
Abstract
We investigated the structure of the chondrocyte cytoskeleton in intact tissue sections of mature bovine articular cartilage using confocal fluorescence microscopy complemented by protein extraction and immunoblotting analysis. Actin microfilaments were present inside the cell membrane as a predominantly cortical structure. Vimentin and tubulin spanned the cytoplasm from cell to nuclear membrane, the vimentin network appearing finer compared to tubulin. These cytoskeletal structures were present in chondrocytes from all depth zones of the articular cartilage. However, staining intensity varied from zone to zone, usually showing more intense staining for the filament systems at the articular surface compared to the deeper zones. These results obtained on fluorescently labeled sections were also corroborated by protein contents extracted and observed by immunoblotting. The observed cytoskeletal structures are compatible with some of the proposed cellular functions of these systems and support possible microenvironmental regulation of the cytoskeleton, including that due to physical forces from load-bearing, which are known to vary through the depth layers of articular cartilage.
T
Articular chondrocytes do not divide or migrate in healthy adult cartilage, even in response to growth factor stimulation. Therefore, the likely roles for micro-filaments in chondrocytes are in cell–matrix interactions, cell signaling, differentiation, intracellular transport, control of secretion/endocytosis and in resiliency of cell shape. Microfilaments (MFs) have been clearly implicated in control of chondrocyte phenotype (Brown and Benya 1988; Mallein–Gerin et al. 1991; Martin et al. 1999). In chondrocytes, a most likely function for microtubules (MTs) is intracytoplasmic transport, whereas intermediate filaments (IFs) of chondrocytes could be involved in mechanical as well as transport and signaling functions. Mechanical functions of IFs in chondrocytes are supported by previous observations of increased IFs in weight-bearing regions of articular cartilage (Eggli et al. 1988) and in compression-bearing regions in fibrocartilage (Ralphs et al. 1991, 1992). A potential role of IFs in osteoarthritis was also suggested by the loss of IF expression in a mouse model of osteoarthritis before any other detectable extracellular changes (Benjamin et al. 1995).
Given the clear significance of cytoskeletal architecture in the physiology of chondrocytes and cartilage, it is important to develop techniques to assess the presence and organization of cytoskeletal components in chondrocytes in situ. Fluorescence confocal microscopy has been used after cryofixation to obtain confocal images of microfilaments and intermediate filaments, but not microtubules, in intact rat articular cartilage (Durrant et al. 1999). There have been no previous studies reporting the maintenance and observation of all three cytoskeletal systems in chondrocytes in intact articular cartilage (for review see Benjamin et al. 1994). Therefore the purpose of this study was to describe the distribution and physiological 3-D organization of chondrocyte cytoskeletal networks (MFs, MTs, and vimentin IFs) in situ, which is essential for a better understanding of the roles of the cytoskeleton in cartilage physiology, mechanotransduction, and disease, such as osteoarthritis. Distinct cell and extracellular matrix (ECM) differences are known to exist among different depth zones of articular cartilage (Muir et al. 1970; Speer and Dahners 1979; Bayliss et al. 1983; Schenk et al. 1986; Eggli et al. 1988; Wong et al. 1996), as are depth-dependent patterns of the mechanical parameters of stress and strain (Kempson et al. 1973; Woo et al. 1976; Roth and Mow 1980; Guilak 1995; Schinagl et al. 1996; Jurvelin et al. 1997; Kolmonen et al. 1997). Given these known cell, ECM, and mechanical zonal dependencies, we hypothesized that cytoskeletal structure and composition may also display zonal dependencies. We therefore required that the cytoskeleton be assessed in each of the classically defined noncalcified zones: superficial (or tangential), transitional, and radial (just before the calcified cartilage zone). Confocal immunofluorescence and epifluorescence techniques were used to characterize the morphology and zonal dependence of the chondrocyte cytoskeletal components actin, tubulin, and vimentin. The observed zonal distributions and morphology of the three cytoskeletal filament systems lend support to some of their potential functional roles in chondrocytes and provide a reference point with which alterations can be compared.
Materials and Methods
Tissue Explant Isolation and Culture
A recently described cartilage-bone tissue explant system (Dumont et al. 1999) was used in these studies. The unique features of this disk explant system compared to most previously used cartilage explant systems (Grodzinsky 1990) are the presence of an intact articular surface and a thin layer of subchondral bone, thus minimizing tissue cutting and swelling in culture. The lack of swelling allows easier maintenance of homeostasis by limiting diffusional loss of matrix molecules. We showed stable cell viability, collagen and proteoglycan content, synthesis, and loss during 3-week culture using defined serum-free medium. We used this cultured explant system in the current study to allow future studies involving application of mechanical loads in vitro and to provide a postmortem stabilization period of several days in culture. Briefly, cartilage-bone cores were isolated from the load-bearing areas of the humeral heads of 1–2-year-old steers using an orthopedic coring bit (050.720; Straumann Canada, Toronto, ONT, Canada) under constant cooling irrigation with HBSS (14060; Gibco BRL, Rockville, MD). The location of isolated explants within the load-bearing contact area was noted. In a grid of ∼30 disks taken from one load-bearing region of the humeral head, in contact with the scapular articular cartilage, there were 18 disks circumscribing the periphery and 12 disks clustered more centrally within the load-bearing region. The explants were cut just beneath the bone-cartilage interface with a specialized device to retain a thin layer of subchondral bone, thus preserving the natural cartilage-bone interface. The resulting disks (4-mm diameter, 1.5-mm thick) contained the entire thickness of articular cartilage (∼1.2-mm thick) attached to a layer of subchondral bone (∼0.3-mm thick). Disk diameter was reduced to 3 mm using a biopsy punch (Miltex 15-33-33; Apocom, Montreal, PQ, Canada) cutting from the cartilage surface through the bone. After isolation, the disks were washed five times with HBSS containing antibiotics, transferred to 96-well round-bottom microplates (5850–96; Corning Costar, Cambridge, MA), and cultured in 250 μl/well of DME/F12 (pH 7.4) (D8900) supplemented with 50 μg/ml gentamycin (G1272), 0.01% BSA (A8412), and 20 μg/ml ascorbate (A4034; all from Sigma, St Louis, MO) at 37C in 95% air, 5% CO2. Medium was changed every 24 hr, with ascorbate prepared fresh immediately before medium change.
Cell Viability
Cell viability was determined as described previously (Dumont et al. 1999). Cartilage sections (75-μm thick) obtained with a Tissue-Chopper (McTwain; Brinkmann, Westbury, NY) were submitted to an assay solution consisting of PBS with 1 μM calcein AM, and 1.2 μM ethidium homodimer-1 (L-3224 LIVE/DEAD viability/Cytotoxicity Kit; Molecular Probes, Eugene, OR) to detect green fluorescing live and red fluorescing dead cells. Samples were incubated for 30 min or longer at 37C, after which green (live) or red (dead) cells were visualized with a Zeiss inverted fluorescence microscope and documented using a digital camera and Northern Eclipse software from Empix Imaging (Mississauga, ONT, Canada). Image manipulation and 3D reconstruction were achieved using Scion Image from Scion (Frederick, MD).
