
Abstract
There is a great need for rapid but reliable assays to determine quantitatively effects of xenobiotics on biological systems in environmental research. Hepatocytes of European flounder are sensitive to low-dose toxic stress. Glucose-6-phosphate dehydrogenase (G6PDH) is the major source of NADPH in cells and is therefore of major importance for NADPH-dependent xenobiotic biotransformation and defense against toxic injury. These facts prompted us to develop a sensitive cytochemical method to detect G6PDH activity in living isolated flounder hepatocytes using the tetrazolium salt method. The intact plasma membrane did not appear to be a barrier for substrate, co-enzyme, and dye molecules because the intracellular enzyme reaction started immediately when incubation medium was added and could be monitored in real time per individual cell using image analysis. The reaction was effectively stopped for end point measurements by using 4% formaldehyde in 0.1 M phosphate buffer (pH 5.3). The final reaction product, formazan, was stable in hepatocytes for at least 12 days at 4C. This is the first time that a chromogenic histochemical assay is applied to living cells. This approach provides an easy tool for large-scale screening of xenobiotic metabolism and cellular stress defense. (J Histochem Cytochem 49:1025–1032, 2001)
Keywords
I
Flounder (Platichthys flesus L.) is frequently used as a sentinel species in international biomonitoring programs of biological effects of chemical pollution in coastal and estuarine areas (Von Landwüst et al. 1996; Broeg et al. 1999; ICES 1999) because this marine bottom-dwelling flatfish is strongly affected by liver cancer due to its close contact with sediments that accumulate hepatotoxic and carcinogenic compounds (Malins et al. 1984, 1985; Myers et al. 1990; Stein et al. 1990; Schiewe et al. 1991; Köhler 1991, 1995; Vethaak and Jol 1995). As in other vertebrates, the liver of fish is the main organ of biotransformation. Therefore, it is the organ of choice for biomonitoring effects of pollution of the sea. Primary cultures of fish hepatocytes provide standardized models despite the fact that isolated cells may not completely reflect the functional integrity and biotransforming activity of intact livers. However, they allow large-scale screening, and inter-individual variations are eliminated by parallel investigations of differently exposed cells of the same animal. Therefore, they are routinely used in xenobiotic research (Braunbeck 1993; Segner and Lenz 1993; Anderson et al. 1996). In the present study, a simple and rapid cytochemical method is described for quantitative detection of G6PDH activity in individual living hepatocytes of flounder.
Materials and Methods
Chemicals
Live/Dead viability/cytotoxicity kit was purchased from Molecular Probes (Eugene, OR). G6P and NADP were obtained from Roche (Mannheim, Germany), 1-methoxyphenazine-methosulfate (PMS) from Serva (Heidelberg, Germany), and Biobond from BBI International (Cardiff, UK). Collagenase Type IV, polyvinyl alcohol (PVA), tetranitroblue tetrazolium salt (TNBT), and all other reagents were ordered from Sigma (St Louis, MO).
Animals
Premature flounder with a body length of 17–25 cm were caught in an area of the German Bight (Tiefe Rinne) near Helgoland (North Sea) that is relatively unpolluted in terms of levels of hydrocarbons and heavy metals in fish and sediments (Von Landwüst et al. 1996; Broeg et al. 1999). Catching of fish was conducted with a bottom trawl (opening 1.5 m; mesh width in the cod end 40 mm) and sorted out immediately. Fishing periods were limited to 30 min to minimize sampling stress. To allow adaptation, fish were kept in tanks at 15C with permanent water flow-through and aeration for at least 2 weeks before they were used for the experiments. They were fed ad libitum with standard protein food (Trouvit; Milkivit-Werke, Burgheim, Germany). For each parameter, cells of 10 fish were tested.
Primary Hepatocyte Cultures
Animals were sacrificed by destruction of the brain with a knife followed by severing of the spines, and livers were removed immediately. Isolation and culture of hepatocytes were performed at 10C under sterile conditions. Hepatocytes were isolated by dissociation of the freshly dissected organs. Collagenase treatment of the pieces of liver was performed according to Lowe et al. (1992) with the following modifications.
After washing in Hanks' salt solution, liver pieces were incubated for 40 min under gentle shaking conditions in Hanks' salt solution containing 20 mg collagenase per liver. The cell suspensions were passed through 250-μm and 50-μm gauzes to complete cell isolation. After washing three times in Hanks' salt solution, hepatocytes were plated at a concentration of 2 × 106 cells per well in 24-well plates (Greiner; Fickenhausen, Germany) in vitamin E-containing Medium 199 supplemented with 20 mM Hepes and 1% penicillin/streptomycin in the presence or absence of 10% fetal bovine serum (FBS). Cells were kept in the dark for 48 hr at pH 7.5 and sampled at 24 hr after isolation. Before use they were washed with 100 mM Sorensen phosphate buffer (pH 7.2).
Cell suspensions after isolation and culture contained not only hepatocytes but also non-parenchymal cells, such as erythrocytes. However, morphological discrimination of these cell types was easy using image analysis.
Cell Viability
Cell viability was determined by using the Live/Dead viability/cytotoxicity kit. Aliquots (50 μl) of hepatocytes in 0.1 M PBS (pH 7.2) were incubated for 5 min in 100 μl of a solution of 2 μM calcein-bisacetyloxylmethylester (calcein AM) and 4 μM ethidium homodimer-1 diluted in 0.1 M PBS (pH 7.2). Calcein AM penetrates intact cells and is hydrolyzed by intracellular esterases to green fluorescent calcein, whereas ethidium homodimer-1 does not stain viable cells. It can penetrate only damaged cell membranes of dying or dead cells and stains their nucleic acid red. Fluorescence was measured with 470-nm excitation light. Emission was determined at 530 nm for living cells and at 585 nm for dying or dead hepatocytes with a red fluorescent nucleus. Cells were counted in a hemocytometer and viability was calculated as percentage of the total number of cells. Only suspensions of hepatocytes were used that showed more than 80% viability immediately before experimental use (data not shown). Dead cells were easy to distinguish with image analysis on the basis of disruption of the cell membrane, and were excluded from measurements.
