent contraction (Rochlin et al. 1995). The mesh network of actin filaments also appears to span the space between the dorsal and ventral membranes in the peripheral lamellipodium, enabling it to contract intracellular proteins involved in adhesive contracts. Thus, it could provide a link for production of traction forces mediated by receptors to substrate adhesion molecules. Another requirement of the dynamic network contraction model is that myosin II must be concentrated in the transition zone and organized into bipolar filaments. Experiments conducted by Rochlin et al. (1995), using isoform-specific antibodies to the myosin IIA and B heavy chains, demonstrated by immunofluorescence that myosin IIB was at its greatest intensity in the transition/marginal region, whereas myosin IIA was most concentrated in the central region. The punctate staining was often arranged in rows along actin bundles indicating a possible series or sarcomere-like arrangement. Much of this punctate staining probably represents biopolar minifilaments similar to those found in non-neuronal cells (Svitkina et al. 1997). These minifilaments could be labeled with antibodies to either myosin IIA or B, supporting a contractile role for both isoforms in growth cone motility (Bridgman 2002). With all of the necessary components of the dynamic network contraction model in place, it is easy to envision a contraction model involving myosin II in growth cones that differs only in its more transient formation of adhesion sites, i.e., a clutch mechanism that engages periodically in local regions of the growth cone in response to regulation factors. In fact, work by Bridgman et al. (2001) measuring the force production of individual filopodia in normal and myosin IIB KO growth cones verified that the KO growth cones had, on average, reduced filopodia-mediated traction force (Figure 2). Therefore, myosin IIB contributes to traction force but is not the only myosin responsible. The other most likely candidate contributing to traction force generation via a means similar to that described by the dynamic network contraction model is myosin IIA. It therefore seems possible that the dynamic network contraction model may explain many of the force-dependent mechanisms of growth cone locomotion. This relationship has also been suggested by others (Heidemann and Buxbaum 1998). However, the final proof awaits the functional consequences of the specific inhibition of all myosin II isoforms present in growth cones.
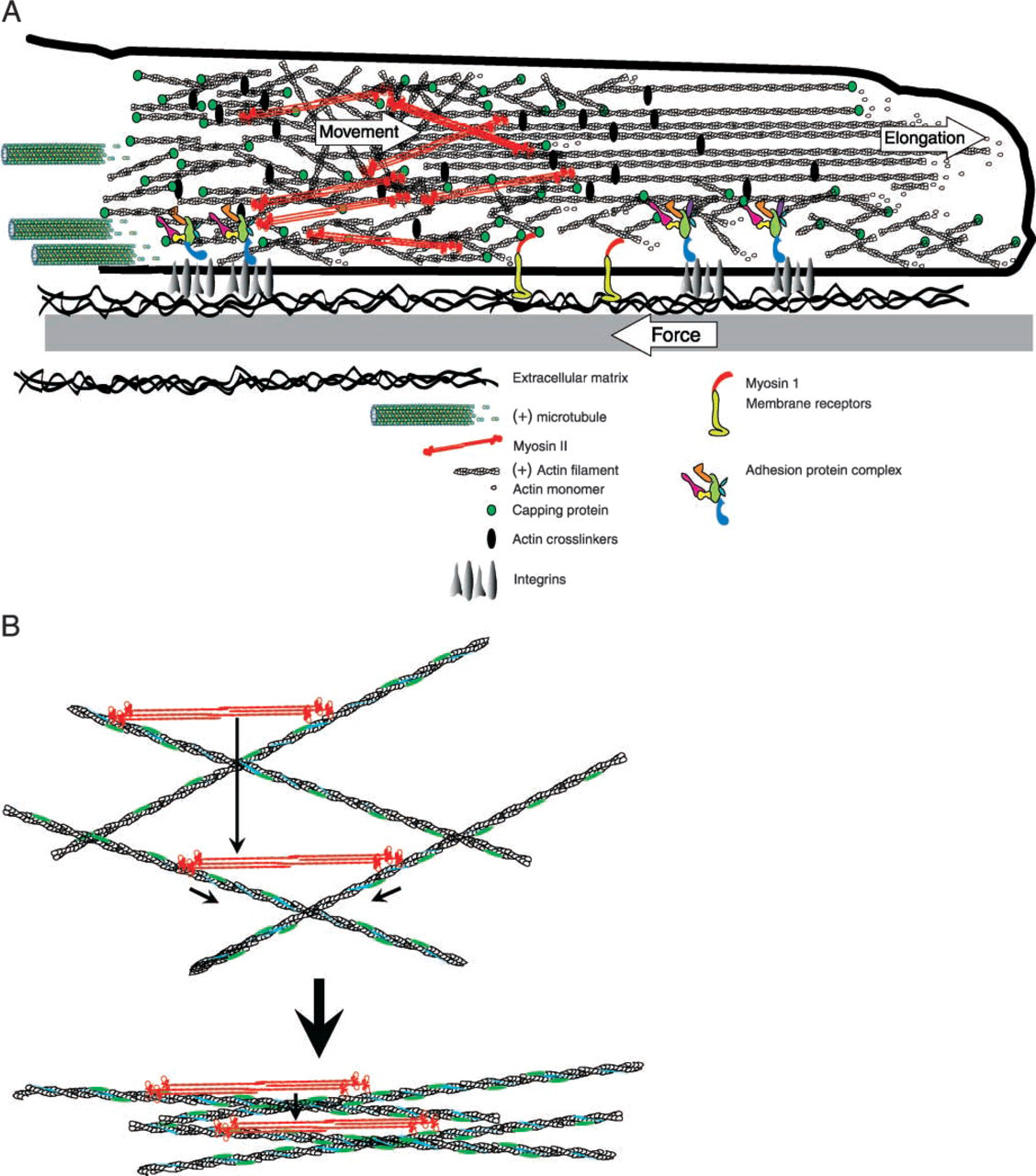