Tissue Preparation for Fluorescence Microscopy
The cytoskeletal stabilization medium during processing was a modified Hank's balanced salt solution (mHBSS): 136.9 mM NaCl, 5.36 mM KCl, 2 mM MgCl2, 0.336 mM Na2HPO4, 0.44 mM KH2PO4, 4 mM NaHCO3, 2 mM EGTA, 5.55 mM
Confocal scanning microscopy was performed using argon laser excitation on a Leica TCS 4-D CLSM (Leica; Heidelberg, Germany) equipped with PL Apo 100 × and Apochromat 63 × 1.4 NA oil immersion objectives. Optical sections at 0.5 μm increments were obtained throughout the 100-μm section or were obtained for regions limited to particular cells and cell groups. Epifluorescence images were acquired using an inverted florescence Zeiss microscope equipped with a digital camera and image acquisition software (see above). To reduce background for epifluorescence, thinner 50-μm sections from the tissue chopper were used. Cartilage-bone core samples (before cutting off of the subchondral bone) were also sectioned using a vibratome (Vibratome 1000; Micle Lab Engineering, Gomshall, Surrey, UK) because this sectioning procedure was seen to be gentler and to reduce cell death due to sectioning. In that case, the long core sample was held in place by the bone core with a custom mini-vise installed on the stage of the vibratome. Slices 25 and 50 μm thick were taken, slicing from the articular surface through a thin layer of bone. A razor blade was then used to detach the slices from the remaining bone core. During slicing with the vibratome, the sample was bathed in mHBSS at room temperature (RT).
Protein Extraction and Western Blotting Analysis
Western blotting analysis on individual disks was performed to complement observed depth-dependent fluorescence labeling. Cultured disks were glued bone surface down with cyanoacrylate on the stage of the vibratome and bathed in mHBSS, pH 6.5, at RT. The disks were cut transversely into 100-μm circular serial sections, the first section representing the articular surface and the last the deep radial zone. Sections were placed in mHBSS, pH 6.5, in individual wells of a 96-well plate at RT. Each disk yielded 8–12 tangential sections, which were subsequently divided into three equal pools (top ≅ tangential, middle ≅ transitional, bottom ≅ radial) representing the three zones of articular cartilage. Pooled sections were weighed and immediately flash-frozen with liquid nitrogen. Each pool of sections was rendered into powder by pulverizing (BioPulverizer; Biospec Products, Rijswik, The Netherlands) over liquid nitrogen, and the powder extracted with 100 μl RIPA buffer [0.1% SDS (4095–02; JT Baker, Phillipsburg, NJ), 1.0% desoxycholate (AC2849; Anachemia, Montreal, PQ, Canada), 1% Triton X-100 (161–0407; Bio-Rad, Hercules, CA), 10 mM Tris (161–0716; Bio-Rad), pH 7.4, 150 mM NaCl] with protease inhibitors aprotinin (Sigma; A4529), PMSF (Sigma; P7626), iodoacetamide (Sigma; I6125), pepstatin A (Sigma; P5318), TPCK (Sigma; T4376), at concentrations recommended by the manufacturer. Extracts were cleared at 15,800 × g (13 krpm) at 4C for 10 min. Approximately equal volumes of RIPA-extracted supernatant (85 ± 3 μl) were obtained from each group of layers of all disks analyzed. Proportionally equal volumes of supernatant were loaded on three 10% SDS-PAGE gels and transferred to Immobilon-P membrane (IPVH00010; Millipore, Bedford, MA). To generate protein extracts of RIPA-insoluble proteins, the pellets from cleared RIPA extracts were extracted in 100 μl 4 M guanidine-HCl (4078–01; JT Baker) with protease inhibitors for 60 hr at 4C, followed by Centricon concentration with RIPA as the exchange buffer, and volumes were made to equal 100 μl with RIPA before subjecting equal proportions of each sample to SDS-PAGE and immunoblotting transfer. In parallel, a smaller volume of each extract was subjected to SDS-PAGE followed by Coomassie Blue staining to control for total protein loaded from each extract. The resulting membranes were immunoblotted with one of three antisera, including anti-vimentin mouse MAb V9 (V6630, Sigma; 1:2500), anti-β-tubulin MAb (1 111 876, Boehringer; 1:350), or anti-actin MAb (A4700, Sigma; 1:500), followed by anti-mouse HRP-conjugated secondary MAb (PI-2000; Vector, Burlingame, CA, 1:2500) and ECL chemiluminescence (RPN 2106; Amersham).
Time Lapse Imaging
A custom-made microscopy chamber was used to observe chondrocyte morphology in permeabilization and fixation solutions to ensure a minimum of structural alterations during these procedures. The microscopy chamber consisted of a stainless steel plate with the dimensions of a slide. The plate was pierced in the middle and two coverslips were used as transparent bottom and top for the chamber that could be used on an inverted microscope. Thick slices (100 μm) were taken from fresh and cultured (up to 3 days) disks using a tissue chopper as described above and were placed in mHBSS in the microscopy chamber. Differential interference contrast (DIC) images were taken of the fresh and cultured sections using an inverted fluorescence Zeiss microscope equipped with a ×100 oil immersion objective. DIC time-lapse images of cultured sections were taken during the 20-min permeabilization period (2% Octyl Poe and 0.125% glutaraldehyde in mHBSS) and the 30-min fixation period (1% gluteraldehyde in mHBSS) to observe potential alterations in chondrocyte morphology due to those procedures.
Results
Reported results for cell viability, low-magnification epifluorescence microscopy, confocal microscopy, and time-lapse imaging represent common characteristics seen in groups containing four to eight samples.
Cell Viability
Chondrocytes were viable in all zones at the time of tissue isolation and throughout the culture period of 1–3 days, with the exception of a thin layer of cells at the articular surface and the cut radial edge of the disks (as seen previously in Figure 3 of Dumont et al. 1999). The occasional dead cells in zones other than the most superficial layer were found to be those cut by the tissue chopper during sectioning.
Labeling Intensity and Western Blotting Analysis Showed a Heterogeneous Content of Cytoskeletal Components in the Different Depth Zones of Articular Cartilage
Low-magnification epifluorescence images of the immunostained cartilage sections displayed distinctive zonal dependencies of labeling intensity of the components of the chondrocyte cytoskeleton (Figure 1). Vimentin IF and MT signal intensity was higher in the superficial zones compared to deeper zones, whereas signal intensity for actin MFs was more uniform. Sharp vimentin and tubulin filament network gradients were observed in those disks situated near the periphery of the load-bearing region, whereas more centrally located disks showed only slight depth-dependent expression gradients. These zonal characteristics were observed both in sections taken from disks using the tissue chopper and in those prepared by the more gentle cutting of the vibratome on cartilage–bone cores containing long bone stems.