Permeabilization of Cell Membranes
Various fixatives and permeabilizing agents that are used in routine histochemistry were tested before the G6PDH assay to increase the permeability of intact cell membranes and consequently allow optimal staining of hepatocytes with the tetrazolium salt procedure. Cells were incubated at 4C for 1 hr with (a) 0.025% glutaraldehyde (GA) [50 μl of a 50% GA solution in 100 ml 0.1 M Sorensen phosphate buffer (pH 7.2)] (Van Noorden et al. 1982), (b) 1% DMSO [100 μl DMSO in 9.9 ml 0.1 M Sorensen phosphate buffer (pH 7.2)], (c) 3.7% formaldehyde in 0.1 M Sorensen buffer (pH 7.2) (Lin and Leise 1996), (d) 0.05% saponin [1 ml of a 0.5% stock solution of saponin in 0.1 M Sorensen phosphate buffer (pH 7.2)], (e) Bouin solution (7.5 ml of freshly filtrated pikrin acid, 2.5 ml of 37% formaldehyde, 0.5 ml 100% acetic acid), or (f) 0.05% Triton X-100 (50 μl Triton X-100 in 100 ml 0.1% sodium citrate buffer, pH 7.8). Enzyme detection followed immediately after washing in 0.1 M Sorensen phosphate buffer (pH 7.2). To protect the active site of G6PDH during fixation in GA, cells were preincubated for 10 min in a solution of 20 mM NADP in 0.1 M Sorensen phosphate buffer (pH 7.2) (Van Noorden et al. 1982). After Bouin fixation, hepatocytes were dehydrated in 50% and 70% ethanol for 10 min each. Enzyme detection followed immediately after a short period of drying.
Cytochemical End Point Detection of G6PDH Activity
Tetrazolium salt methods are established precipitation reactions for the detection of the activity of dehydrogenases, reductases, and oxidases in cryostat sections of tissues. The enzyme-catalyzed oxidation of the substrate frees protons that are picked up by the co-enzyme (co-enzyme reduction). The reduced co-enzyme itself reduces electron carriers such as PMS which, in turn, transfers the electrons to the tetrazolium salt (TNBT) as final electron acceptor. In this way, water-insoluble formazan is rapidly formed (Lojda et al. 1976; Seidler 1991; Van Noorden and Frederiks 1992).
In the present study, G6PDH activity was detected cytochemically with a tetrazolium salt method described by Van Noorden and Frederiks (1992) for cryostat sections after slight modification. Untreated or fixed hepatocytes were incubated in Eppendorf tubes for 15 min at 10C or room temperature in 0.1 M phosphate buffer (pH 7.4) containing a low concentration of PVA. A concentration of PVA of 2% instead of 18% was used to facilitate the washing off of the viscous medium at the end of the incubation period. The incubation medium was prepared by adding 100 mg PVA to 5 ml 0.1 M phosphate buffer (pH 7.4), stirring, and heating the medium to 60C. After cooling of the medium to 37C, a concentrated substrate (G6P) solution (stock solution of 170 mg G6P in 500 μl distilled water) was used to prepare media containing 0, 2, 4, 6, 8, and 10 mM substrate. The following components were added in 50 μl per 5 ml incubation medium to give a final dilution of 0.8 mM NADP: 0.45 mM PMS, 4 mM MgCl2, and 5 mM sodium azide. To prevent precipitation of TNBT in solution in the low-viscosity medium, we selected a final concentration of 1 mM instead of 5 mM. TNBT was first dissolved in 100 μl ethanol and 100 μl DMSO before adding it to the medium. Reactions were carried out in the dark to prevent light-induced formazan formation (Van Noorden and Frederiks 1992). Different substrate concentrations were tested to optimize the G6PDH reaction.
Controls were run in the absence of substrate or substrate and co-enzyme (Van Noorden and Frederiks 1992). In addition, controls were performed using 6 mM G6P in the absence and presence of NADP to test the permeability of hepatocyte membranes for NADP. Hepatocytes were also tested for the activity of NADP-dependent malate dehydrogenase (EC 1.1.1.37) and isocitrate dehydrogenase (EC 1.1.1. 42) using optimal substrate concentrations (100 mM) according to Van Noorden and Frederiks (1992) to estimate their interference in the G6PDH reaction by converting their endogenous substrates. The activity of these dehydrogenases was demonstrated in a similar way as the G6PDH assay described above.
Fixation of Incubated Hepatocytes
G6PDH reactions were stopped by adding 0.1 M phosphate buffer (pH 5.3) to the cells as is described for histochemical assays on cryostat sections. In addition to this approach, fixation with 4% glutaraldehyde or 4% formaldehyde in 0.1 M phosphate buffer (pH 5.3) was tested to prevent formazan production after incubation. Furthermore, formaldehyde (4%) fixation in phosphate buffer (pH 7.2) and additional heating (70C) in phosphate buffer (pH 5.3), as well as 1% trichloric acid (TCA), were tested. Cells were kept in the stopping buffer at 4C until end point measurements were made using image analysis. Results in cells that were kept for 2, 5, 12, or 19 days at 4C were compared.
