
Abstract
Tripeptidyl peptidase I (TPP-I) is a lysosomal peptidase with unclear physiological function. TPP-I deficiency is associated with late-infantile neuronal ceroid lipofuscinosis (NCL), a fatal neurodegenerative disease of childhood that is characterized by loss of neurons and photoreceptor cells. We have developed two novel fluorogenic substrates, [Ala-Ala-Phe]2-rhodamine 110 and [Arg-Nle-Nle]2-rhodamine 110, that are cleaved by TPP-I in living cells. Fluorescence of liberated rhodamine 110 was detected by flow cytometry and was dependent on the level of TPP-I expression. Rhodamine-related fluorescence could be suppressed by preincubation with a specific inhibitor of TPP-I. When investigated by fluorescent confocal microscopy, rhodamine signals colocalized with lysosomal markers. Thus, cleavage of these rhodamide-derived substrates is a marker for mature enzymatically active TPP-I. In addition, TPP-I-induced cleavage of [Ala-Ala-Phe]2-rhodamine 110 could be visualized in primary neurons. We conclude that [Ala-Ala-Phe]2-rhodamine 110 and [Arg-Nle-Nle]2-rhodamine 110 are specific substrates for determining TPP-I activity and intracellular localization in living cells. Further, these substrates could be a valuable tool for studying the neuronal pathology underlying classical late-infantile NCL. This article contains online supplemental material at http://www.jhc.org. Please visit this article online to view these materials.
Keywords
T
TPP-I shows homology to the sedolisin family of carboxyl proteinases (Wlodawer et al. 2003) and is presently the only known eukaryotic member of this proteinase family. The enzyme is ubiquitously expressed in mammalian tissues. TPP-I-deficient cells contain auto-fluorescent storage material that is the characteristic feature in patient cells of all types of NCL. Further, loss of TPP-I activity results in a selective degeneration of neuronal cells and retinal photoreceptor cells in NCL patients that is macroscopically characterized as cortical and cerebellar atrophy (Hofmann and Peltonen 2001). Current research is focused on the pathogenetic mechanisms that lead to the specific neuronal death.
The activity of TPP-I in cell lysates can be measured with the peptide substrate Ala-Ala-Phe-7-(4-methyl)-coumarylamide (Vines and Warburton 1998) or with fluorescein isothiocyanate-labeled hemoglobin (Sohar et al. 2000). Recently, several other fluorescent substrates have been developed that are specifically cleaved by TPP-I in cell homogenates (Oyama et al. 2005; Tian et al. 2006) or used for histochemical TPP-I detection (Dikov et al. 2004). To study TPP-I activity in living cells, we have designed two new fluorogenic substrates, [Ala-Ala-Phe]2-rhodamine 110 and [Arg-Nle-Nle]2-rhodamine 110. Rhodamine-110-based peptide substrates are commonly used for in vivo detection of serine and cysteine proteases in mammalian cells (Leytus et al. 1983; Klingel et al. 1994; Hug et al. 1999). Ala-Ala-Phe and Arg-Nle-Nle are tripeptides that are known to precede TPP-I cleavage sites within other substrates (Vines and Warburton 1998; Tian et al. 2006). Our experiments demonstrate that the substrates [Ala-Ala-Phe]2-rhodamine 110 and [Arg-Nle-Nle]2-rhodamine 110 are preferentially cleaved by TPP-I and that the resulting fluorescent rhodamine 110 colocalizes with lysosomes.
