
Abstract
Fluorogenic substrates [Ala-Pro]2-cresyl violet and Ala-Pro-rhodamine 110 have been tested for microscopic detection of protease activity of dipeptidyl peptidase IV (DPPIV) in living cells. DPPIV activity is one of the many functions of the multifunctional or moonlighting protein CD26/DPPIV. As a model we used Jurkat cells, which are T-cells that lack CD26/DPPIV expression, and CD26/DPPIV-transfected Jurkat cells. Ala-Pro-rhodamine 110 is not fluorescent, but after proteolytic cleavage rhodamine 110 fluoresces. [Ala-Pro]2-cresyl violet is fluorescent by itself but proteolytic cleavage into cresyl violet induces a shift to longer wavelengths. This phenomenon enables the simultaneous determination of local (intracellular) substrate and product concentrations, which is important for analysis of kinetics of the cleavage reaction. [Ala-Pro]2-cresyl violet, but not Ala-Pro-rhodamine 110, appeared to be specific for DPPIV. When microscopic analysis is performed on living cells during the first minutes of the enzyme reaction, DPPIV activity can be precisely localized in cells with the use of [Ala-Pro]2-cresyl violet. Fluorescent product is rapidly internalized into submembrane granules in transfected Jurkat cells and is redistributed intracellularly via internalization pathways that have been described for CD26/DPPIV. We conclude that [Ala-Pro]2-cresyl violet is a good fluorogenic substrate to localize DPPIV activity in living cells when the correct wavelengths are used for excitation and emission and images are captured in the early stages of the enzyme reaction.
Keywords
N
Various types of synthetic fluorogenic substrates are available for detection of activity of proteases in living cells (for review see Boonacker and Van Noorden 2001). Usually the substrate consists of one or more sequences of amino acids and a leaving group. The amino acid sequence(s) in the substrate determine(s) to a large extent the specificity for a protease on the basis of interactions between the amino acid sequence and the active site of the enzyme. Specificity of synthetic substrates is obtained by studying variations in the specificity constant on substitutions or alterations of single amino acids in the substrate (Leytus et al. 1983b). Figure 1 shows two examples of fluorogenic substrates to detect dipeptidyl peptidase IV (DPPIV) activity. Substrate specificity is based on the amino acid sequence alanine-proline (Ala-Pro) (Demuth and Heins 1995) and the leaving groups are either cresyl violet (Figure 1A) or rhodamine 110 (Figure 1B). Rhodamine 110 is not fluorescent when amino acids are attached, as it is completely quenched (Leytus et al. 1983a) and becomes fluorescent after proteolytic release, whereas cresyl violet is always fluorescent even when amino acid sequences are attached but fluorescence is shifted to a longer wavelength when the amino acid sequences are removed (Boonacker and Van Noorden 2001). However, the amino acid sequence does not completely determine substrate specificity because intrinsic chemical properties of fluorophores in synthetic substrates may also affect the reactivity of the substrate with the protease (Chase and Shaw 1969; Boonacker and Van Noorden 2001; Lorey et al. 2002). This phenomenon can be due to steric hindrance or to particular chemical properties of the fluorophore. Assay conditions also determine reactivity of a synthetic substrate with the enzyme. When intact cells or tissues are used, homologous proteases may interfere with the cleavage of the synthetic substrate.
Proline is a unique amino acid because of its cyclic structure. This specific conformation of proline imposes many restrictions on cleavage of peptides and proteins that contain proline. A large series of physiologically important biomolecules contain proline in the penultimate position and their biological properties are highly regulated by this proline motif. Only a limited number of proteases can cleave proline-containing peptides (Yaron and Naider 1993; Demuth and Heins 1995). Until recently, it was believed that CD26/DPPIV was one of the very few proteases that can cleave off a terminal dipeptide from proteins with proline in the penultimate position, but it has been recently shown that this is not the case (Boonacker and Van Noorden 2003). It appeared that a series of DPPIV homologues exist. For example, DPPII acts preferentially at acidic pH (Lojda et al. 1976) and prolyl carboxypeptidase shows similar activity as CD26/ DPPIV (Rawlings and Barrett 1996; Sedo and Malik 2001). Recently, it was found that specific inhibitors of post-proline-cleaving proteases cause apoptosis in quiescent lymphocytes independently of CD26/DPPIV. These results lead to the discovery of another proline-specific peptidase, quiescent cell proline dipeptidase (Chiravuri et al. 1999; Underwood et al. 1999). Quiescent cell proline dipeptidase was cloned from a Jurkat T-cell line that lacks CD26/DPPIV expression. The putative active site residues serine, aspartate, and histidine of quiescent cell proline dipeptidase shows an ordering of the catalytic triad similar to that in the post-proline-cleaving exopeptidases prolyl carboxypeptidase and CD26/DPPIV (David et al. 1993; Hooper et al. 2001). DPPIV homologues are listed in Sedo and Malik (2001) and in Boonacker and Van Noorden (2003). To study activity of DPPIV and its homologues in a specific manner, the characteristics of fluorogenic substrates have to be known.
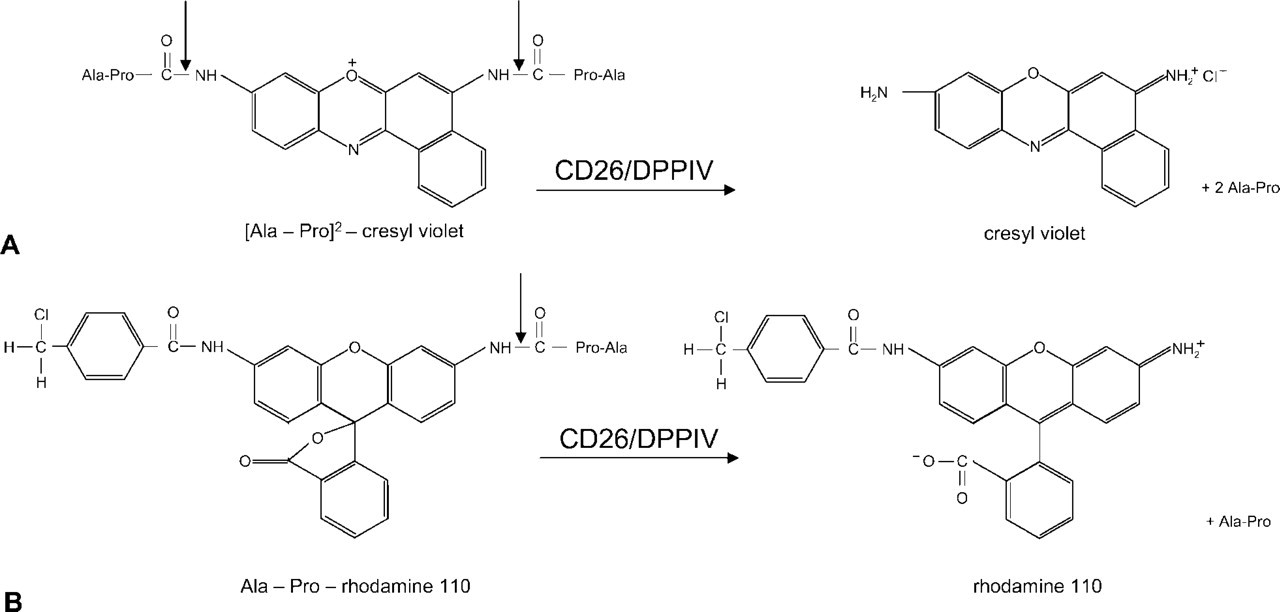
Structural formulas of the bisubstituted [Ala-Pro]2-cresyl violet (
In this study we compared the reactivity of two synthetic substrates that contain the same amino acid sequence but different fluorogenic leaving groups, cresyl violet (Van Noorden et al. 1997b) and rhodamine 110 (Leytus et al. 1983a,b; Figure 1), in the analysis of DPPIV activity after purification and in living Jurkat cells lacking CD26/DPPIV and CD26/DPPIV-transfected Jurkat cells (Hegen et al. 1993a,b; Tanaka et al. 1993) to establish to what extent the Ala-Pro-containing substrates are specific for DPPIV. We found that the two substrates show different specificities towards DPPIV-like proteases, depending on the leaving group. [Ala-Pro]2-cresyl violet is specific for DPPIV activity whereas Ala-Pro-rhodamine 110 is not. We concluded that [Ala-Pro]2-cresyl violet is the preferred substrate for microscopic analysis of DPPIV activity in living cells. Because of the highly dynamic character of (enzymatic) processes in living cells and to avoid diffusion of cresyl violet from sites in cells where it is formed, microscopic images have to be taken during the first few minutes of incubation of cells.