Kinetic Measurements of G6PDH Activity
In addition to end point measurements, enzyme reactions were monitored in real time in living cells attached to slides. Hepatocytes were allowed to attach to Biobond-pretreated glass slides in suspensions (40 μl in Medium 199) for 20 min. To prevent drying and death of the hepatocytes, this procedure was performed in a humid chamber at 10C. Cell preparations were rinsed with PBS (pH 7.2) to remove culture medium and those cells that were not attached properly. G6PDH activity was analyzed quantitatively using image analysis and a measurement was taken every 5 min over a period of 30 min using a water-immersion objective (C-Apochromat, ×40; Zeiss, Oberkochen, Germany).
Influence of Temperature on G6PDH Activity
G6PDH activity was measured at room temperature (22C), 37C, and 10C to determine cell adaptation to culture temperatures. Incubations were performed in the presence or absence of 6 mM G6P and/or NADP.
Image Analysis of the G6PDH Reaction
Cytophotometric analysis was performed by quantitative video microscopy with a CCD color video camera (Sony; Aalen, Germany) connected to an Axioskop light microscope (Zeiss) and linked via a frame grabber (maximal size of 786 × 512 pixels) to an image analysis system (Zeiss) with the KS300 software package. Details of camera signal and setup, conversion of gray levels to absorbance values, and analysis of absorbance in real time in cells, as well as the calculation of initial velocities, have been described elsewhere (Chieco et al. 1994; Jonker et al. 1995, 1997; Van Noorden et al. 1997). Absorbance of formazan was measured in individual hepatocytes using monochromatic light of 580 nm. According to Van Noorden and Jonges (1995), areas of the cytoplasm of cells were measured and concentrations of the reaction product formazan (c) were calculated using the Lambert-Beer law
>A = ∊ · c · d,
with A being measured absorbance, є being the extinction coefficient of the reaction end product at the specific wavelength (= 19,000), and d being the height of the hepatocytes. An average cell height of 7.5 μm was assumed. Enzyme activity was calculated on the basis of the fact that one molecule of formazan is formed by the conversion of two molecules of G6P.
Statistical Analysis
All data were analyzed with non-parametric statistics (Kruskal-Wallis test and post-hoc Nemenyi test; Statistica 5.0, StatSoft, Tulsa, OK). p = 0.05 was taken as level of significance.
Results and Discussion
This article is the first report on the application of a chromogenic cytochemical assay for demonstration of enzymatic activity in living cells. Only slight modifications of existing histochemical methods enabled the detection, localization, and quantification of G6PDH activity in unfixed living hepatocytes of flounder.
Permeabilization of Cell Membranes