Immunoblotting analysis showed that, in general, RIPA-solubilized cytoskeletal subunit levels were elevated in the most superficial layer relative to the middle and deep zones (Figure 2). No major differences were observed between central and peripheral disks for the RIPA extracts. However, GuCl extracts from RIPA-insoluble pellets contained amounts of vimentin that appeared to match the histochemical vimentin gradients observed in adjacent disks. In the GuCl extracts of centrally located disks, vimentin levels were higher in the tangential and radial layers compared to the transitional region, similar to the immunohistochemical signals (Figure 1). For peripherally located disks, there was a monotonic gradient in vimentin distribution which was also similar to the immunohistochemical signals (Figure 1), with increasing levels of vimentin content from the radial to the tangential zone. Neither actin nor tubulin was detected in the GuCl extracts. The SDS-PAGE stained with Coomassie Blue showed protein loading to be globally equal for extracts from different layers (data not shown), where several bands appeared uniformly present in different depths, whereas others manifested gradients of either decreasing (as for cytoskeletal proteins) or increasing intensity with depth. An example of the latter was observed with blotting with an antibody specific for the C-propeptide of collagen Type II (data not shown), confirming the efficiency of protein extraction and recovery from different layers and the presence of physiological gradients in protein concentrations vs depth from the articular surface.
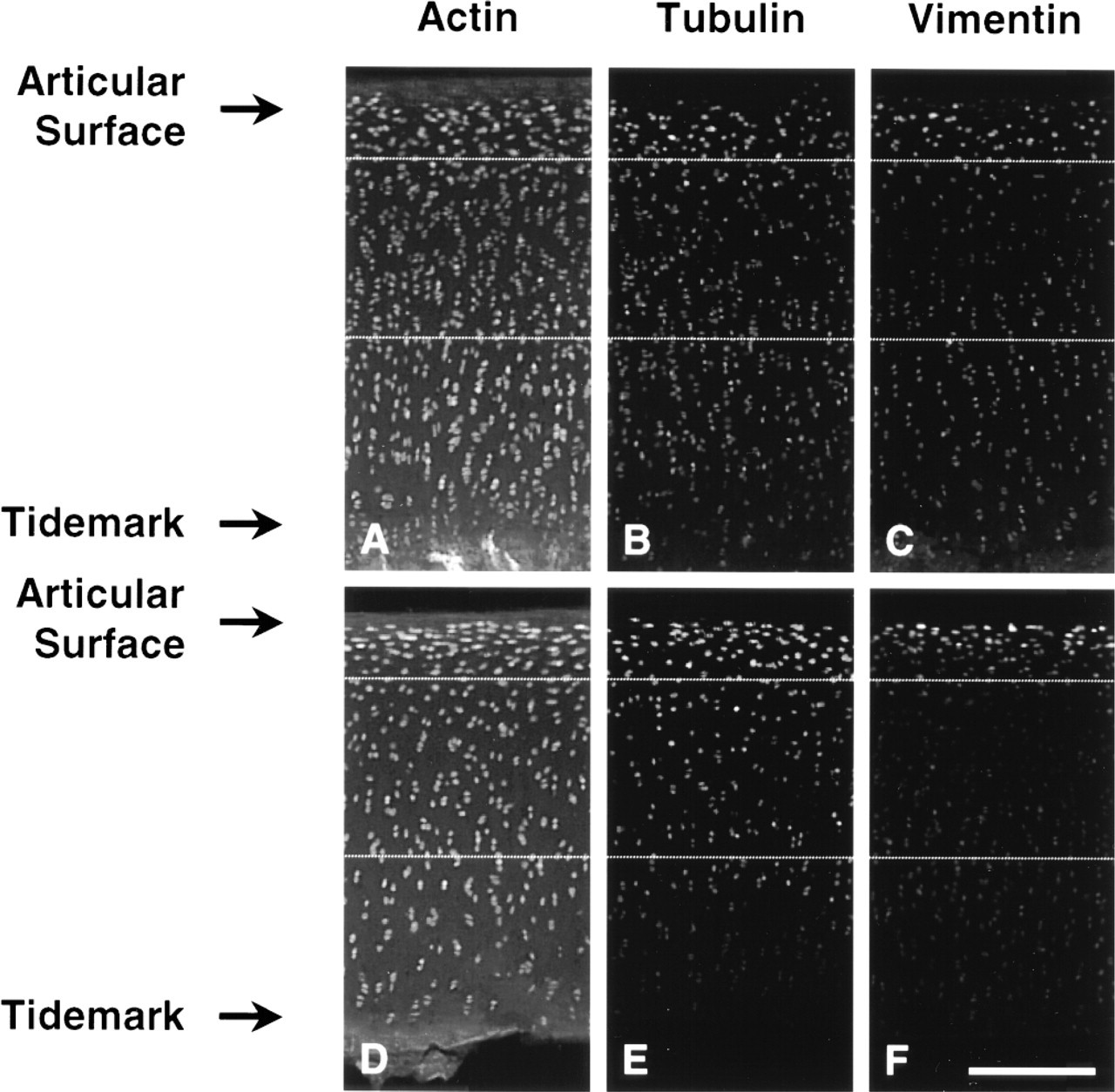
Low-magnification epifluorescence images of cartilage sections labeled for actin microfilaments (
Specific Structural Morphologies for Actin, Tubulin, and Vimentin Filament Systems in Chondrocytes In Situ Were Observed Using Confocal Microscopy
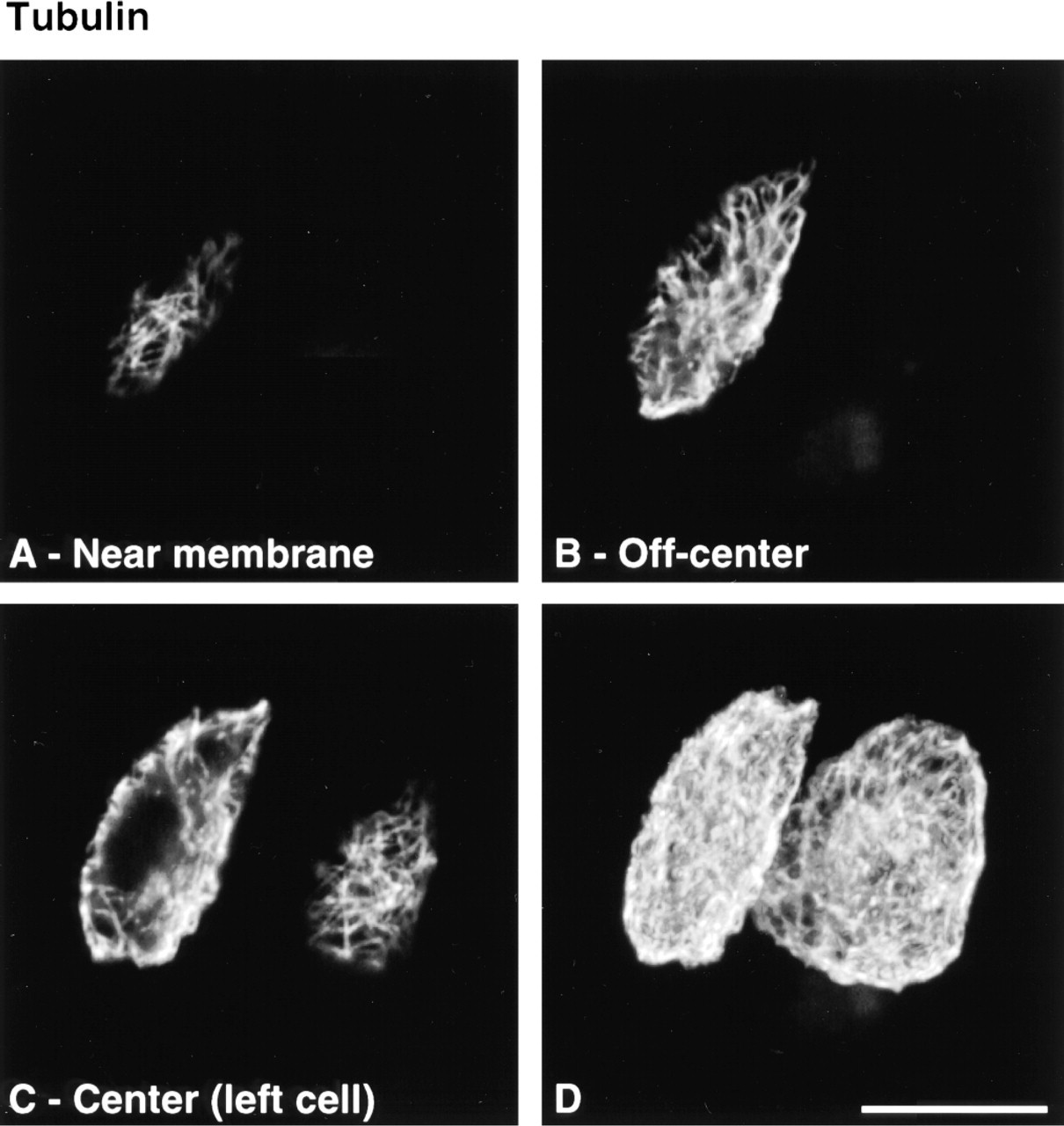
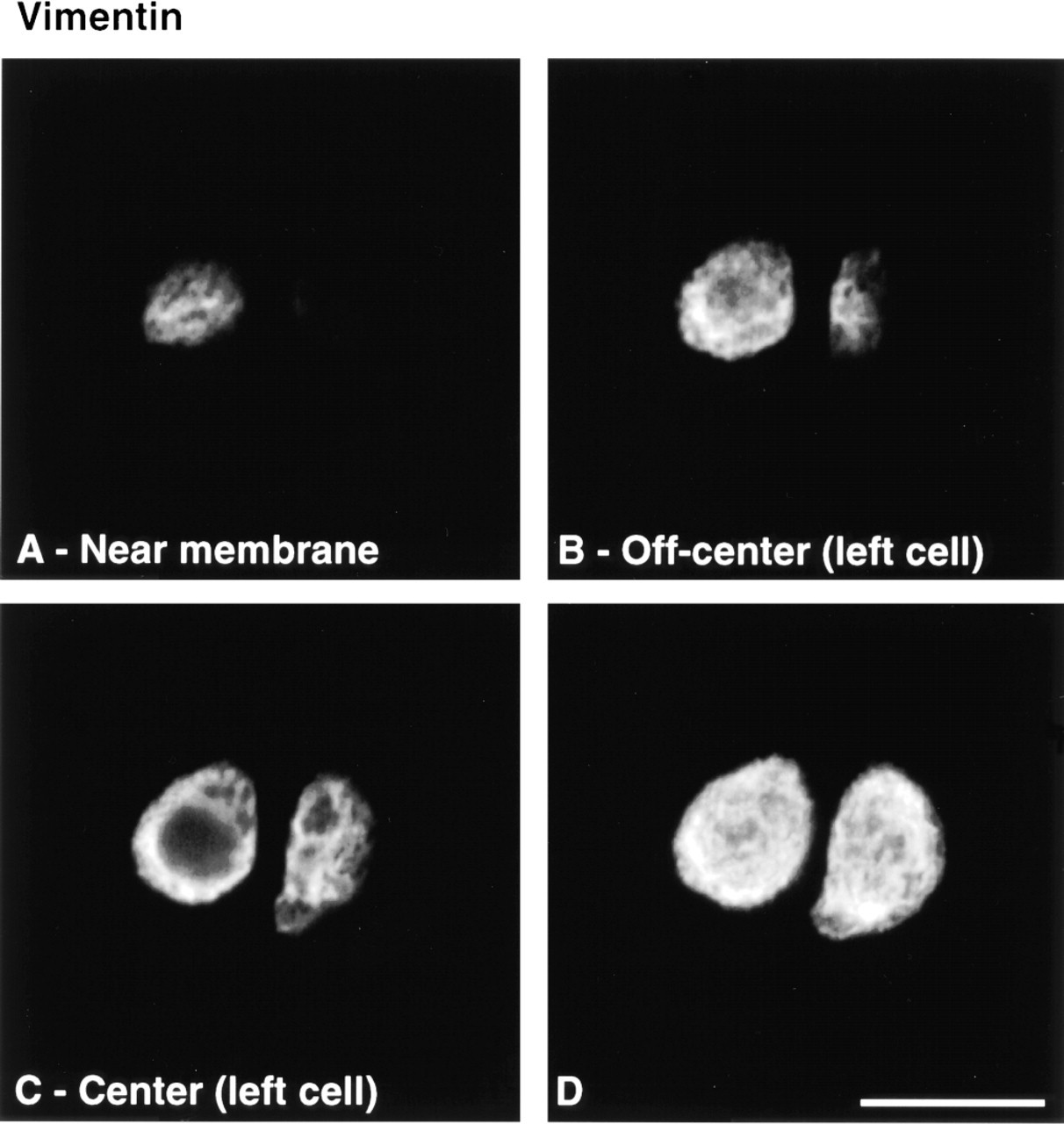
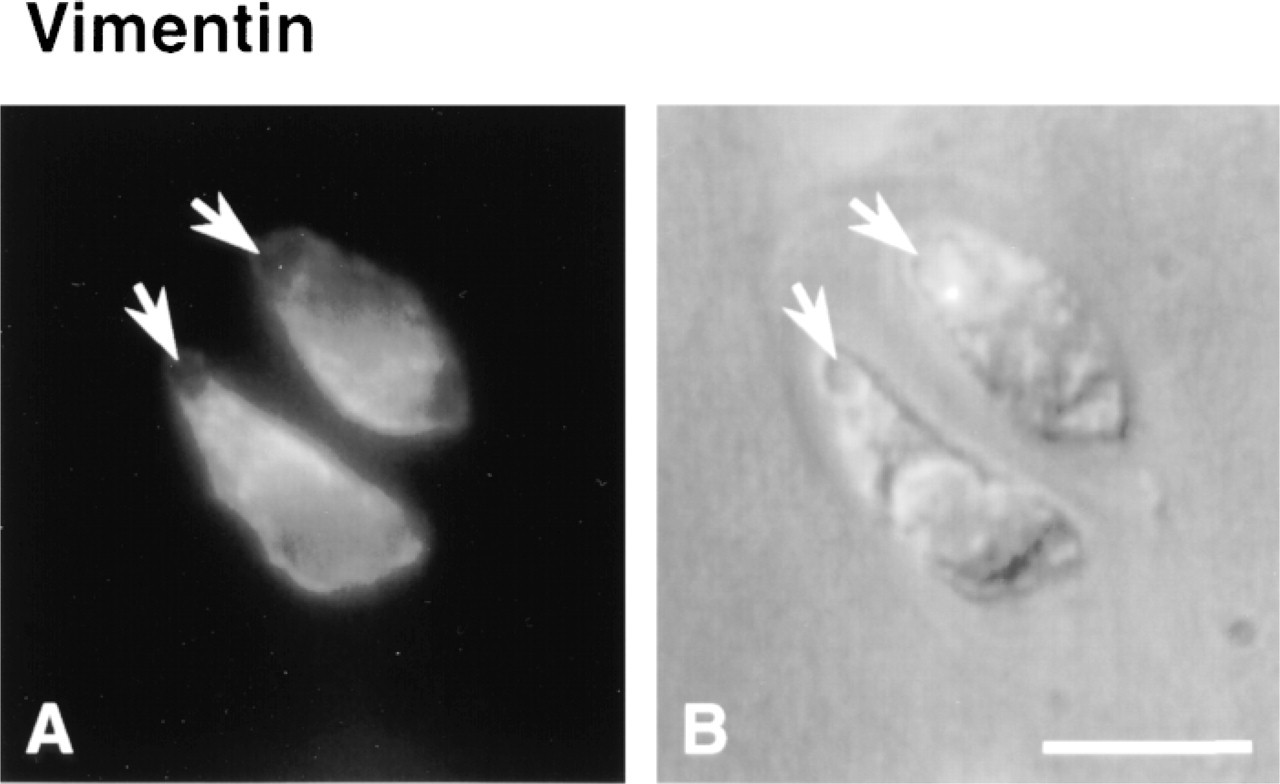
Cytoskeletal morphology within chondrocytes indicated specific structural characteristics for the three filament systems in all depth zones (Figure 3). Actin labeling was dense, punctate, and