Materials and Methods
Reagents
[Ala-Ala-Phe]2-rhodamine 110 and [Arg-Nle-Nle]2-rhoda-mine 110 were synthesized by Thermo Electron, Ulm, Germany according to published methods (Klingel et al. 1994). Aliquots of the [Ala-Ala-Phe]2-rhodamine 110 substrate were dissolved in DMSO (5 mM) and stored at −80C. The peptide inhibitors Ala-Ala-Phe-chloromethylketone and Pro-Phe-Arg-chloromethylketone and the substrate Ala-Ala-Phe-[7-amido-4-methylcoumarin] were purchased from Bachem, Weil am Rhein, Germany. Rat anti-lamp-1 antibody was obtained from BD Pharmingen (Erembodegem, Belgium); mouse antisynaptophysin (svp38) antibody came from Sigma-Aldrich (Munich, Germany), and mouse anti-MAP-2 antibody was purchased from Chemicon (Hampshire, UK). Anti-secretogranin II antibody was a gift from W. Huttner (Kromer et al. 1998). Secondary goat anti-rabbit, anti-rat, and anti-mouse antibodies conjugated to Alexa Fluor 546 or 633 were purchased from Molecular Probes (Leiden, The Netherlands). Rhodamine 110, protease inhibitors PMSF, pepstatin A, E64, leupeptin, bestatin, and apstatin, and all other reagents were purchased from Sigma-Aldrich.
Cells
Primary lymphocytes derived from NCL patients carrying the two most common pathogenic mutations, R208X and g.3556G>C in the CLN2 gene, and control lymphocytes derived from healthy individuals were immortalized by Epstein-Barr virus (EBV) transformation.
K562 cells (American Type Culture Collection; Manassas, VA) were transfected with a pcDNA3-TPP-I construct as described previously (Steinfeld et al. 2004). Stably TPP-I-expressing clones were selected in G418-containing medium. Single clones were achieved by the limited dilution procedure and extended for further analysis. Mouse fibroblasts and primary hippocampal neurons from newborn mice were prepared as described previously (Fuhrmann et al. 2002; Kasper et al. 2005).
TPP-I activity in cellular homogenates was assayed using the substrate Ala-Ala-Phe-[7-amido-4-methylcoumarin] as described previously (Vines and Warburton 1998). TPP-I-overexpressing K562 cells revealed a 16-fold higher TPP1 activity than wild-type K562 cells (data not shown).
For cytofluorometric enzyme assays, EBV-transformed lymphocytes and wild-type, TPP-I, or mock-transfected K562 cells were grown in DMEM/10% fetal calf serum (FCS) at 37C and 5% CO2 before analysis. EBV-transformed lymphocytes were washed twice in Hepes-buffered saline (HBS/ 2 mM EDTA) and then incubated with 5 μM of either [Ala-Ala-Phe]2-rhodamine or [Arg-Nle-Nle]2-rhodamine for 10 min at 37C. Washed wild-type or transfected K562 cells were preincubated with either Ala-Ala-Phe-chloromethylketone or Pro-Phe-Arg-chloromethylketone at concentrations of 0.2, 1, 5, and 25 μM for 10 min at 37C before the substrate [Ala-Ala-Phe]2-rhodamine (5 μM) was added and incubated for 10 min at 37C. After a short incubation period at 37C, the cells were cooled, the incubation volume was increased 10-fold by the addition of cold PBS, and the cells were kept at 8C for up to 8 hr before analysis in a FACSORT flow cytometer (Becton Dickinson; Mountain View, CA). Excitation was done at 488 nm and emission scanned between 500 nm and an emission peak at 529 nm (Leytus et al. 1983). Data were further analyzed and graphically displayed with the program Cell Quest (Becton Dickinson; Franklin Lakes, NJ). Additional control experiments were performed with 5 min preincubation of the following protease inhibitors: PMSF (0.5 mM), pepstatin A (150 μM), E 64 (10 μM), leupeptin (2 μM), bestatin (200 μM), and apstatin (120 μM).
