
Abstract
We present here evidence of in vivo epithelial endocytosis and trafficking of non-lipid-modified Sonic hedgehog (ShhN) when infused into rat efferent ducts via microinjection. Initially, exogenous ShhN is detected in endocytic vesicles and early endosomes located near the apical plasma membrane of non-ciliated cells. Within 30-60 min following infusion, ShhN can be detected in lysosomes and at basolateral regions of non-ciliated cells. Basolaterally, ShhN was observed along the extracellular surfaces of interdigitated plasma membranes of adjacent cells and in the extracellular compartment underlying the efferent duct epithelium. Uptake and subcellular trafficking of infused ShhN by non-ciliated cells could be blocked by either anti-megalin IgG or the megalin antagonist, RAP. Ciliated cells, which do not express megalin, displayed little if any apical internalization of ShhN even though they were found to express Patched-1. However, ShhN was found in coated pits of lateral plasma membranes of ciliated cells as well as in underlying endocytic vesicles. We conclude that megalin-mediated endocytosis of ShhN can occur in megalin-expressing epithelia in vivo, and that the internalized ShhN can be targeted to the lysosome or transcytosed in the plane of the epithelium or across the epithelium. These findings highlight the multiple mechanisms by which megalin may influence Shh morphogen gradients in vivo.
S
Megalin (gp330/LRP-2) is an endocytic receptor belonging to the low-density lipoprotein receptor family (May and Herz 2003; McCarthy and Argraves 2003). During early development, megalin is expressed by absorptive epithelial cells at sites in which morphogenic patterning is under the influence of Shh, such as in the neural tube (McCarthy et al. 2002; Spoelgen et al. 2005). Recently, megalin has been shown to bind and mediate endocytosis of ShhN (McCarthy et al. 2002), the N-terminal fragment of Shh that lacks lipid modifications but retains most of the signaling activity of ShhNp (Marti et al. 1995; Roelink et al. 1995). The physiological significance of megalin-mediated endocytosis of ShhN is not certain. Of potential importance in this regard is the observation that ShhN endocytosed via megalin is not efficiently targeted to the lysosome (McCarthy et al. 2002), as is the case with other megalin ligands (Morales et al. 1996). This raises the possibility that ShhN is transcytosed following megalin-mediated endocytosis. This possibility is consistent with the fact that megalin mediates transcytosis of other ligands including thyroglobulin, retinal-binding protein, vitamin B12, and the 39-kDa receptor-associated protein (RAP) (Marino et al. 2000,2001; Marino and McCluskey 2000; Nielsen et al. 2001; Pan et al. 2004). Evidence for the involvement of megalin in Shh transcytosis could be important in the process by which Shh mediates long-and short-range control of growth and patterning during development.
Our previous studies have shown that megalin is abundantly expressed on the apical plasma membrane of non-ciliated cells lining the efferent ducts of the male reproductive tract (Morales et al. 1996; Hermo et al. 1999). We have also demonstrated that megalin expressed by these cells mediates endocytosis and lysosomal targeting of both endogenously expressed clusterin/apolipoprotein J (apoJ) and RAP introduced into efferent ducts by microinjection (Morales et al. 1996). The relative ease of introducing ligands into rat efferent ducts by microinjection and tracking them by immunocytological techniques makes efferent ducts an ideal system for studying epithelial endocytosis and trafficking of megalin ligands in vivo. Here we evaluate megalin-mediated trafficking of ShhN by efferent duct epithelial cells in vivo.
Materials and Methods
Antibodies
The mouse monoclonal ShhN antibody, 5E1 (Ericson et al. 1996), was produced from a hybridoma obtained from the Developmental Studies Hybridoma Bank at the University of Iowa, Iowa City, Iowa. 5E1 IgG was purified by protein G-Sepharose chromatography. Rabbit polyclonal anti-megalin IgG (rb6286) has been described previously (Kounnas et al. 1993). A rabbit polyclonal antiserum was produced against a carboxy terminal polypeptide from mouse patched-1 (Ptc-1) (amino acid residues 1158-1434). The Ptc-1 polypeptide was expressed in bacteria as a glutathione S-transferase (GST) fusion protein and the GST portion removed by thrombin cleavage prior to immunization. IgG was purified from the resulting antiserum (rb2160) by affinity chromatography on a column of the GST-Ptc-1 fusion protein coupled to Sepharose followed by protein G-Sepharose. The antibody was shown to react with a polypeptide corresponding to the size of Ptc-1 in extracts of Chinese hamster ovary (CHO) cells transfected to express full-length mouse Ptc-1. The antibody showed no reactivity in immunoblotting of extracts from non-transfected CHO cells. Goat anti-rabbit IgG and goat anti-mouse IgG conjugated to horseradish peroxidase (HRP) for use in light microscopy (LM) or to colloidal gold for use in electron microscopy (EM) were purchased from Cedarlane Labs (Hornby, ON).