None of the pretreatments tested to increase membrane permeability of the living flounder hepatocytes for substrate, co-enzyme, and dye molecules resulted in increased formazan production due to a faster reaction of G6PDH (Table 1). Triton X-100 and saponin pretreatment almost abolished all G6PDH activity in the hepatocytes. Formaldehyde prefixation also decreased G6PDH activity, whereas Bouin fixation had a detrimental effect on cell morphology and strongly reduced G6PDH activity. GA fixation in combination with NADP pretreatment and DMSO treatment did not significantly alter G6PDH activity in hepatocytes. A tetrazolium salt technique without membrane permeabilization has also been used by Chikamori and co-workers (Chikamori et al. 1998a, b; Higashijima et al. 2000) for in situ demonstration of succinate dehydrogenase activity in Paramecium caudatum, but the way of uptake of dyes and substrates was not specified. For reliable localization and quantification of G6PDH activity in fish hepatocytes, the cell membrane apparently does not need to be permeabilized. Therefore, permeabilization procedures were omitted in all further experiments.
Activity of G6PDH (expressed as μM G6P converted per min per ml cytoplasm; median ± quartile range) a
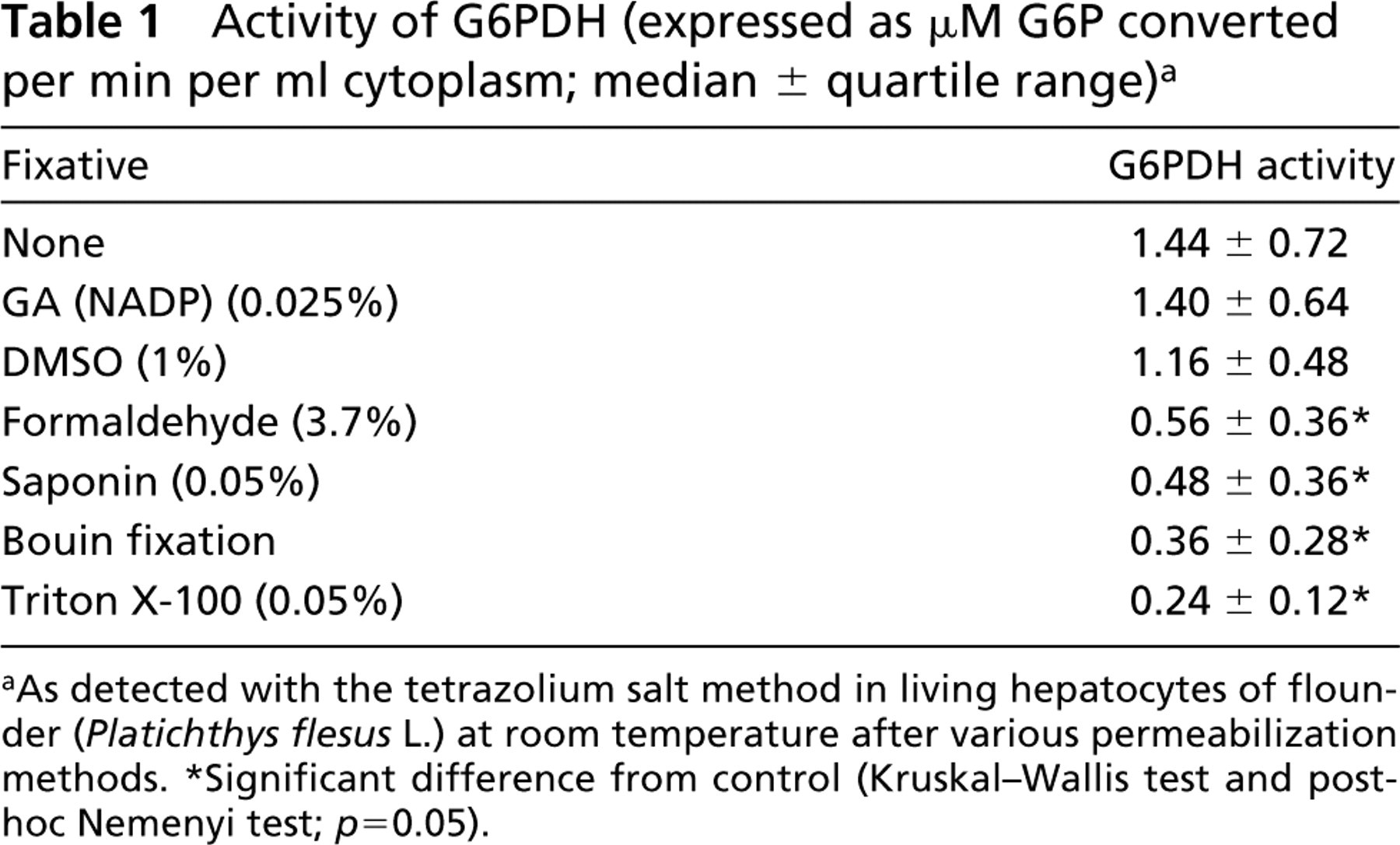
As detected with the tetrazolium salt method in living hepatocytes of flounder (Platichthys flesus L.) at room temperature after various permeabilization methods.
Significant difference from control (Kruskal-Wallis test and post-hoc Nemenyi test; p = 0.05).
Cytochemical End Point Detection of G6PDH Activity
Figure 1 shows a light microscopic image of living flounder hepatocytes after staining for G6PDH activity. The final reaction product, formazan, is localized in the cytoplasm of cells, leaving the nucleus unstained.
End point measurements showed that 6 mM G6P was the optimal substrate concentration (Figure 2). This concentration is identical to the optimal substrate concentration suggested for cryostat sections of flounder liver (Van Noorden et al. 1997; Köhler and Van Noorden 1998). Relatively strong substrate inhibition occurred at 8 and 10 mM G6P.
Kinetic Measurements of G6PDH Activity
When kinetic measurements were performed in the presence and absence of 6 mM G6P, specific formazan production in the cytoplasm of cells increased linearly during the first 15 min (Figure 3). During the second 15 min of incubation, leveling off of G6PDH activity was observed. To ensure linearity in further experiments, an incubation period of 15 min is suggested for kinetic and end point studies.
Compared to histochemical data obtained from cryostat sections, the reaction appears to be slower in intact hepatocytes, possibly caused by the penetration of components into living cells, whereas the target enzyme in cryostat sections is directly accessible to components of the incubation medium. Consequently, enzyme activity in living cells (1.9 ± 0.7 μM G6P converted per ml cytoplasm per min) of healthy flounder differed by a factor of 3–5 from histochemical data obtained in cryostat sections of livers of healthy juvenile flounder (6.7 ± 3.3 μM G6P converted per ml cytoplasm per min; A. Köhler, unpublished data), but data show the same range of variation. The control reaction in the absence of substrate and coenzyme produced low amounts of formazan (Table 2; Figure 3A).