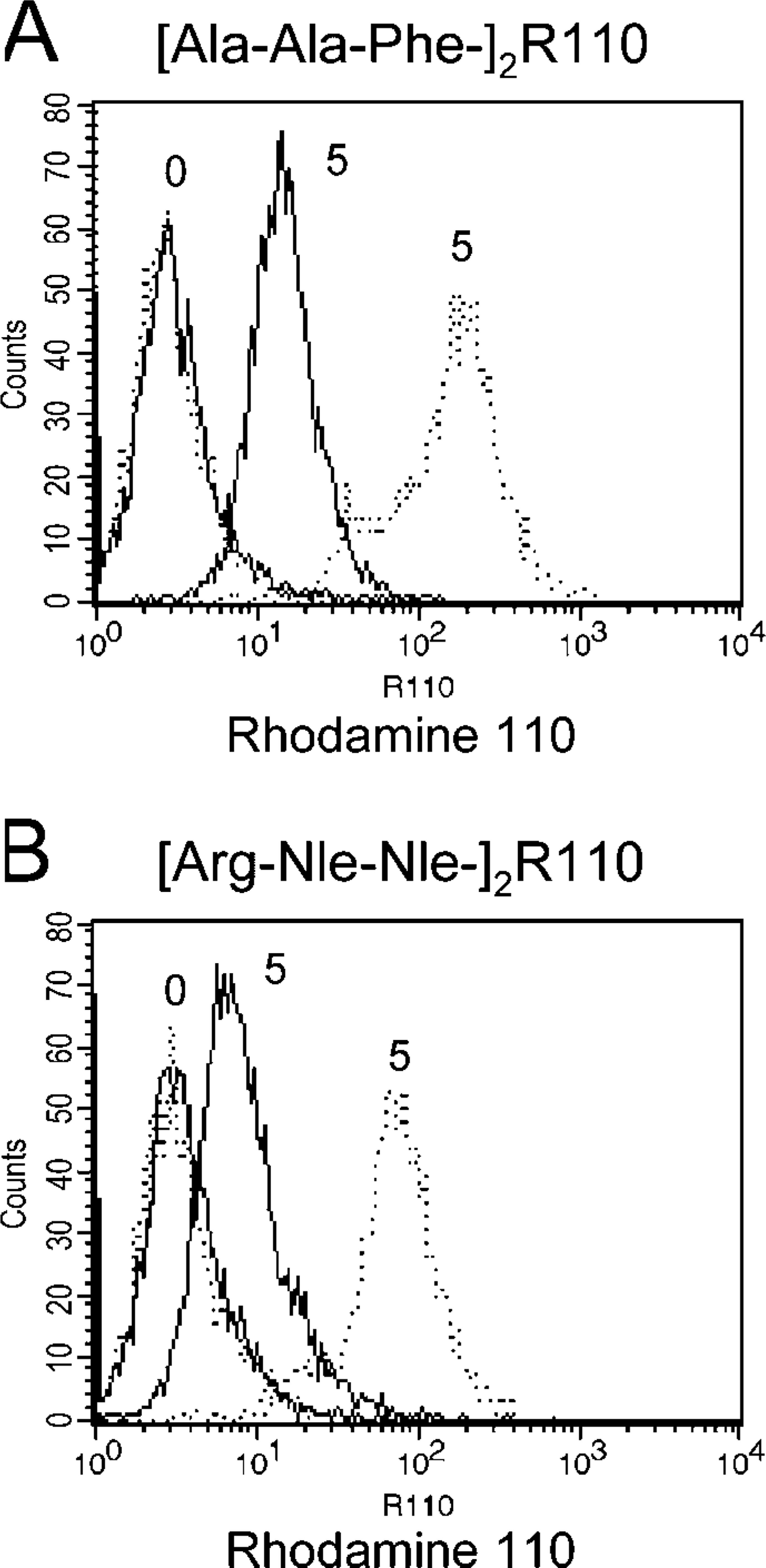
Detection of intracellular substrate cleavage in tripeptidyl peptidase I (TPP1)-/- and control lymphocytes. (
Fluorescent Confocal Microscopy
Mouse fibroblasts were cultured on glass coverslips in DMEM/10% FCS at 37C and 5% CO2. The substrate was added at a concentration of 5 μM, and cells were incubated for 60 min at 37C. Fixation and staining were carried out at 4C to minimize diffusion of the rhodamine dye. After washing with PBS, cells were fixed with 4% (w/v) paraformaldehyde in PBS for 15 min. Antibody incubation was performed for 30 min for primary and 30 min for secondary antibodies, both in PBS with 3% (w/v) bovine serum albumin and 0.1% (w/v) saponin. After three washes with PBS, coverslips were mounted with Fluoromount G (Science Services; Munich, Germany) and analyzed with a Leica NTSC laser confocal microscope (Leica; Bensheim, Germany). Rhodamine 110 was excited using the 488 nm laser line, and emission was analyzed from 500-530 nm. Primary mouse neurons were grown on coverslips coated with polyornithine. At day 9, neurons were incubated with 15 μM [Ala-Ala-Phe]2-rhodamine for 60 min at 37C. Cells were fixed, stained, and mounted for fluorescence confocal microscopy as indicated above.