Proteins
Recombinant amino terminal fragment of mouse Shh (ShhN) was expressed as a fusion protein in bacteria and purified as described previously (McCarthy et al. 2002). The fusion protein was composed of GST followed by a thrombin cleavage site (LVPRGS), a five-amino acid phosphorylation target site (RRASV), and amino acids residues 20-198 of mouse ShhN. The ShhN fusion protein was adsorbed onto a Detoxigel Endotoxin Removing Gel (Pierce Biotechnology; Rockford, IL), and its biological activity was assayed in C3H10T1/2 cells using the method of Williams et al. (1999). Bacterially expressed human RAP was purified as previously described (Kounnas et al. 1992,1995).
Radiolabeling of GST-ShhN
GST-ShhN was labeled with [γ-32P]-ATP using heart muscle kinase (HMK) as described in McCarthy et al. (2002). Briefly, 50 μg of protein was incubated with 125 U of HMK (catalytic subunit; Sigma, St Louis, MO) in 1X HMK buffer (20 mM PIPES, pH 6.5, 1 mM DTT, 20 mM NaCl, 12 mM MgCl2) plus 0.1% denatured BSA and 50 μCi [γ-32P]-ATP (Amersham Biosciences; Baie d'Urfé, QC) for 1 hr. Labeled fusion protein was purified by size exclusion chromatography using PD-10 columns.
In Vivo Infusion of ShhN
[32P]-GST-ShhN (1 × 106 CPM) in 250 μl of PIPES buffer (20 mM PIPES, 20 mM NaCl, 12 mM MgCl2, pH 6.5) or non-labeled ShhN (2 μg/μl) in PIPES buffer was infused into the efferent ducts of anesthetized 4-month-old Sprague Dawley rats (Charles River Laboratories; Montreal, QC) via a microinjection into the rete testis (Morales and Hermo 1983). To block megalin-mediated interaction with ShhN, ShhN was coinjected with either anti-megalin IgG (5 μg/μl) or the megalin antagonist, RAP (2 μg/μl). At varying time intervals following microinjection (i.e., 5, 30, and 60 min), efferent ducts were removed by dissection, fixed, and either embedded in paraffin for LM or in Lowicryl K11M (Polysciences; Warrington, PA) for EM. Each experimental group/end point was composed of efferent ducts from a minimum of three animals. Institutional guidelines on the use of animals were followed in all experimentation involving rats.
Autoradiography
Autoradiography was performed on 5-μm-thick deparaffinized tissue sections essentially as described by Qi et al. (2002). After a passage in alcohol, slides were air dried, dipped in LM-1 emulsion (Amersham Biosciences; Buckinghamshire, UK), and then exposed in the dark at 4C for 8 days. Slides were then developed at 15C by treating with Kodak D-170 developer (Kodak; Rochester, NY) for 7 min, fixed for 4 min in Kodak Rapid Fixer, and counterstained with hematoxylin and eosin.
Immunoblot Analysis
Efferent ducts were homogenized on ice in sucrose buffer (250 mM sucrose, 10 mM HEPES, 5 mM EDTA, 1 mM PMSF, 10 μg/ml leupeptin, 10 μg/ml aprotinin, pH 8.0) and the extracts centrifuged for 6 min at 3020 × g. Supernatant was centrifuged for 40 min at 48,000 × g and the resulting pellet dissolved in buffer A (2 mM CaCl2, 1mM MgCl2, 10 mM HEPES, and 140 mM NaCl, pH 7.8) containing 1% 3-[(cholamidopropyl)-dimethylammonio-l-propanesulfonate] (CHAPS; Boehringer Mannheim, Mannheim, Germany). Buffer A was added to give a final concentration of 0.25% CHAPS. Insoluble debris was removed by centrifugation at 48,200 × g. As a control, detergent extracts from CHO cells transfected with a vector expressing full-length mouse Ptc-1 (pRK5mptcNEN; provided by Phillip A. Beachy, Johns Hopkins University School of Medicine, Baltimore, MD) were prepared in a similar manner. Protein concentration in extracts was determined by BCA (Pierce Biotechnology). Aliquots containing 5 μg of protein were run under reducing conditions on 4-12% NuPAGE gels (Invitrogen; Carlsbad, CA), transferred to PVDF membranes, and probed using rabbit anti-Ptc-1 or control rabbit IgG. Bound antibody was detected using HRP-conjugated anti-rabbit IgG and ECL Plus Detection Kit (Amersham Pharmacia; Piscataway, NJ).