cortical, predominantly located just inside the cell membrane (Figure 4). MTs formed a loose basket-like mesh uniformly distributed in the cytoplasm (Figure 5). Vimentin IFs displayed a tighter mesh that also traversed the cytoplasm from the plasma to the nuclear membrane (Figure 6). Regions of cells that were devoid of any label (including double-labeled specimens) were often recognized (Figure 7). This absence of staining for cytoskeletal components co-localized with vacuolar structures seen using differential interference contrast (DIC) optics.
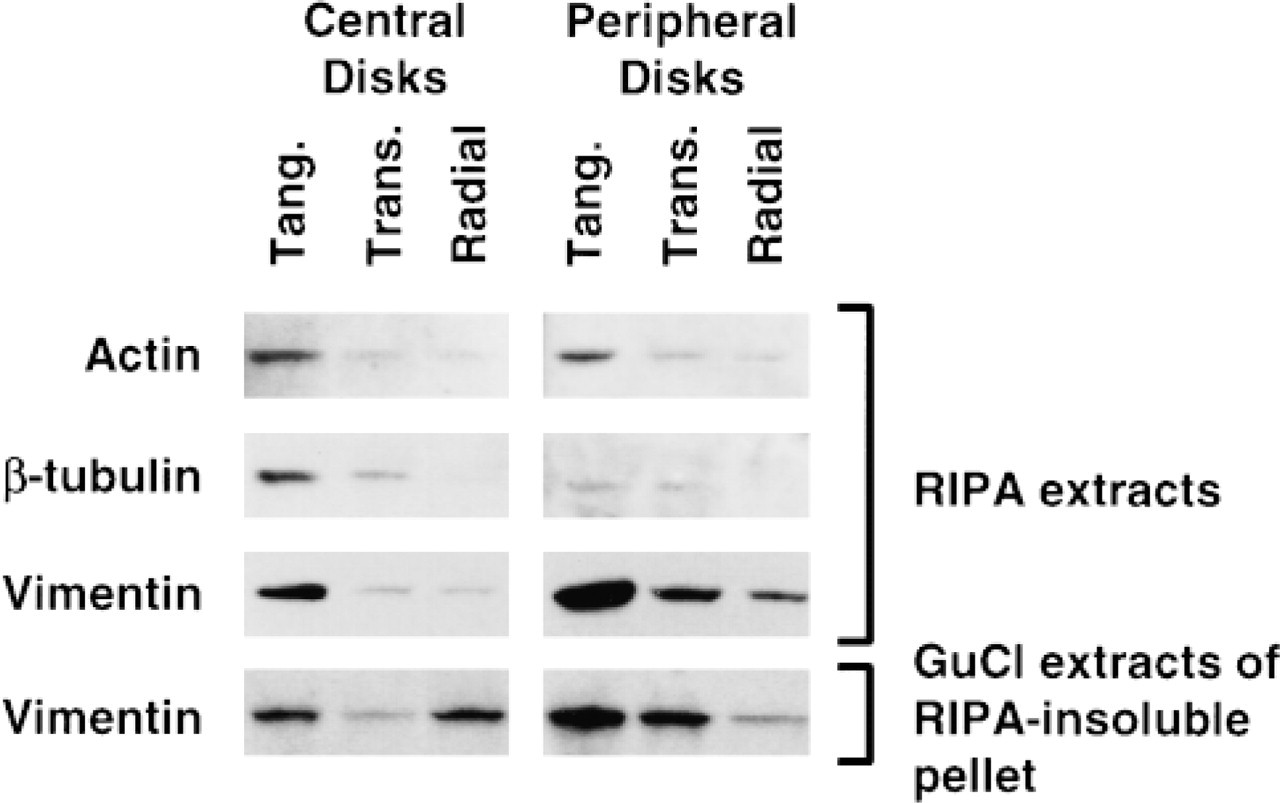
Western blotting analysis of the three different zones (tangential, transitional, and radial) performed on disks situated in the load-bearing contact region, with more central (left) or more peripheral locations (right). Disks were chosen adjacent to the disks used for low-magnification epifluorescence images (Figure 1). Equal proportions of RIPA extracts and GuCl extracts of RIPA-insoluble proteins were subjected to SDS-PAGE and immunoblotting transfer. Membranes were immunoblotted for actin, β-tubulin, and vimentin.