Results
Using cytofluorometry, we measured the enzymatic conversion from the non-fluorescent bis-amide substrates [Ala-Ala-Phe]2-rhodamine 110 and [Arg-Nle-Nle]2-rhodamine 110 into the fluorescent free rhodamine 110. Immortalized lymphocytes derived from late-infantile NCL patients carrying pathogenic CLN2 mutations showed considerably less fluorescent signal intensity when compared with immortalized lymphocytes from control individuals (Figure 1). Incubation of control and TPP1-/- lymphoblasts with the cell supernatant containing free rhodamine 110 resulted in only minor diffusion of rhodamine 110 into cells (Supplemental Figure 1). K562 cells overexpressing TPP-I showed an increase in fluorescent signal intensity when compared with wild-type K562 cells. Preincubation of wild-type and TPP-I-overexpressing K562 cells with the specific inhibitor Ala-Ala-Phe-chloromethylketone caused a dose-dependent decrease in fluorescent signal intensity (Figures 2A and 2B). Extension of the incubation period at low temperature resulted in a considerable increase in the fluorescent signal intensities (Figures 2C and 2D). Preincubation with a non-inhibitory tripeptide-chloromethylketone compound (Pro-Phe-Arg-chloromethylketone) had only a minor effect on the fluorescent signal intensity (Figures 2E and 2F). Preincubation with the protease inhibitors PMSF, pepstatin A, E64, leupeptin, bestatin, and apstatin did not significantly change the resulting fluorescent signal intensity (data not shown). These results underline that the substrate [Ala-Ala-Phe]2-rhodamine is preferentially cleaved by intracellular TPP-I.