Materials and Methods
Jurkat Cell Lines
Jurkat cells (clone E6-1; American Type Culture Collection, Manassas, VA), which lack CD26/DPPIV expression, were used as well as Jurkat cells transfected with CD26/DPPIV (Hegen et al. 1993a,b). This model system enables the determination of specificity of substrates for DPPIV. Clone E6-1 was cultured in Iscove's modified Dulbecco's medium (IMDM; Bio-Whitaker, Walkersville, MD) supplemented with 10% fetal calf serum (FCS), whereas the CD26/DPPIV transfectants were grown in Dulbecco's modified medium supplemented with 10% FCS, containing glutamine (1 mM) and Geneticin G418 (1 mg/ml; Invitrogen, Carlsbad, CA) to maintain the selection for the CD26/DPPIV construct (Hegen et al. 1993a,b; Tanaka et al. 1993). Before DPPIV activity measurements, cells were washed twice in cold PBS. Intact cells were kept on ice before mixing with the incubation media. Suspensions of 4 × 106 cells/ml were used. Permeabilization of cells was performed by ultrasonic treatment (three times, 5 sec each).
Western Blotting and Zymography of DPPIV Activity
To test specificity of substrates, samples containing soluble CD26/DPPIV (sCD26/DPPIV) derived from human seminal fluid were run on polyacrylamide gels and submitted to Western blotting or were incubated in the presence of a series of synthetic DPPIV substrates. sCD26/DPPIV was enriched by isolating prostasomes from human seminal fluid, as described by Ronquist and Brody (1985). The pelleted prostasome fraction was resuspended in 20 mM Tris-HCl (pH 7.4) containing 1% Triton X-100 for 1 hr at 4C. Samples were treated ultrasonically three times for 5 sec each. Subsequently, 3 × Laemmli buffer, consisting of 150 mM Tris-HCl (pH 6.8), 30% glycerol, 6% SDS, and 0.3% bromphenol blue, was added and the samples were heated to 37C for 5 min. Equal amounts of proteins were transferred onto the gels. After electrophoresis at 20 mA per gel at 4C, gels were washed twice at room temperature (RT) with PBS containing 2.5% Triton X-100 for 30 min to remove SDS.
DPPIV activity was detected with the following substrates: 20 μM [Ala-Pro]2-cresyl violet (Enzyme Systems Products and Prototek, Livermore, CA; Van Noorden et al. 1997b), 20 μM Ala-Pro-AFC (Enzyme Systems Products; Smith et al. 1997), or 20 μM Ala-Pro-MNA (Enzyme Systems Products; Smith et al. 1997) in 100 mM cacodylate buffer (pH 7.4), or 20 μM Ala-Pro-rhodamine 110 (Molecular Probes, Eugene, OR; Leytus et al. 1983a,b) in 10 mM Tris-HCl buffer (pH 7.5). In the case of Ala-Pro-MNA, a coupling agent was added as well, either 1 mM nitrosalicylaldehyde (Merck, Darmstadt, Germany; Van Noorden and Frederiks 1992) or Fast Blue B (FBB4; Serva, Heidelberg, Germany; Van Noorden and Frederiks 1992). After 20 min of incubation, DPPIV activity was determined using a STORM Fluor-imager (Molecular Dynamics; Sunnyvale, CA) and Image Quant Software Package (Molecular Dynamics) or, in the case of the chromogenic substrates, scanned on a flatbed scanner using white light.
Samples were also subjected to Western blotting to determine CD26/DPPIV protein expression. Proteins were blotted at 4C and 100 V to nitrocellulose paper for 1 hr. After washing in PBS containing 0.05% Tween, blots were blocked overnight with 5% BSA in PBS. Blots were incubated with anti-CD26 antibody Ta1 (Central Laboratory for Blood Transfusion; Amsterdam, The Netherlands) at a dilution of 1:100 in the blocking buffer containing 0.05% Tween for 1 hr, then washed twice in PBS containing 0.05% Tween, followed by 1-hr incubation with monoclonal horseradish peroxidase-conjugated goat anti-mouse IgG (Nordic; Tilburg, The Netherlands) in a dilution of 1:200 in blocking buffer. Then blots were washed again in PBS. Finally, after 10 min of incubation with Lumi-Light Western blotting substrate (Roche Diagnostics; Mannheim, Germany), chemiluminescence was detected by the Lumi-Imager (Roche Diagnostics).
Fluorescence Spectra of [Ala-Pro]2-Cresyl Violet Substrate and Cresyl Violet Product
Fluorometric analysis of concentrations of [Ala-Pro]2-cresyl violet and cresyl violet acetate (Enzyme Systems Products) was performed on a LS 50 fluorescence spectrometer (Perkin–Elmer; Gouda, The Netherlands). [Ala-Pro]2-cresyl violet (20 μM) and cresyl violet acetate (20 μM) were measured separately and mixed in 1:1 and 1:4 ratios. Excitation was performed at 496 nm and 591 nm and fluorescence emission was monitored at wavelengths ranging from 500 nm to 700 nm.
Thin-Layer Chromatography of [Ala-Pro]2-Cresyl Violet Substrate and Cresyl Violet Product
To demonstrate fluorescent components in batches of substrate and product, separation by thin-layer chromatography (TLC) was performed, using silica gel-coated TLC plates (Merck; Darmstadt, Germany) as the stationary phase and methanol as the mobile phase. Equal amounts of substrate and product molecules were dissolved in methanol and applied to TLC plates. The plates were placed upright in running fluid and ran until the front of the running fluid had reached the end of the plate. The plates were dried and stored for further analysis. Images of the plates were made using white light and a digital camera (Coolpix; Nikon, Tokyo, Japan) to demonstrate the color change from orange to violet as is also observed with the naked eye when living cells are incubated with the substrate in a cuvette. Then the plates were illuminated with UVA light (320–380 nm), and photographed with a UV-blocking filter (>500 nm) using the same camera.