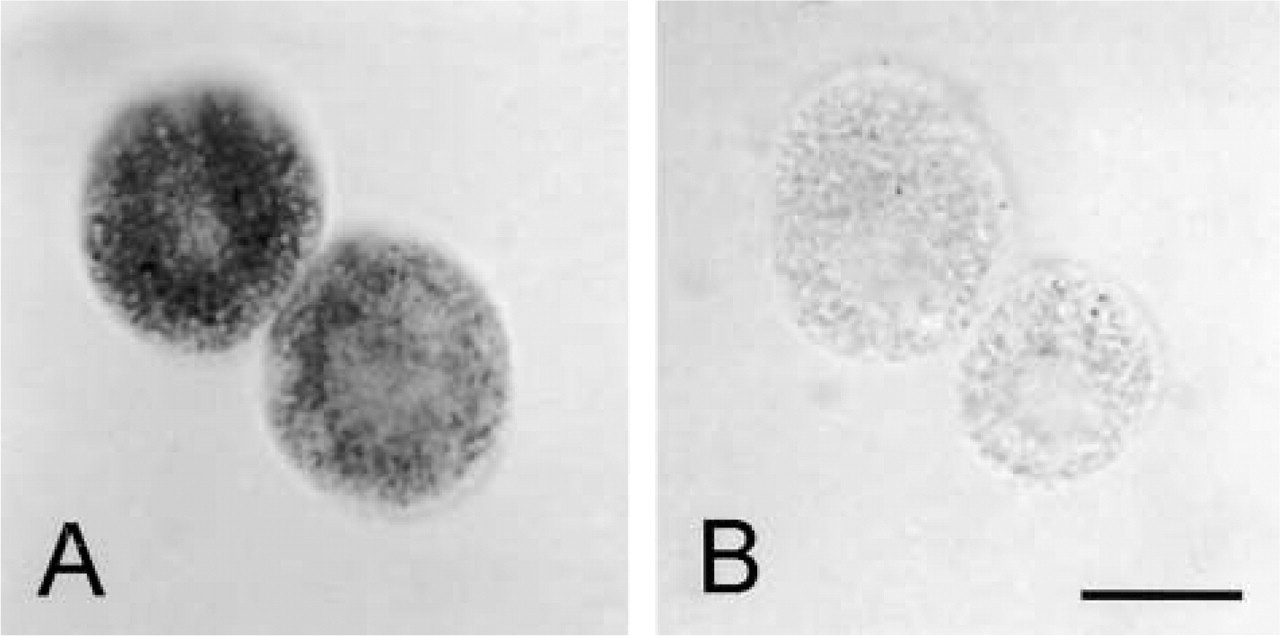
Light microscopic images of G6PDH activity in living isolated hepatocytes of flounder (Platichthys flesus L.) incubated (
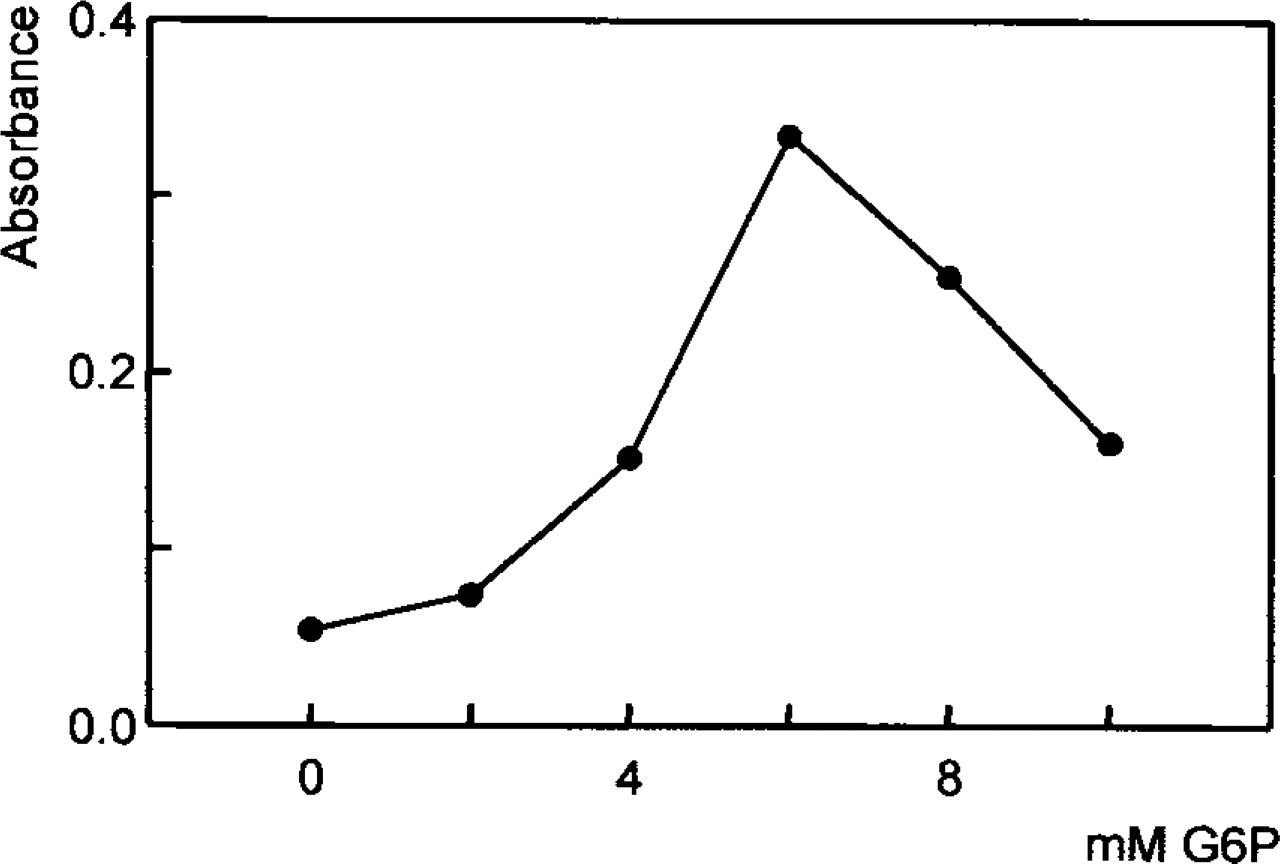
Absorbance of formazan produced by G6PDH activity (median) at room temperature in isolated living hepatocytes of flounder against a series of substrate concentrations (0–10 mM G6P).
Specificity of Formazan Production
G6PDH activity was significantly lower in the absence of co-enzyme in the incubation medium than in the presence of co-enzyme (Table 2), which shows that the intact cells took up the exogenous co-enzyme in the incubation medium and that the plasma membrane did not act as a barrier to NADP. Moreover, kinetic measurements (Figure 3) demonstrated that the enzyme was active from the moment when the incubation medium was added.