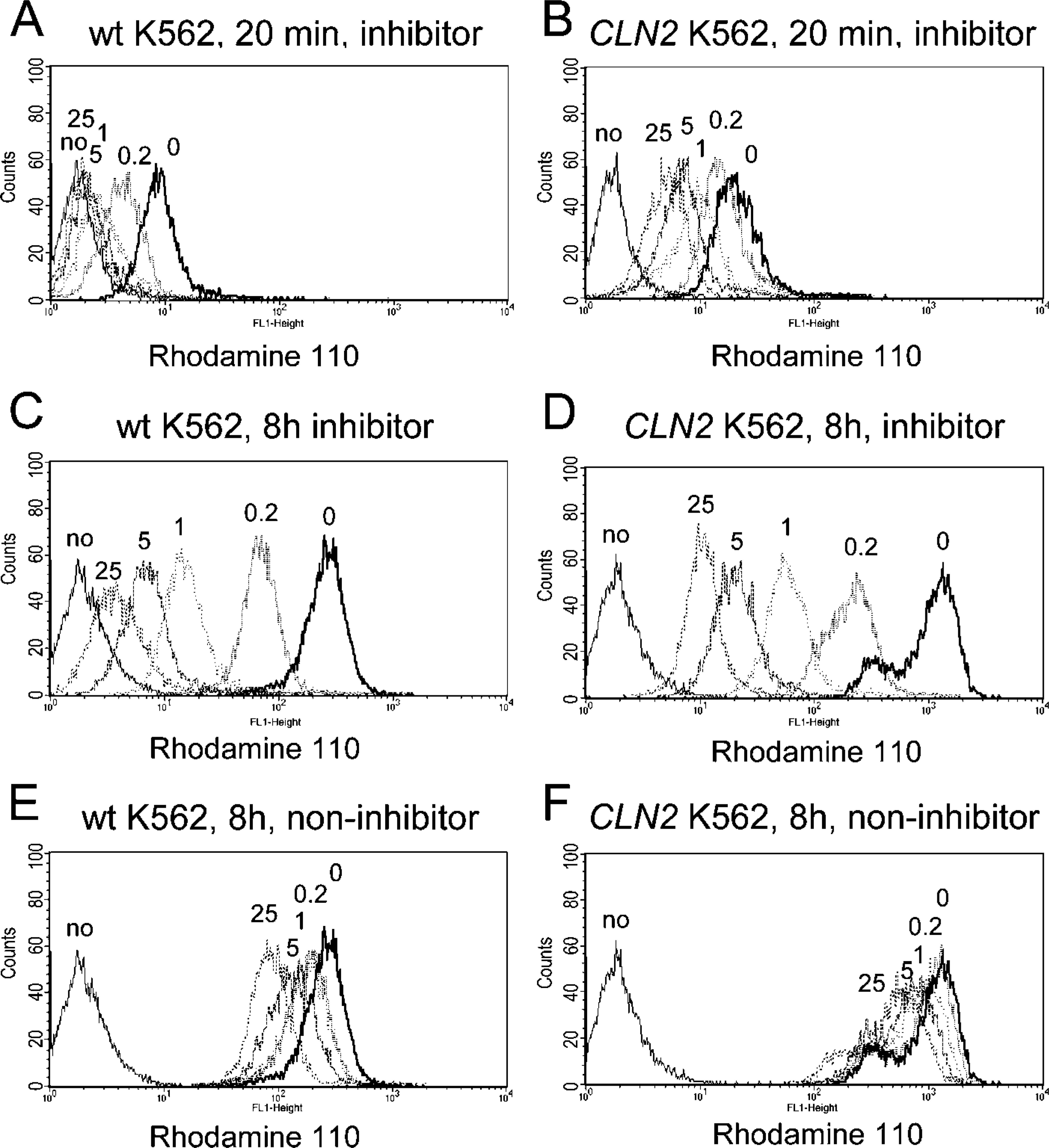
Cytofluorometric measurement of intracellular TPP-I activity. Wild-type (wt) and CLN2-transfected (CLN2) K562 cells were preincubated with the inhibitor Ala-Ala-Phe-chloromethylketone or the non-inhibitor Pro-Phe-Arg-chloromethylketone at concentrations of 0, 0.2, 1, 5, and 25 μM before addition of 5 μM substrate([Ala-Ala-Phe]2-rhodamine). An-alyses were done after 20 min (
To further confirm our findings, we investigated by confocal microscopy the intracellular localization of the cleavage of the rhodamine substrates. Mouse fibroblasts were incubated with the substrate [Ala-Ala-Phe]2-rhodamine or the substrate [Arg-Nle-Nle]2-rhodamine before they were immunostained with markers specific for subcellular organelles. Rhodamine-dependent fluorescent signals colocalized with the lysosomal marker lamp-1 (Figure 3) and thus are associated with the localization of enzymatically active TPP-I (Steinfeld et al. 2004). In addition, we demonstrated subcellular localization of cleaved [Ala-Ala-Phe]2-rhodamine in primary mouse hippocampal neurons by fluorescence microscopy (Figure 4). Intracellular fluorescent signals were distributed in a punctate pattern covering neuronal cell bodies as well as proximal dendrites. Rhodamine 110 signals colocalized with the late endosomal/lysosomal marker lamp-1, but not with synaptophysin or secretogranin II, markers of synaptic and dense-core vesicles, respectively.