Fluorospectrometric Analysis of DPPIV Activity
Living Jurkat cells and CD26/DPPIV-transfected Jurkat cells were harvested and DPPIV activity was determined by fluorospectrometry. Before DPPIV activity measurements, cells were washed twice in cold PBS. Intact cells were kept on ice before mixing with the enzyme incubation media. Parts of the cells were lysed by ultrasonic treatment three times for 5 sec. Incubations were started at t=0 by suspending Jurkat cells or transfected Jurkat cells in prewarmed PBS supplemented with 1.7 mM CaCl2 and 1 mM MgCl2 at 37C containing 0–40 μM of the DPPIV substrate [Ala-Pro]2-cresyl violet or Ala-Pro-rhodamine 110. Actual amounts of ester bonds that are cleaved are twice as high as that of the free fluorochrome in the case of [Ala-Pro]2-cresyl violet, because two amino acid sequences are spliced off per cresyl violet fluorochrome (Figure 1). Incubations were carried out at 37C by using prewarmed PBS to which the substrate was added just before the start of the incubation. The cell suspension was kept on ice before the incubation and added 30 sec after the start of recording. For each measurement, 4 × 106 living cells, or its equivalent in cell lysates, were incubated in a final volume of 1200 μl prewarmed PBS containing 0–40 μM [Ala-Pro]2-cresyl violet or Ala-Prorhodamine 110 substrate. Fluorometric analysis was performed on an LS 50 fluorescence spectrometer under continuous magnetic stirring to keep cells in suspension. Cuvettes with an excitation light path of 1 cm and an emission light path of 4 mm were used. Excitation for cresyl violet was performed at 591 nm with a slit width of 10 nm and emission was detected at 628 nm with a slit width of 10 nm (Boonacker and Van Noorden 2001). Rhodamine 110 was excited at 491 nm with a slit width of 10 nm and emission was detected at 529 nm with a slit width of 10 nm (Boonacker and Van Noorden 2001). Fluorescence was measured continuously during the first 4 min of incubation. Because both synthetic substrates are not completely stable in aqueous solution, a spontaneous small but continuous increase of fluorescence is detected when the substrates are incubated in an aqueous solution. By starting the incubation with the medium containing the substrate only, fluorescence of Ala-Pro-cresyl violet due to spontaneous formation of product (both cresyl violet and rhodamine 110) was measured. These values were subtracted from fluorescence values obtained after the cells were added to the medium, thus correcting for the spontaneous nonspecific increase in fluorescence. Fluorescence values were plotted against time.
Confocal Microscopic Analysis of DPPIV Activity in Living Cells
Confocal analysis was performed to localize DPPIV-like activity in living Jurkat and transfected Jurkat cells on a Leica SP2 AOBS confocal microscope (Leica Microsystems; Mannheim, Germany). In case of the use of [Ala-Pro]2-cresyl violet, fluorescence of both substrate and product was analyzed with settings of the AOBS for 488-nm excitation of the substrate and 594-nm excitation of the product. Fluorescence was measured at 515–576 nm and 613–734 nm for substrate and product, respectively. Each focal plane was scanned in a sequential manner in time, xyt, or in volume, xyz, for end-point images. In case of the use of Ala-Prorhodamine 110, excitation was performed at 496 nm and emission was measured at 550–580 nm. Living Jurkat cells were incubated on the stage of a confocal microscope in Dulbecco's modified medium containing 0.1 mg/ml Geneticin G418 and 1 mM glutamine and 10% FCS. This medium allowed prolonged incubations for longer periods of time without any significant cell damage. The suspended cells were incubated in 1 ml incubation medium on the microscope stage on a γ-irradiated glass bottom in a poly-
Results
Reactivity of the various Ala-Pro-containing synthetic substrates with CD26/DPPIV was demonstrated with the use of an enriched fraction of sCD26/DPPIV that was submitted to native gel electrophoresis and Western blotting. Figure 2 demonstrates a similarly stained banding pattern with two major bands of DPPIV activity obtained with all substrates tested. Western blotting after staining with the anti-CD26/DPPIV anti-body shows a similar banding pattern as well (Figure 2). These findings indicate that all substrates identify DPPIV activity and demonstrate the different isoforms of DPPIV/CD26 as they occur in prostasomes. It should be noted that intensities cannot be compared directly due to the limited number of excitation wavelengths available on the scanner used and thus to the more or less optimal excitation of the fluorophores.
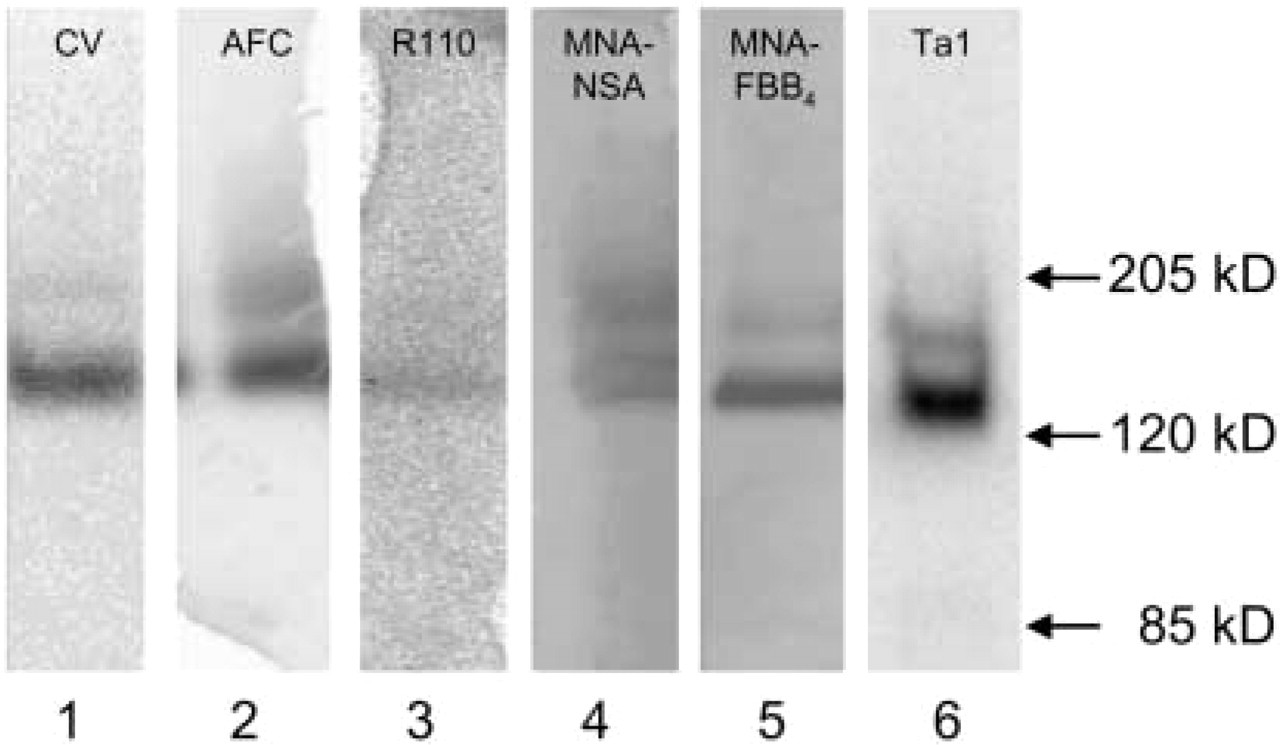
DPPIV activity as detected after gel electrophoresis of sCD26/DPPIV enriched from prostasomes in human seminal fluid with different synthetic substrates for DPPIV (Lanes 1–5) and CD26/ DPPIV protein after Western blotting (Lane 6). DPPIV activity is detected in Lane 1 with [Ala-Pro]2-cresyl violet (CV), in Lane 2 with Ala-Pro-AFC (AFC), in Lane 3 with Ala-Pro-rhodamine 110 (R110), in Lane 4 with Ala-Pro-MNA in combination with NSA as coupling agent (MNA-NSA), in Lane 5 with Ala-Pro-MNA in combination with FBB4 as coupling agent (MNA-FBB4), and CD26/DPPIV is detected in Lane 6 with an anti-CD26 monoclonal antibody (Ta1). Molecular weight markers are presented at right. Native CD26/DPPIV is located just above the arrow of 120 kD, which is in agreement with the molecular weight of 140 kD of the native protein. Due to the limited number of excitation wavelengths on the storm scanner, emission intensities cannot be directly compared with respect to reactivity.