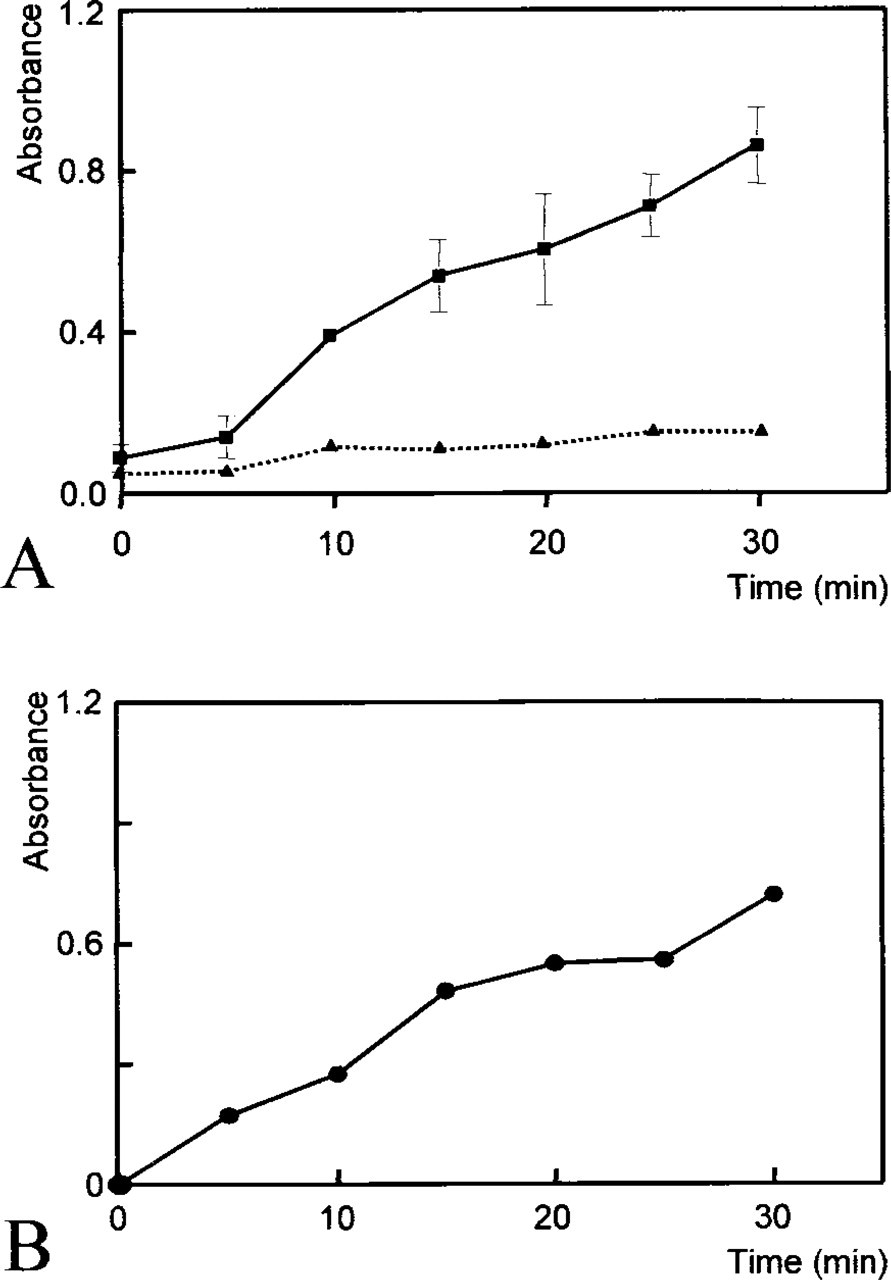
Time-dependent increase in absorbance of formazan produced by G6PDH activity in isolated living hepatocytes of flounder at room temperature as determined kinetically with image analysis (
Activity of G6PDH (expressed as μM G6P converted per min per ml cytoplasm; median ± quartile range) a
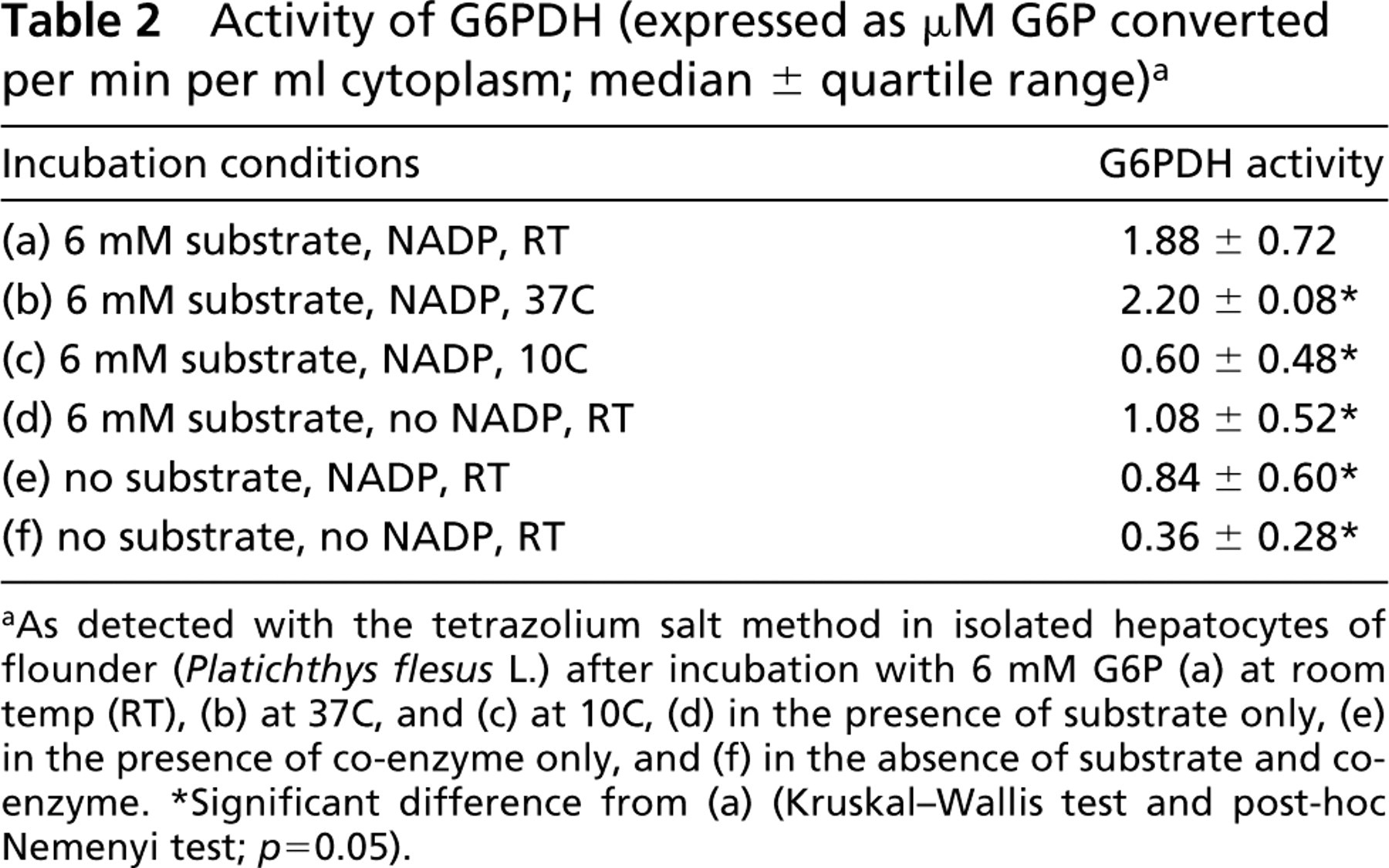
As detected with the tetrazolium salt method in isolated hepatocytes of flounder (Platichthys flesus L.) after incubation with 6 mM G6P (a) at room temp (RT), (b) at 37C, and (c) at 10C, (d) in the presence of substrate only, (e) in the presence of co-enzyme only, and (f) in the absence of substrate and coenzyme.
Significant difference from (a) (Kruskal-Wallis test and post-hoc Nemenyi test; p = 0.05).
Interference of other NADP-dependent enzymes in formazan production in the G6PDH assay was low. Addition of 100 mM malate instead of G6P did not result in substantial activity of malate dehydrogenase (0.32 ± 0.44 μM malate converted per min per ml cytoplasm) in living hepatocytes. NADP-dependent iso-citrate dehydrogenase activity was also relatively low (1.16 ± 0.80 μM isocitrate converted per min per ml) in the presence of 100 mM isocitrate instead of G6P. These findings exclude significant interference in G6PDH-specific formazan production due to activity of these enzymes by converting (low) concentrations of their endogenous substrates in the cytoplasm.
Variability of G6PDH Activity