Discussion
In our studies, we established a method to investigate the intracellular activity of TPP-I in living cells by fluomicroscopy. rescent cytometry and fluorescent microscopy. We present experimental data indicating that TPP-I specifically cleaves the substrates [Ala-Ala-Phe]2-rhodamine and [Arg-Nle-Nle]2-rhodamine. TPP-I-deficient lymphocytes reveal significantly lower fluorescent signal intensity than control lymphocytes. Cells overexpressing TPP-I not only showed a greater signal of free fluorescent rhodamine 110 but also had consistently higher residual enzymatic activity when preincubated with increasing concentrations of the specific inhibitor Ala-Ala-Phe-chloromethylketone. In contrast, preincubation with comparable concentrations of the structurally similar compound Pro-Phe-Arg-chloromethylketone only slightly reduced the released fluorescent signal. Studies with TPP-I purified from rat spleen demonstrated the specific inhibition by Ala-Ala-Phe-chloromethylketone (Vines and Warburton 1998). Further, several known protease inhibitors and, in particular, aminopeptidase inhibitors had no significant effect on the detectable fluorescent signal. Importantly, this included PMSF, a potent serine protease inhibitor that is known to inhibit dipeptidyl peptidase II as well as TPP-II but not TPP-I (Wilson et al. 1993; Vines and Warburton 1998; Lin et al. 2001). Additional evidence that TPP-II is not involved in the intracellular cleavage of [Ala-Ala-Phe]2-rhodamine or [Arg-Nle-Nle]2-rhoda-mine is derived from the preferential lysosomal rather than cytosolic localization of the fluorescence signal when analyzed by confocal microscopy. It should be stressed that rhodamine 110 can penetrate membranes more easily than the non-cleaved bis-amide compounds. Therefore, the intracellular measured fluorescent signals are dependent on the cleavage of rhodamine substrates as well as on the diffusion of released rhodamine 110 into the extracellular space. To minimize the loss of released rhodamine 110, prolonged incubations should be performed at low temperature. However, we cannot exclude that the internalized substrates [Ala-Ala-Phe]2-rhodamine and [Arg-Nle-Nle]2-rhodamine are preferentially targeted to or better retained in lysosomes, and hence, tripeptidyl peptidase activities within non-lysosomal compartments are overall less-efficiently detected than are lysosomal compartments.
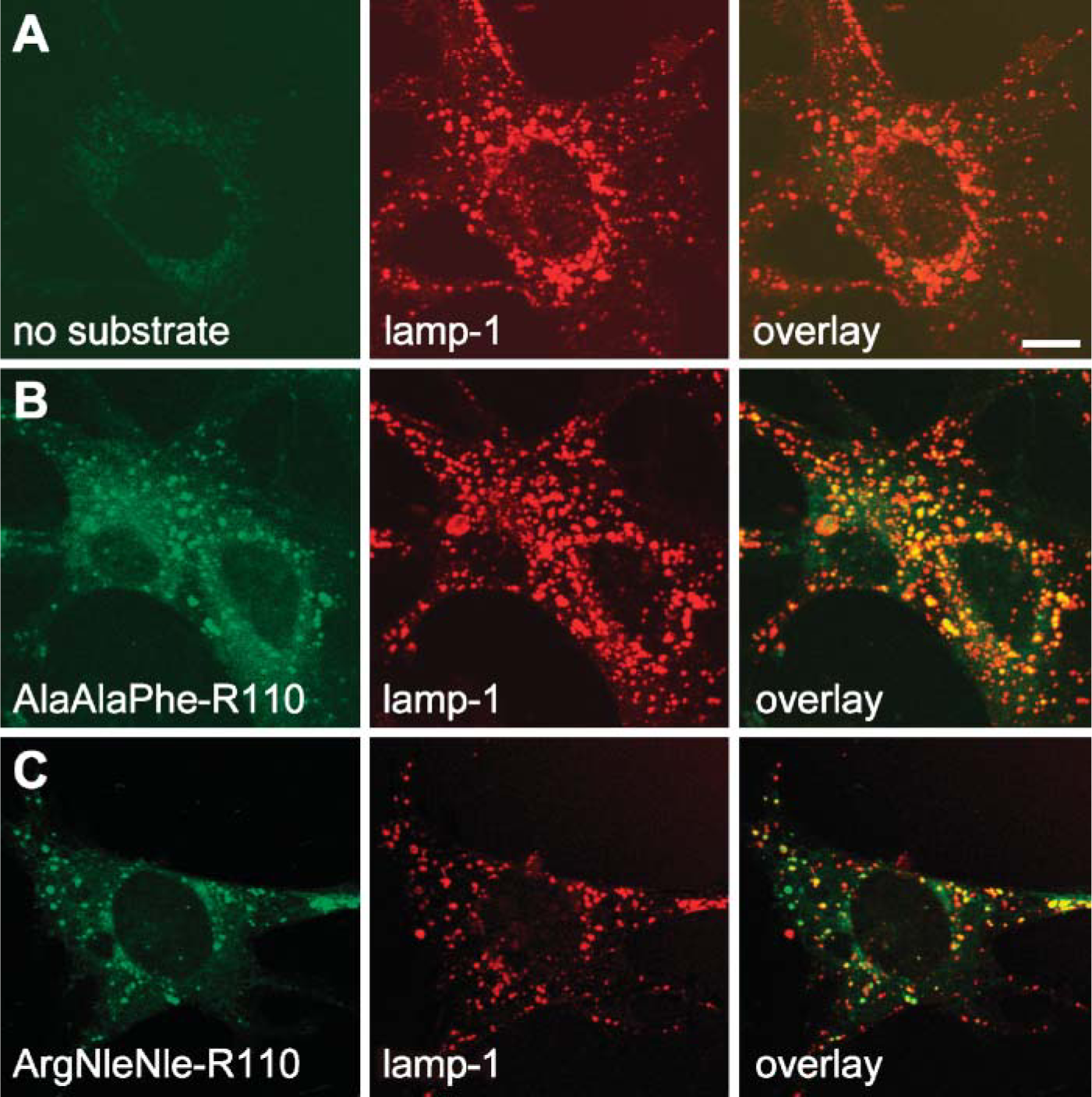
Localization of TPP-I activity by fluorescent confocal Mouse fibroblasts were incubated with the substrate [Ala-Ala-Phe]2-rho-damine or [Arg-Nle-Nle]2-rhodamine for 1 hr at 37C, then cells were fixed and immunostained for the lysosomal marker lamp-1. (