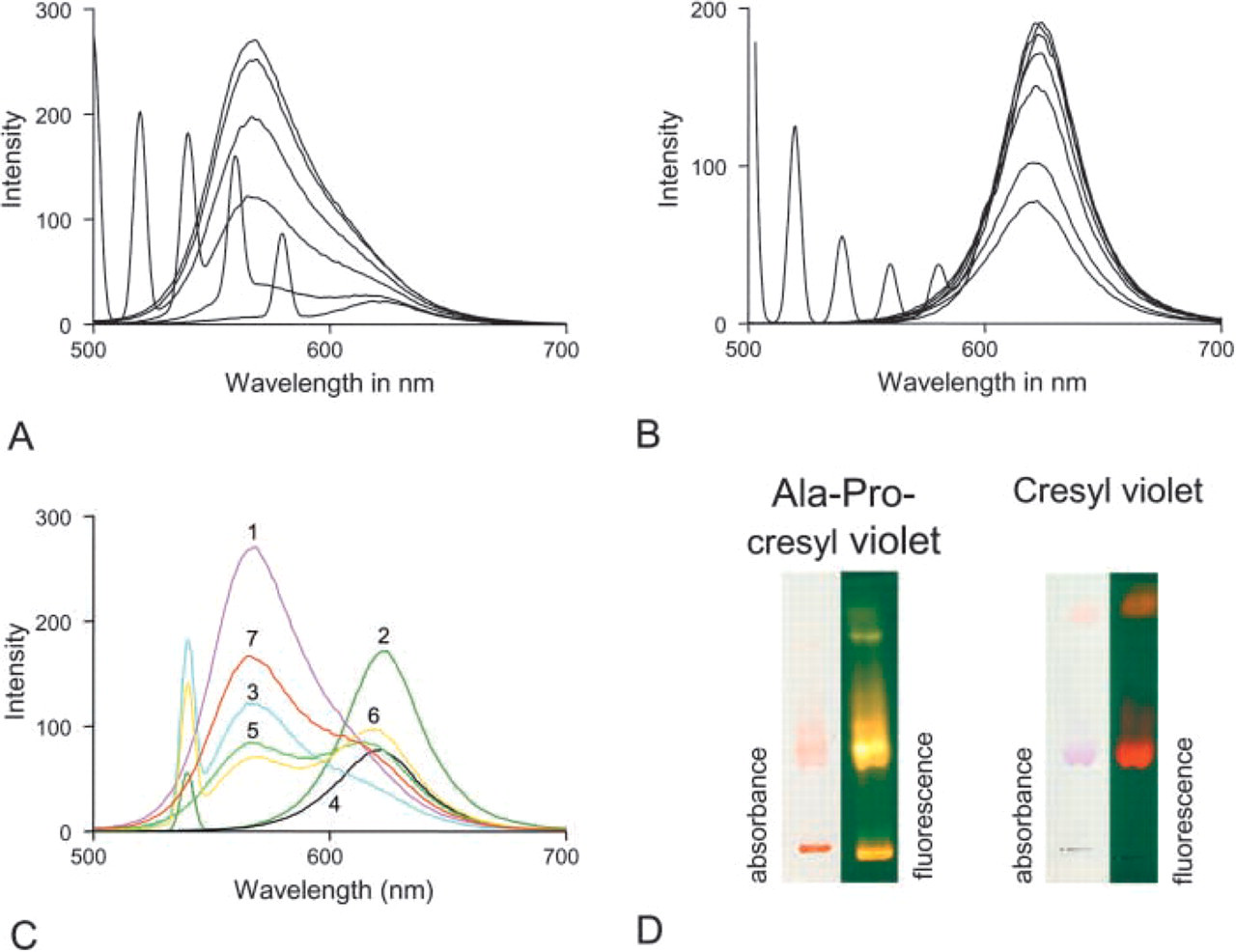
Emission spectra of [Ala-Pro]2-cresyl violet (
To characterize the shift in fluorescence that occurs after proteolytic cleavage of [Ala-Pro]2-cresyl violet into cresyl violet and Ala-Pro dipeptides, emission and excitation spectra were determined for both [Ala-Pro]2-cresyl violet substrate and cresyl violet product separately (Figures 3A and 3B), and in mixtures with different ratios (1:1, 1:4; Figure 3C). Solutions of 20 μM were made of both substrate and product, and emission spectra were determined. When light of 480, 500, 520, or 540 nm was used for excitation, [Ala-Pro]2-cresyl violet showed one emission peak at 570 nm (Figure 3A). When excitation light of 560 or 580 nm was used, emission was at a higher wavelength but insignificant. When light of 480, 500, 520, 540, 560, or 580 nm was used for excitation, the proteolytic product cresyl violet was fluorescent with one emission peak only at 628 nm (Figure 3B). Because of the broad fluorescence spectrum of the substrate [Ala-Pro]2-cresyl violet, fluorescence of the product could not be detected specifically when excitation light was used at a wavelength <570 nm because the substrate caused too high levels of background fluorescence (Figure 3C). In practice, substrate concentrations are much higher than product concentrations in cells when initial stages of enzyme reactions are measured. On the other hand, the 570-nm fluorescence of [Ala-Pro]2-cresyl violet substrate can be used to determine amounts of substrate present in both cells and/or incubation medium to enable the determination of kinetics of [Ala-Pro]2-cresyl violet cleavage on the basis of local substrate concentrations and production of cresyl violet. The spectra illustrate that for specific measurement of fluorescence of the cresyl violet product at 628 nm, excitation at 591 nm must be used to avoid interference by fluorescence of [Ala-Pro]2-cresyl violet (Figure 3C). In conclusion, when an excitation wavelength of 591 nm is used, fluorescence at 628 nm is specific for the product. The substrate concentration can be measured using excitation at 488 nm and emission at 570 nm.