As shown in Figure 4, G6PDH activity, and also endogenous NADP-dependent activity in the G6PDH co-enzyme control, varied considerably in individual hepatocytes of pooled flounder livers. This could be due either to interindividual variation or to intercellular variation caused by zonation of liver parenchyma, differences in cell membrane properties, or genetic/adaptive enzymatic alterations in single hepatocytes or clones of hepatocytes due to environmental or experimental exposure conditions.
Individual fish were found to vary in enzyme activity depending on sex or grade of pollution (data not shown), but high variability was also observed in hepatocytes of single animals. In contrast to mammalian livers (Jonges and Van Noorden 1989), fish liver does not show a zonation of G6PDH activity in lobuli (Köhler et al. 1998). On the other hand, intra-individual variations of activities of G6PDH were observed in histochemical studies of feral flounder populations. The occurrence of enzyme-altered foci (Köhler et al. 1998) or unevenly distributed zones of higher and lower activity (Köhler and Van Noorden 1998) may indicate hepatocellular heterogeneity due to adaptation of enzyme activity to environmental conditions. Because the intercellular variability of G6PDH activity under all conditions tested was independent of the presence of fetal bovine serum in the incubation medium (data not shown), the membrane-stabilizing influence of serum components on isolated hepatocytes (Winzer et al. 2001) can be excluded as a factor for enzymatic variability. On the basis of our experience in practice, a sample size of 30 cells is recommended.