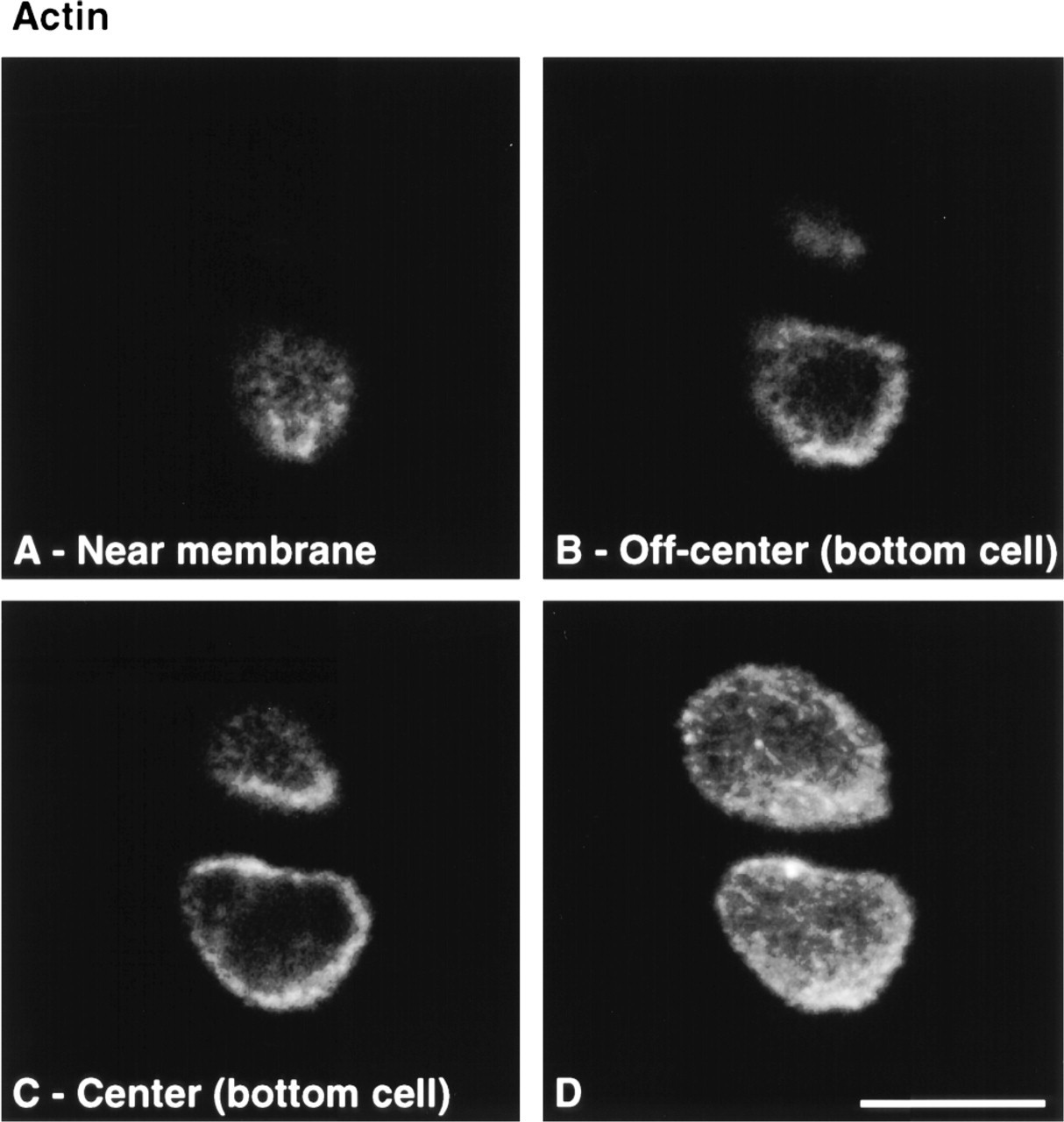
To verify that vacuolar structures were not artifactual, either induced by culture or by permeabilization and fixation procedures, DIC images from sections of fresh and cultured disks, as well as time-lapse imaging of sections undergoing permeabilization and fixation, were obtained. Vacuolar structures were visible on freshly isolated and cultured disks (Figures 8A and 8B). Time-lapse imaging under DIC also revealed that cell morphology, including vacuolar inclusion size and shape, was maintained throughout the permeabilization and fixation procedures (Figures 8C-8F). These results confirm the absence of marked morphological changes caused by culture, permeabilization, or fixation, suggesting that the observed cytoskeletal structures were representative of in situ structures. The observed vacuolar structures could be remnants of lipid droplets (Collins et al. 1965; Stockwell 1967; Eggli et al. 1988) or glycogen particles (Stockwell 1967; Eggli et al. 1988).
Discussion
Structural studies of the cytoskeleton of chondrocytes in intact tissue sections of mature articular cartilage were undertaken to elucidate the in situ morphology of these systems in the different depth zones of articular cartilage. The dependence of morphology and labeling intensity on the different zones, and the potential roles and regulation of these filament systems in chondrocytes within mature articular cartilage, were investigated. A uniform tissue preparation method was found to preserve in situ structures of all three filament systems, based on a non-ionic detergent–glutaraldehyde preparation of thick tissue sections, followed by autofluorescence blocking and glycosaminoglycan digestion to allow for antibody penetration. We observed that each filament system possessed a distinct structure and that the intensity of labeling depended on the depth zone of the labeled chondrocyte. Some functional roles and cytoskeletal regulatory factors are compatible with these observations.
The Specific Structures Pertaining to Actin, Tubulin, and Vimentin Filament Systems Highlight Certain Functional Roles of These Systems in Chondrocytes of Articular Cartilage
The three cytoskeletal filament networks, MFs, MTs, and vimentin IFs, exhibited specific structural characteristics that can be related to their distinctive functions. Actin MF labeling was dense and punctate at the peripheral cortex, similar to that reported by Durrant et al. (1999). The localization of actin just inside the cell membrane has been observed in other cell types and is consistent with roles of secretion and endocytosis. The MF network could further offer structural protection against shear stress at the cell cortex because, at low strains, MF networks offer greater resistance to shear stress compared to IF and MT networks (Janmey et al. 1991). In articular cartilage, fluid flow occurs during dynamic compression, and this flow could impose shear stress on the chondrocyte cortex via the cell membrane (Kim et al. 1995). These focal points of actin at cell membranes of chondrocytes in situ were recently seen to co-localize with vinculin (Durrant et al. 1999) and could therefore represent sites of adhesion to the ECM. This cortical distribution of actin and apparent lack of a direct link between the MFs and the nucleus argues against direct transmission of mechanical deformation to the nucleus via microfilaments (Guilak 1995). [It is also worth noting that cytochalasin D does not specifically alter cytoskeletal actin independently of nuclear actin (Karim et al. 1992) or other cytoskeletal components.] Instead, the linkage between actin MFs and the nucleus could be indirect, involving other cytoskeletal proteins such as IFs. Finally, the importance of microfilament architecture in the control of chondrocyte phenotype cannot be underestimated (Brown and Benya 1988; Mallein–Gerin et al. 1991). Unlike chondrocytes grown in a monolayer, chondrocytes in situ contained no stress fibers, further emphasizing the importance of developing these in situ observation techniques.