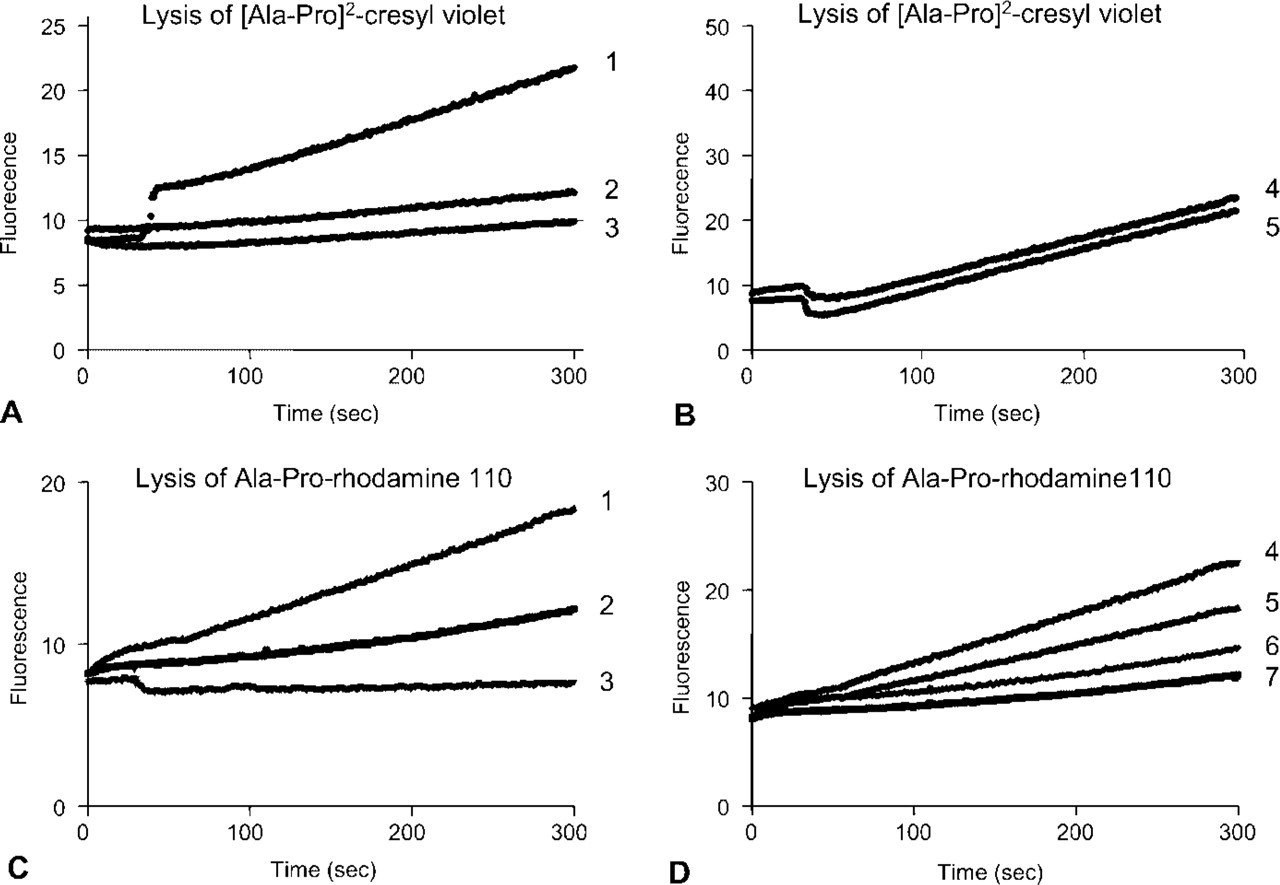
Increase in fluorescence in time at 628 nm after excitation at 591 nm as a measure of production of cresyl violet in a solution of 20 μM [Ala-Pro]2-cresyl violet in the presence or absence of intact living Jurkat cells lacking CD26/DPPIV and CD26/DPPIV-transfected Jurkat cells. The substrate is not stable in the aqueous medium and cresyl violet is generated in the medium with substrate alone (line 3). Jurkat cells produce a similar increase in fluorescence as the medium with substrate alone (line 2), whereas CD26/DPPIV-transfected Jurkat cells produce significantly higher amounts of fluorescence at 628 nm (line 1). (
Ala-Pro-rhodamine 110 substrate is not fluorescent, whereas the DPPIV reaction product, rhodamine 110, has an excitation peak at 491 nm and an emission peak at 529 nm (Leytus et al. 1983a,b).
Comparison of [Ala-Pro]2-cresyl violet and cresyl violet bands after TLC shows the color shift in both absorbance and fluorescence when the substrate is proteolytically cleaved (Figure 3D). A similar color change is observed when CD26/DPPIV is incubated with the substrate. Specificity of [Ala-Pro]2-cresyl violet and Ala-Pro-rhodamine 110 as substrates for CD26/DPPIV was demonstrated in intact and permeabilized wild-type Jurkat cells and CD26/DPPIV-transfected Jurkat cells by incubating the cells for 4 min in the presence of both substrates (Figure 4). A small increase in fluorescence over time was observed when both synthetic substrates, but particularly [Ala-Pro]2-cresyl violet, were incubated in the aqueous medium in the absence of cells, indicating spontaneous disintegration of the substrates. The increase in fluorescence over time in wild-type Jurkat cells was similar to the spontaneous disintegration of [Ala-Pro]2-cresyl violet, indicating that other DPPIV-like proteases did not cleave the substrate. CD26/DPPIV-transfected Jurkat cells produced significantly higher amounts of fluorescence at 628 nm in the presence of [Ala-Pro]2-cresyl violet. Figure 4B shows a similar production of fluorescence at 628 nm in intact and permeabilized transfected Jurkat cells against 20 μM [Ala-Pro]2-cresyl violet, again indicating that intracellular DPPIV-like proteases do not cleave the substrate.