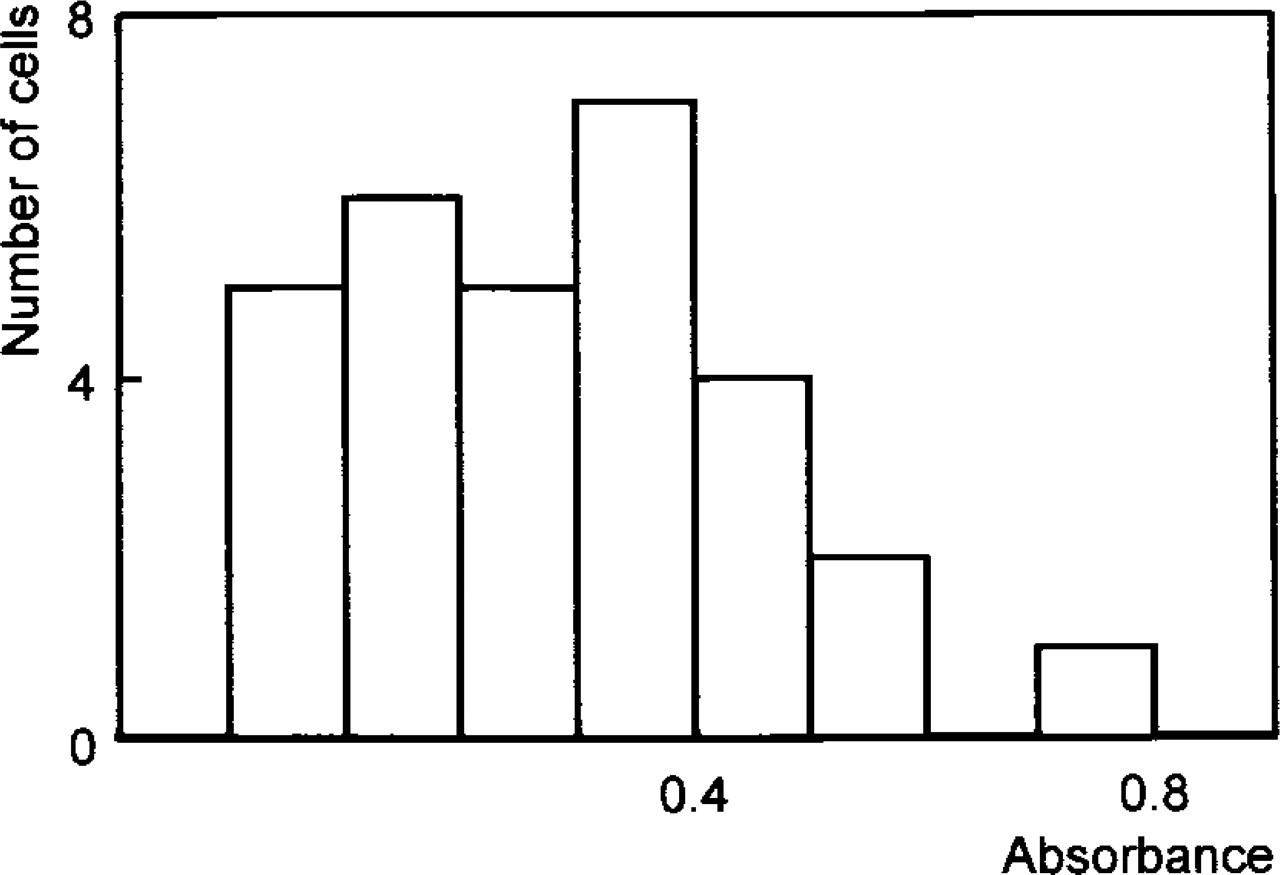
Variation in absorbance of formazan produced by G6PDH activity in single living hepatocytes obtained from five flounder livers.
Effects of Incubation Temperature on G6PDH Activity
G6PDH activity in isolated hepatocytes was significantly lower at 10C (0.60 ± 0.48 μM G6P converted per min per ml cytoplasm) than at room temperature (22C; 1.88 ± 0.72 μM G6P converted per min per ml cytoplasm), whereas at 37C activity was only slightly higher than at 22C (2.20 ± 0.08 μM G6P converted per min per ml cytoplasm) (Table 2). Therefore, G6PDH in hepatocytes of this poikilotherm fish species is specifically modulated within the physiological temperature range (5–22C).
Fixation of Hepatocytes After Incubation
Acid phosphate buffer (pH 5.3) is normally used to stop the incubation, and thus formazan production, in cryostat sections (Van Noorden and Frederiks 1992). This approach was not sufficient to completely stop G6PDH activity in isolated living hepatocytes, and formazan content of cells significantly increased after 24 hr of storage. It appeared that, of all fixatives tested, 4% formaldehyde in acid phosphate buffer (pH 5.3) was the best treatment to stop the reaction immediately. This procedure gave stable results until 12 days after incubation (data not shown). The effective termination of the enzyme reactions allows the setup of end point experiments as a basis for large-scale testing of toxic compounds and other factors in cell culture models. Compared to time-consuming kinetic measurements of G6PDH in single cells, where single samples can be analyzed per 15 min only, end point incubations are more appropriate for a large-scale screening.
On the basis of our study, we recommend the following assay conditions for end point measurements of G6PDH activity in living hepatocytes of flounder. Incubate untreated hepatocytes for 15 min at room temperature in a tetrazolium salt-containing medium containing 2% PVA, 6 mM G6P, and 0.8 mM NADP. Perform control incubations in the absence of substrate and co-enzyme. Stop the reaction with 4% formaldehyde in acid phosphate buffer (pH 5.3) at 4C and measure samples within 12 days.
In conclusion, this modified cytochemical method not only allows the study of G6PDH activity in living flounder hepatocytes but also enables analysis of activity of this sensitive marker enzyme for large-scale testing of early effects of xenobiotics. Results obtained in living cells under clearly defined conditions, such as temperature, UV irradiation, and hormone concentrations, may help to assess the toxicity of single chemicals and their combinations and to understand the initial mechanisms of cellular adaptations and mutations that lead to xenobiotic-induced carcinogenesis.
Footnotes
Acknowledgments
This article is based on a doctoral study by Katja Winzer which was conducted in the Department of Ecophysiology and Ecotoxicology, Alfred-Wegener-Institute for Polar and Marine Research, Germany (Dr A. Köhler, co-promotor) in cooperation with the University of Amsterdam, The Netherlands (Prof Dr C.J.F. Van Noorden, promotor, Academic Medical Center), supported by a PhD grant of the Biologische Anstalt Helgoland in the Alfred-Wegener-Institute.
We thank Prof W. Becker (Zoological Institute, University of Hamburg, Germany) for his support. We also thank Captain Lührs and his crew of the MV “Uthörn” for the sampling of flounder.