MTs formed a loose, basket-like mesh spanning throughout the cytoplasm. This structure is in agreement with the intracytoplasmic transport functions associated with MTs. In addition, the suggestion that MTs could serve as stabilizing elements for other cytoskeletal systems (Janmey et al. 1991; Maniotis et al. 1997) is compatible with the MT organization observed in our study.
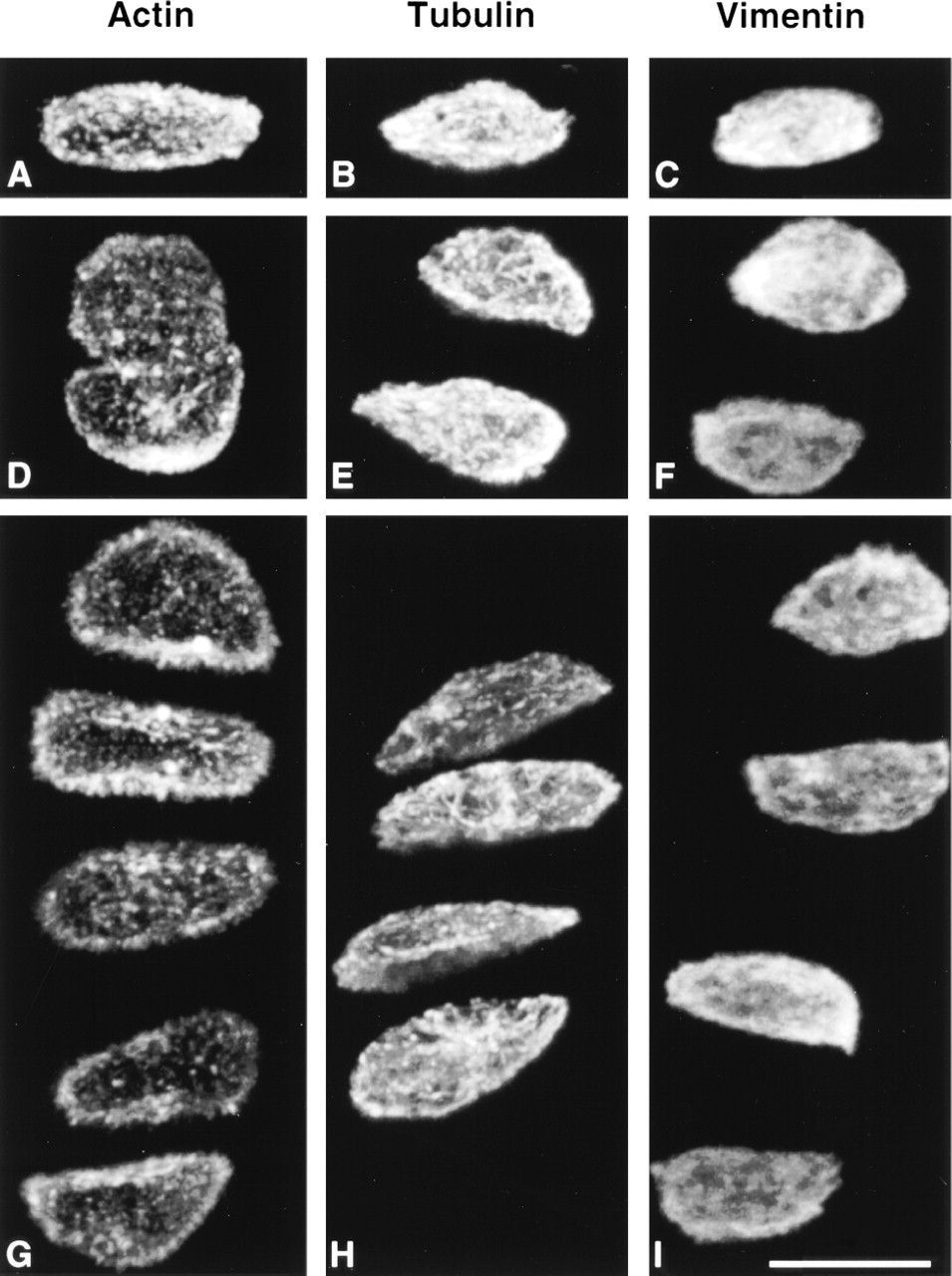
Three-dimensional reconstructions from confocal laser scanning microscopy (CLSM) of cartilage sections labeled for actin microfilaments (
Optical CLSM (
Vimentin IFs displayed a tighter, yet finer, mesh than MTs that also traversed the cytoplasm from the plasma membrane to the nuclear membrane. Intracellular mRNA transport and targeting functions of the IF cytoskeleton are well served by this spanning organization of vimentin, which is also consistent with the notion that IFs may serve as mechanical integrators of cellular space (Lazarides 1980). In agreement with its network organization spanning from the nuclear to the plasma membrane, the vimentin cytoskeleton could be part of an intracellular mechanical signal transduction system in chondrocytes. For example, mechanical forces can be transferred across the cell membrane to the cytoskeleton via β1 integrins (Wang et al. 1993), and mechanical coupling exists between cell surface integrin receptors, cytoskeletal filaments, and the nuclear scaffold, even when MFs and MTs have been destabilized (Maniotis et al. 1997). In chondrocytes, vimentin IFs can be disassembled and reassembled in response to the mechanical stimuli associated with tissue swelling during the generation of cartilage explants containing no subchondral bone (Durrant et al. 1999). In that study, vimentin IFs were disassembled by tissue swelling and then spontaneously reassembled during subsequent culture, and mechanical load was capable of limiting tissue swelling and thereby limiting vimentin IF disassembly. Furthermore, previous studies showed that changes in the synthesis of the main proteoglycan of the extracellular matrix in articular cartilage, aggre can, strongly correlated with alterations of cellular and especially nuclear structure in statically compressed cartilage explants (Buschmann et al. 1996). The vimentin cytoskeleton could therefore be implicated in mechanotransduction by directly transducing cell deformation to nuclear deformation.