Kinetic parameters of cleavage of [Ala-Pro]2-cresyl violet (CV) and [Ala-Pro]-rhodamine 110 (R110) by intact and permeabilized Jurkat cells and CD26/DPPIV-transfected Jurkat cells
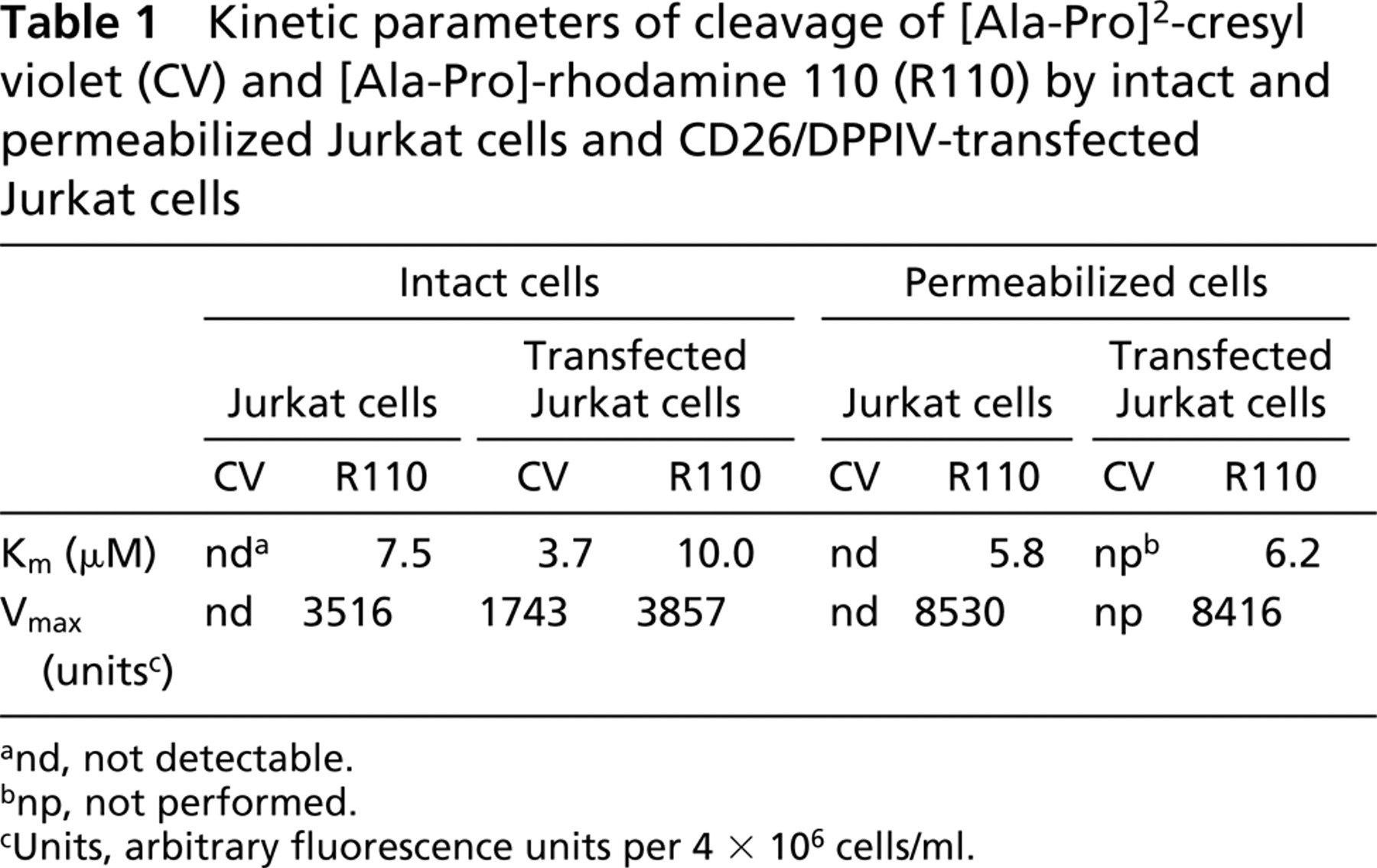
and, not detectable.
bnp, not performed.
cUnits, arbitrary fluorescence units per 4 × 106 cells/ml.
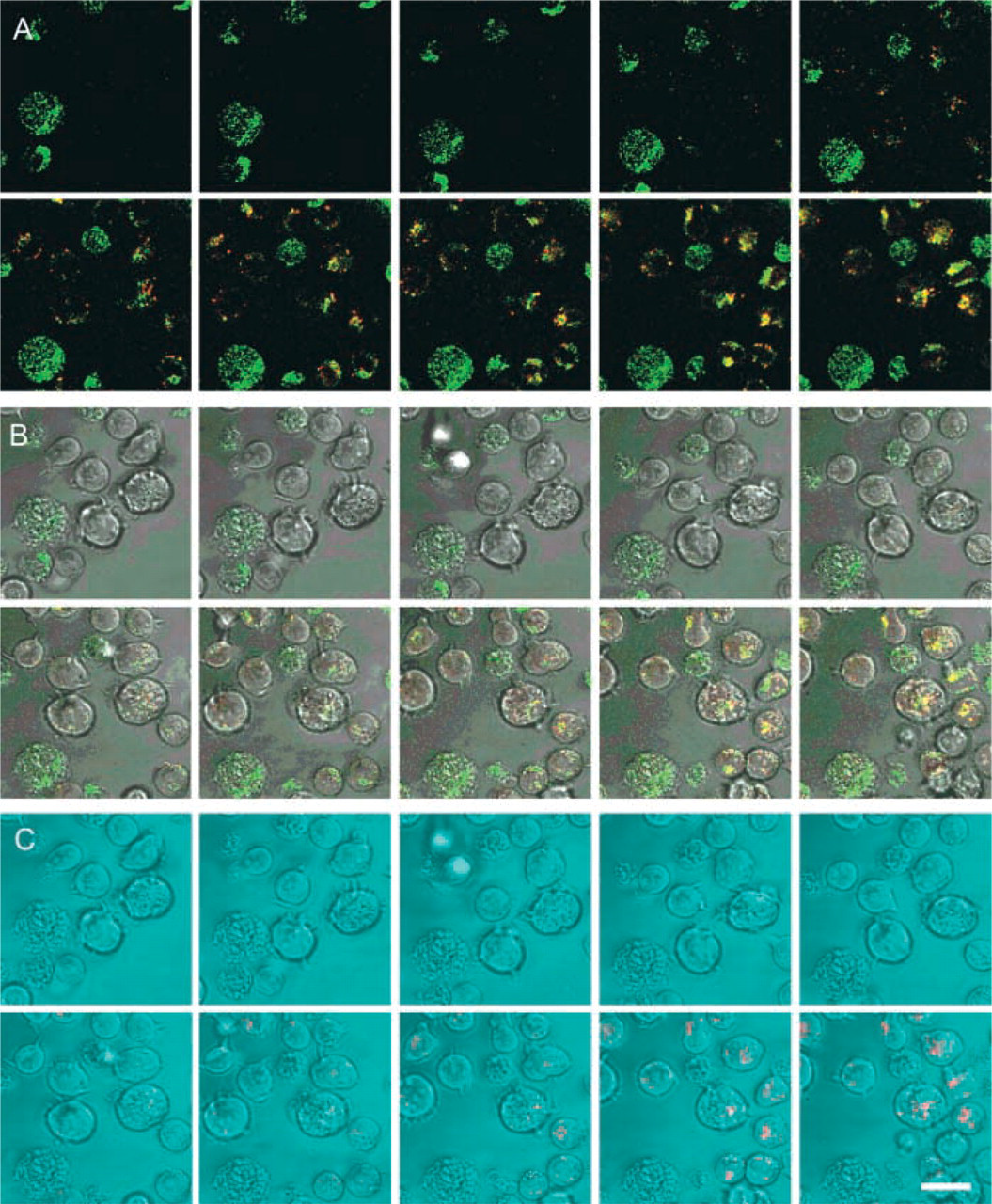
Galleries of confocal images of fluorescence in time in optical sections of CD26/DPPIV-transfected intact living Jurkat cells using 20 μM [Ala-Pro]2-cresyl violet synthetic substrate to localize DPPIV activity. The three galleries are sets of images of the same cells captured at every 30 sec after substrate was added to the incubation medium. (
Wild-type Jurkat cells lacking CD26/DPPIV process Ala-Pro-rhodamine 110 at a considerable rate, but the reaction velocity in CD26/DPPIV-transfected Jurkat cells against 20 μM Ala-Pro-rhodamine 110 was considerably higher than in Jurkat cells lacking the enzyme (Figure 4C). Permeabilization increases the reaction rate in both wild-type Jurkat cells and transfected Jurkat cells, indicating that intracellular DPPIV-like proteases cleave the rhodamine 110-containing substrate (Figure 4D). Please note that inner filter effects did not play a significant role because all plots are linear with time, showing that saturation does not occur due to inner filter effects. Km values of cleavage of [Ala-Pro]2-cresyl violet and Ala-Pro-rhodamine 110 by intact living and permeabilized wild-type Jurkat cells and CD26/DPPIV-transfected Jurkat cells were similar in the range of 3–10 μM (Table 1). Remarkably, Vmax values of Ala-Pro-rhodamine 110 cleavage were similar in wild-type Jurkat cells and transfected Jurkat cells, and permeabilization of the cells increased Vmax almost threefold (Table 1). Please note that V
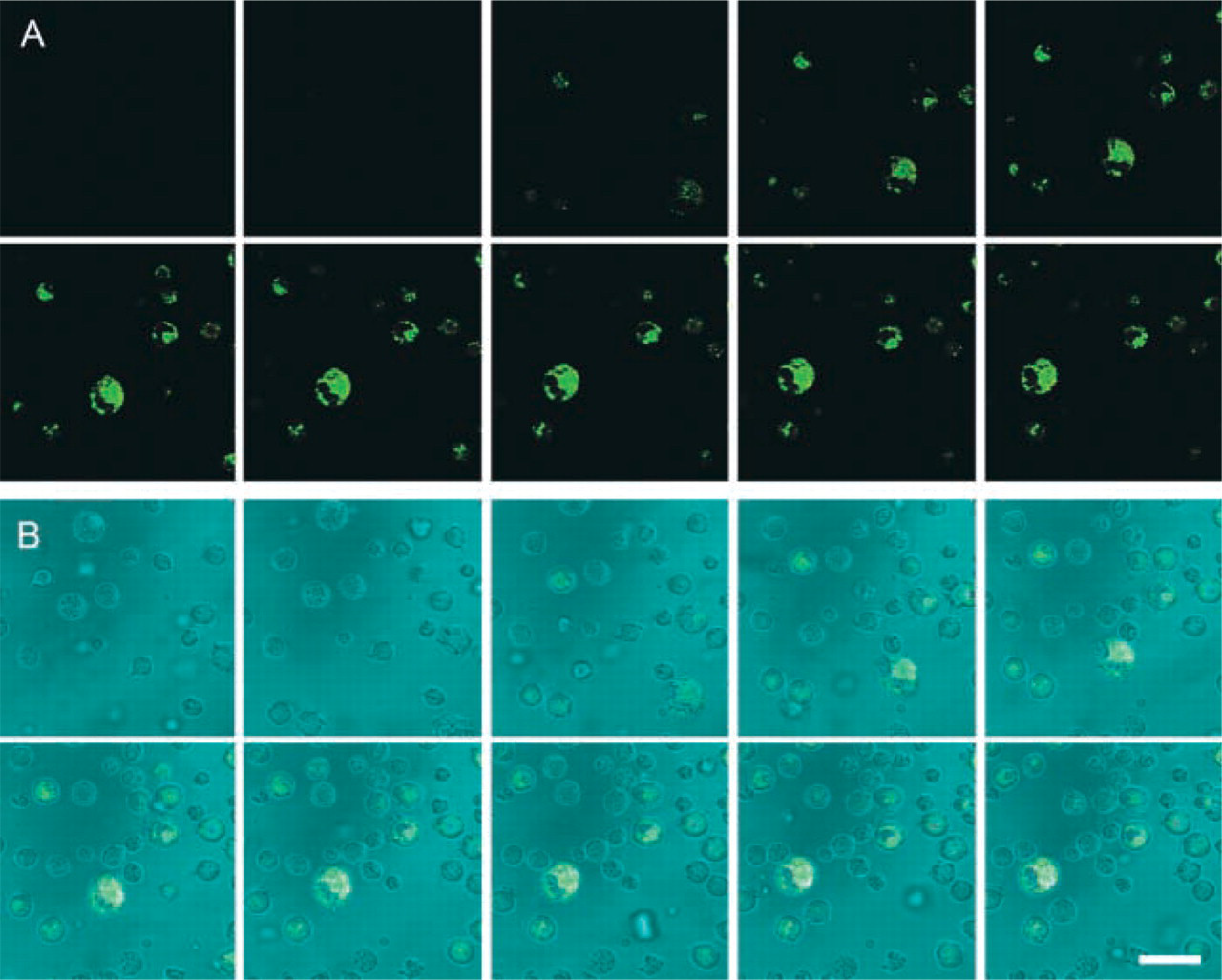