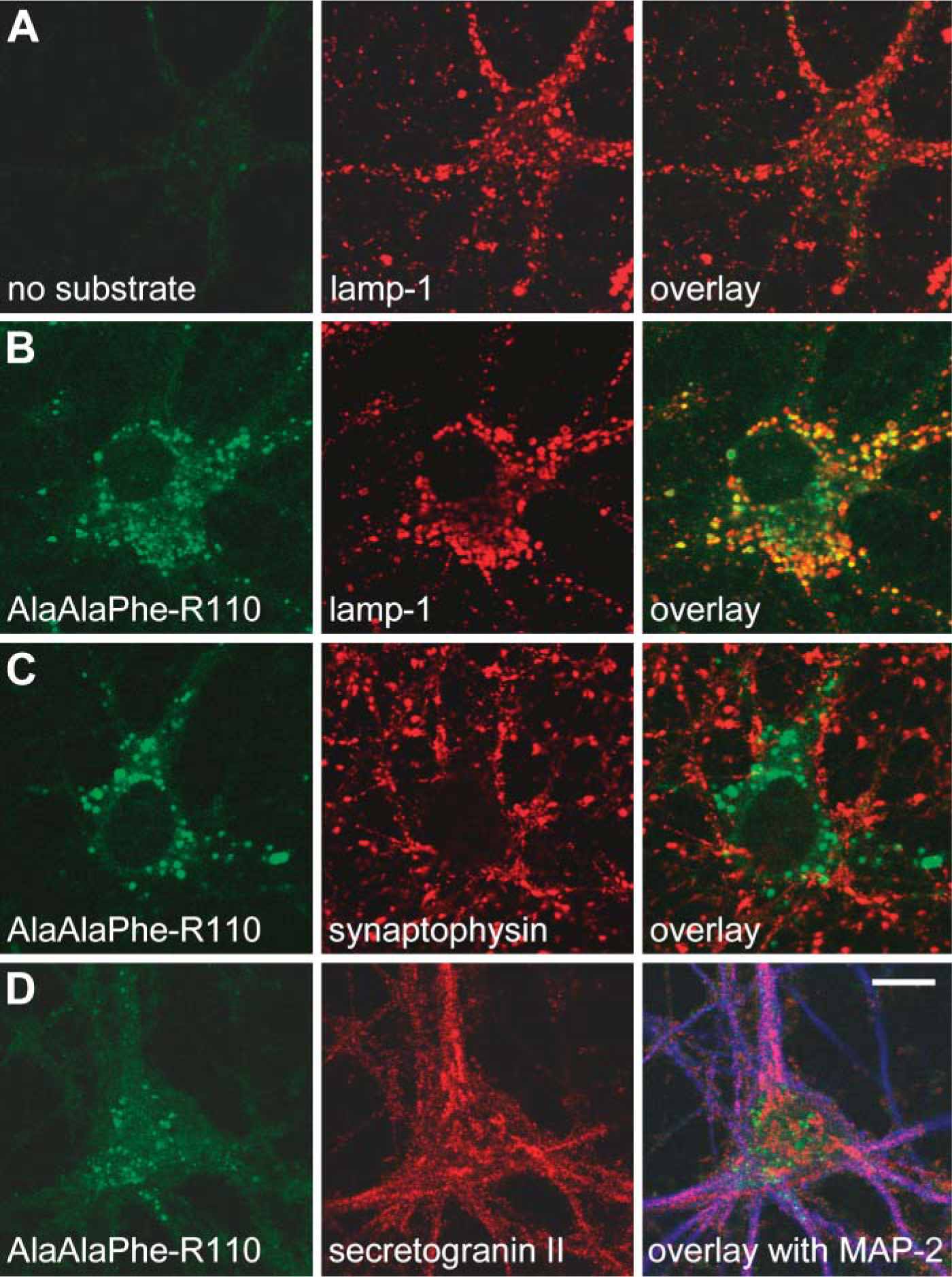
Localization of TPP-I activity in mouse hippocampal neurons. Primary mouse neurons were incubated with substrate [Ala-Ala-Phe]2-rhodamine for 1 hr at 37C, immunostained, and analyzed by fluorescent confocal microscopy. (
In summary, we have developed novel fluorogenic substrates for the analysis of intracellular TPP-I activity that are helpful in the study of the enzymatic activity of TPP-I in living cells and also its intracellular localization. These novel substrates are not only applicable to patients' lymphocytes and cultured fibroblasts but also to primary neurons. Thus, they will be an important future tool in the further elucidation of the specific pathomechanisms of neuronal degeneration associated with TPP-I deficiency and a key feature in understanding the disease pathogenesis of late-infantile NCL.
Footnotes
Acknowledgements
We thank Prof. Dr. Schneppenheim and Mrs. Grabowski, Department of Pediatric Oncology, University Hospital Eppendorf, Hamburg for their support and their technical assistance in the usage of the FACSORT flow cytometer. We are grateful to Thomas Jentsch, in whose lab part of this work was done, and to W. Huttner for the secretogranin II antibody.