(A) Diagram based on the dynamic contraction model (Svitkina et al. 1997), showing how the interaction of myosin and actin could lead to retrograde flow or traction force generation. Two populations of actin filaments are present. One consists of relatively long filaments oriented with their fast-growing ends (+) towards the leading edge. The second consists of shorter filaments with mixed polarity. These two populations of actin filaments are interconnected and crosslinked. Actin filaments of roughly opposite orientations become bundled in the transition zone through interaction with myosin II bipolar filaments. The actin network interacts with integrins in the membrane through a complex of adhesion proteins that form a clutch mechanism. When the clutch is engaged, traction force develops if the peripheral adhesion sites outweigh those more proximal. When the clutch mechanism is not engaged, retrograde flow occurs at a maximal rate. States between the two extremes result in some traction and slowed retrograde flow. In addition, there may be interactions between monomeric myosin I proteins, membrane receptors, and the actin network. This could also lead to retrograde flow or traction, provided that the myosin I is anchored. (
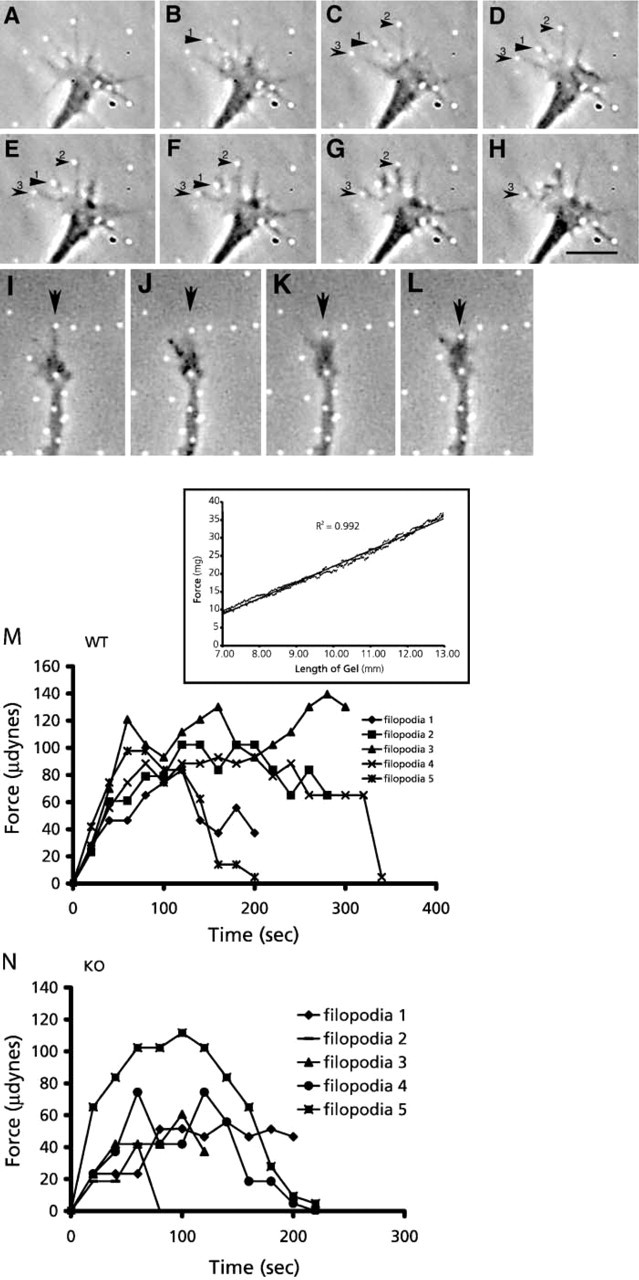
Interaction of growth cone filopodia and lamellipodia with the 3.75% acrylamide gel surface causes displacement of fluorescent beads (white dots) embedded in the gel. (
In conclusion, from the research presented here and the many works that came before them, a more complete model of neurite outgrowth that is dependent on the motile activity of the growth cone is coming into focus. The actin polymerization rate at specific sites on the leading edge temporarily exceeds the local retrograde flow rate, allowing filopodial/lamellipodial extension. Transitory adhesion sites regulated by myosin IIA and microtubules can either bind tightly to the substrate-bound extracellular matrix proteins (engaging the clutch) or quickly release, allowing the rapid remodeling characteristic of growth cones. If the clutch has been engaged and the focal complexes attach to the substrate, this then couples (through the actin cytoskeleton) the motor located in the transition zone to an immobile surface. Tension develops in the cytoplasm as the motor pulls on the peripherally immobilized actin network. This allows the growth cone cytoplasm to move forward, exerting traction forces on the surface as it advances. This is necessarily coupled to elongation of the trailing neurite through continued polymerization of microtubules and other cytoskeletal components. New extensions form at the leading edge and the cycle is repeated.
Footnotes
Acknowledgements
P.C.B. acknowledges the continued support from NIH to projects related to the work described in this review.
We thank Dr. Mike Brown for comments on the manuscript.