Galleries of confocal images of fluorescence in time in optical sections of intact living Jurkat cells using 20 μM [Ala-Pro]2-cresyl violet synthetic substrate to localize DPPIV activity. The two galleries are sets of images of the same cells captured at every 30 sec after substrate was added to the incubation medium. (
Confocal microscopical analysis of cleavage of [Ala-Pro]2-cresyl violet and Ala-Pro-rhodamine 110 in living wild-type Jurkat cells and CD26/DPPIV-transfected Jurkat cells is shown in Figures 5 and 6. Accumulation of cresyl violet in time was observed only in transfected Jurkat cells (Figure 5) and not in Jurkat cells lacking CD26/DPPIV (Figure 6) in small vesicles near the plasma membrane during the first few minutes of incubation. After longer incubation periods, the fluorochrome was transported to other organelles in transfected Jurkat cells (Figure 7), mimicking the pathways of internalization that have been described for CD26/DPPIV (Boonacker and Van Noorden 2003).
When Ala-Pro-rhodamine 110 was used as substrate, fluorescence occurred in both transfected and wild-type living Jurkat cells. Although fluorescence appeared to be more diffuse, accumulation was also observed in small vesicle-like structures near the plasma membrane (Figure 7).
Discussion
For visualization of specific enzyme reactions in living cells in general and activity of CD26/DPPIV in particular, it has to be established whether other enzymes interfere in the reaction and whether substrate is available at all sites of the enzyme (Boonacker and Van Noorden 2001). Therefore, we compared synthetic fluorogenic substrates that are considered to be specific for DPPIV activity (Lojda et al. 1976). These synthetic substrates differ in fluorochrome and the number of peptides attached. The rhodamine-containing substrate is monosubstituted, whereas the cresyl violet-containing substrate is bisubstituted.