Optical CLSM (
Optical CLSM (
Epifluorescence (
Depth-dependent Labeling Intensity Suggests Microenvironmental Regulation of the Chondrocyte Cytoskeleton, Including That Due to Physical Forces from Load-bearing
We verified whether histochemical gradients observed in low-magnification epifluorescence images could be confirmed by Western blotting analysis of the three zones dissected from the explants, being careful to process pairs of neighboring disks, one for immunohistochemistry and one for Western blotting. β-Tubulin in the articular cartilage explants possessed biochemical and immunohistochemical gradients that self-conferred. The gradient of actin distribution in the Western analysis was discordant with the observed homogeneous phalloidin staining pattern. It should be noted, however, that actin detected by immunoblotting will represent both filamentous and unpolymerized actin, whereas phalloidin will detect only polymerized actin MFs remaining after permeabilization. Therefore, the higher Western signal in the superficial zone could represent a higher content of actin in the soluble pool compared to deeper regions, potentially corresponding to greater remodeling in superficial zones. The distribution of RIPA-soluble vimentin in the three zones did not completely correlate with the histological staining pattern observed immunohistochemically. However, vimentin extracted with GuCl from the insoluble pellets revealed a distribution that clearly followed the histological staining intensity, suggesting that the vimentin histological staining pattern follows the protein content obtained from its least soluble fraction (RIPA-insoluble GuCl extracted). An important point in the interpretation of these Western blotting results is that although cell density decreases with depth, an increase in individual cell volume renders total cell volume (or cell volume fraction) depth-independent (Wong et al. 1996). Therefore, the intensities of protein bands in our Western blots represent total protein normalized to total cell volume in a particular depth zone. Finally, the observation by Durrant et al. (1999) concerning vimentin zonal distribution may appear to contradict our observed zonal distribution of vimentin. In that study, vimentin was concentrated in the deeper zones rather than the superficial zones as depicted in our study. Several explanations, including different fixation procedures, are possible for this different observation. However, in particular, the rat tissue in Durrant et al. (1999) is only ∼60-μm thick compared to our 1200-μm-thick bovine tissue, potentially changing significantly the regulatory (mechanical and humoral) signals seen in the various zones.
Axial deformation under static load is higher in superficial regions of mature cartilage (Schinagl et al. 1996; Kolmonen et al. 1997), where we also observed more intense labeling of cytoskeletal components. This suggests that a denser cytoskeleton may be developed in chondrocytes that are subjected to greater levels of cell deformation. Chondrocytes in these regions may require a more robust cytoskeleton to satisfy special structural and stability roles. In deeper layers, high levels of hydrostatic pressure rather than macro-molecular deformation are expected to be present, at least during dynamic loading, so that cell mechanical requirements may be fundamentally different. It is also important to note that loading conditions can change across joint surfaces, further modulating form and content of the chondrocyte cytoskeleton, as observed in our study, in which differences were observed in histochemical gradients between peripheral and more centrally located disks on the humeral head. Our disk explant system is amenable to further investigation of the roles and dynamics of the chondrocyte cytoskeleton and, in particular, its alteration due to load. Such studies may have implications for the ability of chondrocytes to structurally withstand certain levels of mechanical load and subsequently to properly maintain a functional tissue or lead to a cellular malfunction and net degeneration of articular cartilage. Furthermore, the techniques developed in our study could be applied to observe the chondrocyte cytoskeleton in cartilage tissue from animal models of disease or from humans, to aid understanding of cartilage pathology.
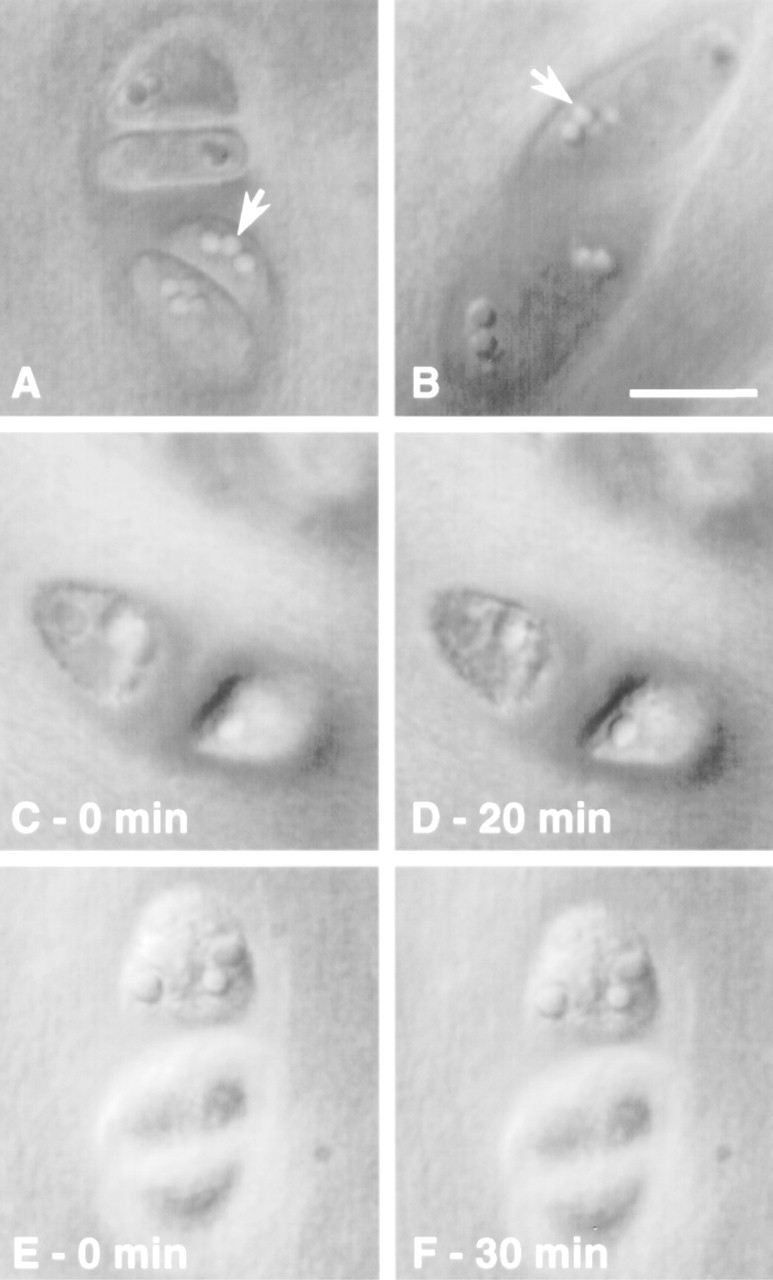
DIC images of cartilage sections. Vacuolar structures (arrows) are visible on freshly isolated disks (
Footnotes
Acknowledgements
Supported by the Arthritis Society (Canada), by Fonds pour la Formation de Chercheurs et l'Aide à la Recherche (scholarship to EL), by the Swiss National Foundation for Scientific Research, by the M.E. Müller Foundation of Switzerland, and by the Canton Basel Stadt.