Surprisingly, the two substrates behaved in rather different ways with respect to specificity. Ala-Prorhodamine 110 appeared to react with other DPPIV activity homologues, such as lysosomal DPPII, DPPVIII, and attracktin, whereas [Ala-Pro]2-cresyl violet reacted with DPPIV only. The most likely explanation is that the catalytic cleft of DPPIV only is suitable to give access to cresyl violet. The 3D configuration of the tunnel in the molecule of CD26/DPPIV and activity homologues that gives access to the active site is rather tight (Brandt 1997; Gorrell et al. 2000) and must provide steric hindrance for the cresyl violet-based substrate in the DPPIV activity homologues. As both substrates are built up rapidly in T-helper cells, it is unlikely that differences in permeability of the cells for the substrates cause the differentiation in specificity (Boonacker and Van Noorden 2001).
Excitation and emission spectra of cresyl violet and rhodamine 110 are not the same. Therefore, differences in absolute amounts of product formation on the basis of increases in fluorescence cannot be compared. Probably, rhodamine 110 is excited more intensely than cresyl violet, because light of 488 nm is more intense than light at 591 nm. The illumination source is not similarly intense throughout the spectrum, as light (lasers) does not produce similar amounts of photons at each wavelength. Furthermore, hydrolysis of monosubstituted and bisubstituted fluorochromes is not comparable either, especially when over 15% of the substrate is hydrolyzed during an assay (Leytus et al. 1983b). Lastly, fluorescence generated during incubation can differ on the basis of the microenvironment of the fluorophore.
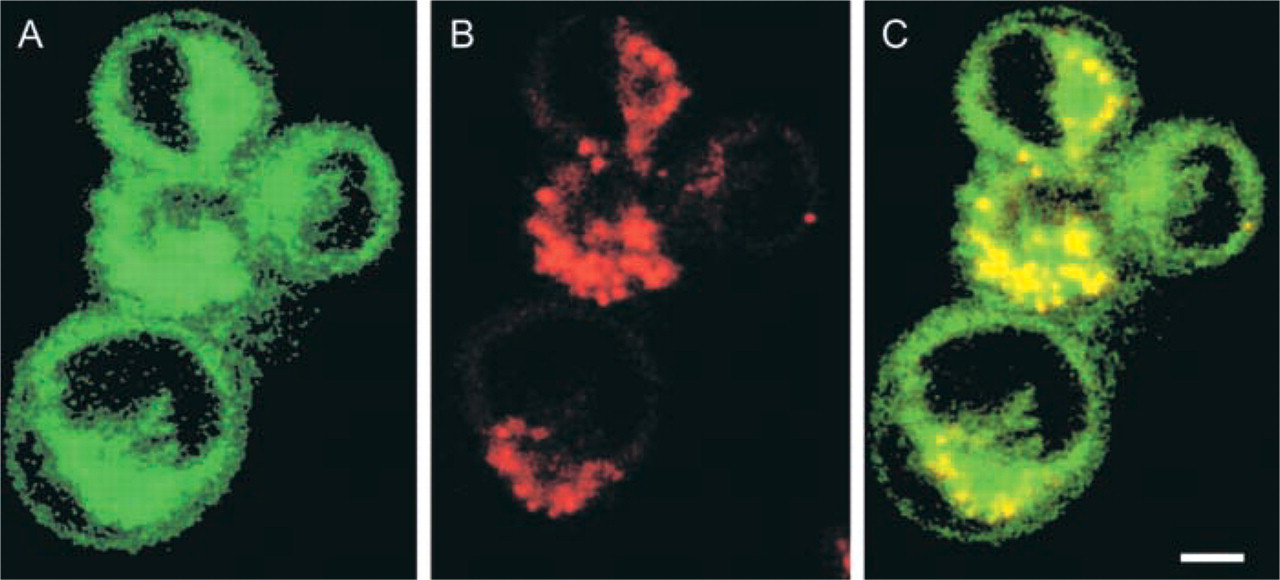
Higher magnification of CD26/DPPIV-transfected intact living Jurkat cells incubated for 5 min in a medium containing 20 μM [Ala-Pro]2-cresyl violet to demonstrate DPPIV activity. (
With respect to cytotoxicity, it can be stated that low amounts of excitation light should be used to avoid damage to the cells. Preferentially, laser power lower than 10 mW/cm2 should be used (E. Manders, personal communication). Illumination by laser light is more toxic for cells containing fluorochromes than cells without fluorochromes. Because of the lower energy of light at longer wavelengths, [Ala-Pro]2-cresyl violet is also to be preferred over Ala-Pro-rhodamine 110 because cresyl violet has to be excited at 591 nm and rhodamine at 488 nm.
A disadvantage of the cresyl violet-based substrate over the rhodamine-based substrate is the rather high instability of the former in aqueous solutions. However, the great advantage of [Ala-Pro]2-cresyl violet is its specificity for DPPIV activity. Wild-type Jurkat cells do not produce more fluorescence than incubation medium only, whereas CD26/DPPIV-transfected Jurkat cells produce distinctly more fluorescence. The same incubations were also performed on permeabilized cells. After sonification, cells were incubated as described above. Permeabilization of wild-type Jurkat cells and of CD26/DPPIV-transfected Jurkat cells did not lead to an additional increase in fluorescence. Apparently, [Ala-Pro]2-cresyl violet does not react with other DPPIV activity homologues.
In intact cells, green fluorescence of the [Ala-Pro]2-cresyl violet substrate is converted into red fluorescence of the liberated cresyl violet. Strikingly, damaged or apoptotic cells showed hardly any enzymatic activity but did accumulate [Ala-Pro]2-cresyl violet rapidly, as if the enzyme was already inactivated.
Although [Ala-Pro]2-cresyl violet is specific for DPPIV, localization of the cleavage product is another matter. Accumulation of the fluorochrome at first in small submembranous granules and later in larger intracellular compartments may be due to lipophilicity, the charge of cresyl violet, or transport in intact CD26/DPPIV-expressing cells, because intracellular localization patterns are in agreement with the recycling pathway of the M6P receptor, which can bind CD26/DPPIV and molecules associated with it (Boonacker and Van Noorden 2003).
Ala-Pro-rhodamine 110 also shows some spontaneous hydrolysis when dissolved in incubation medium. When incubations were performed with intact living Jurkat cells lacking CD26/DPPIV, hydrolysis of Ala-Pro-rhodamine 110 was distinctly higher, demonstrating its lack of specificity. CD26/DPPIV-positive transfected Jurkat cells produced an additional increase of fluorescence over time. Permeabilization of cells almost doubled the production of fluorescence, indicating that diffusion into the cells of the rhodamine 110-based substrate is a limiting step in the accessibility of this substrate for intracellular proteases other than CD26/DPPIV. Therefore, we conclude that the nature of the fluorophore significantly affects interactions of these synthetic substrates with the active site of DPPIV as has been demonstrated before (Chase and Shaw 1969; Wong and Shaw 1976; Castillo et al. 1979; Lorey et al. 2002).
Footnotes
Acknowledgements
We are grateful to Ms Trees Pierik and Mr Jan Peeterse for careful preparation of the manuscript and figures, respectively.