Immunohistochemical Staining
Efferent ducts were fixed by perfusion with Bouin's fixative through the abdominal aorta as described previously (Morales et al. 1996). Efferent ducts were then removed and immersed in Bouin's fixative for an additional 24 hr. Tissues were dehydrated in graded ethanol and embedded in paraffin. Immunostaining was accomplished using a microwave antigen-retrieval protocol described by Tacha and Chen (1994) in conjunction with the Zymed SP immunohistochemical staining kit (Zymed; South San Francisco, CA). Briefly, 5-μm-thick paraffin sections were deparaffinized in toluene and rehydrated with ethanol (90%-50%). Endogenous peroxidase activity was blocked by incubation in 3% H2O2 in methanol for 30 min. After the antigen-retrieval technique was performed and the slides cooled, nonspecific binding sites were blocked using 10% goat serum for 30 min. Sections were then incubated for 90 min at room temperature with antibodies against Shh (diluted to 0.28 μg IgG/μl) or Ptc-1 (diluted to 0.16 μg IgG/μl). Sections were then washed in PBS and incubated with biotinylated secondary antibody for 10 min and then with streptavidin-peroxidase for 10 min. Biotin/ streptavidin-peroxidase complexes were stained using the chromogen, diaminobenzidine. Counterstaining was performed using hematoxylin for 30 sec. As controls, Shh antibody (0.28 μg IgG/μl) was incubated with sections in the presence of recombinant ShhN (30 μg/μl), or sections were incubated with non-immune serum (1:100) and antibodies detected by immunoperoxidase reaction.
Immunogold Electron Microscopy
After infusion of ShhN, efferent ducts were perfused with 4% paraformaldehyde and 0.5% glutaraldehyde in 0.05 M phosphate, pH 7.5. Small segments of the fixed efferent ducts (~1-mm long) were dehydrated in ascending concentrations of ethanol (30, 50, 70, 90, and 100%) for 1 hr each, placed in 1:1 ethanol/Lowicryl for 1 hr, and then in a 2:1 Lowicryl/ethanol solution overnight. The tissue was then placed in pure Lowicryl for 4 hr before being embedded within gelatin caplets. Ultrathin sections were cut and placed on formvar-coated nickel grids as described previously (Sylvester et al. 1989). Subsequently, 40-μl drops of 10% goat serum were placed on a rubber mat within a Petri dish, and the grids with the tissue side down were incubated on the drops for 15 min. Grids were then placed on drops of anti-Shh antibody [1.24 μg IgG/μl in TBS (20 mM Tris-HCl, pH 7.4, 150 mM NaC)] and incubated for 1 hr. Four washes of 5-min each with 0.1% Tween-20 were followed by a second 15-min incubation with goat serum. Grids were then incubated in secondary anti-mouse antibody (18.5 μg/μl) conjugated to 10- to 15-nm colloidal gold particles (Cedarlane Labs), followed by four 5-min washes with 0.1% Tween-20 in TBS and two washes in distilled water. Finally, sections were counterstained with uranyl acetate for 2 min and lead citrate for 30 sec and then viewed on a Philips 400 electron microscope (Philips Electronics; Toronto, Ontario, Canada). For double-immunogold labeling studies, grids were incubated with antibodies to megalin and Shh, both diluted in TBS and 10% goat serum. Grids were washed with TBS and then incubated with goat anti-rabbit IgG conjugated to 10-nm colloidal gold particles and with goat anti-mouse IgG conjugated to 15-nm gold particles as per the protocol of Morales et al. (1996).
RT-PCR
Extraction of total RNA was performed with a Qiagen RNeasy kit (Mississauga, ON), and reverse transcription was done with the Invitrogen Superscript first-strand synthesis kit (Burlington, ON). PCR amplification reactions were conducted with Taq polymerase using the following cycling parameters: 30 cycles at 95C, 45 sec; 53C, 45 sec; 72C, 60 sec. Deoxyoligonucleotide primers for rat Ptc-1 were 5′-CTTGATGTGGCCCTTGTT-3′ (forward) and 5′-AAGGAGCAGAGGCCCAAT-3′ (reverse), generating an amplicon of 503 bp; amplification of rat β-actin was accomplished with the mouse β-actin primers 5′-CGGGACCTGACAGACTACCTC-3′ (forward) and 5′-AACCGCTCGTTGCCAATA-3′ (reverse), generating an amplicon of 218 bp.
Results
Efferent Duct Epithelial Cells Mediate ShhN Uptake
Efferent ducts were infused with [32P]-labeled ShhN and, after 5-min postinfusion, radioactive ShhN was observed in the form of silver grains associated with the apical surfaces of the epithelial cells as viewed by dark-field microscopy (Figure 1A). Few silver grains were apparent in the connective tissue underlying the efferent duct epithelium at this time. At 30-min postinfusion, large numbers of silver grains were observed within the epithelium (Figure 1B). Furthermore, the connective tissue surrounding the ducts also contained numerous silver grains, significantly more than were apparent at 5-min postinfusion (Figure 1B). Coinjection of [32P]-labeled ShhN along with the megalin antagonist RAP effectively reduced both the number of intracellular silver grains as well as the number of silver grains present in the tissue surrounding the ductules (Figure 1C). These findings indicate that the uptake of infused ShhN by ductal epithelial cells and delivery of the radioactive moiety to the subepithelial compartment is dependent on a RAP-sensitive receptor.
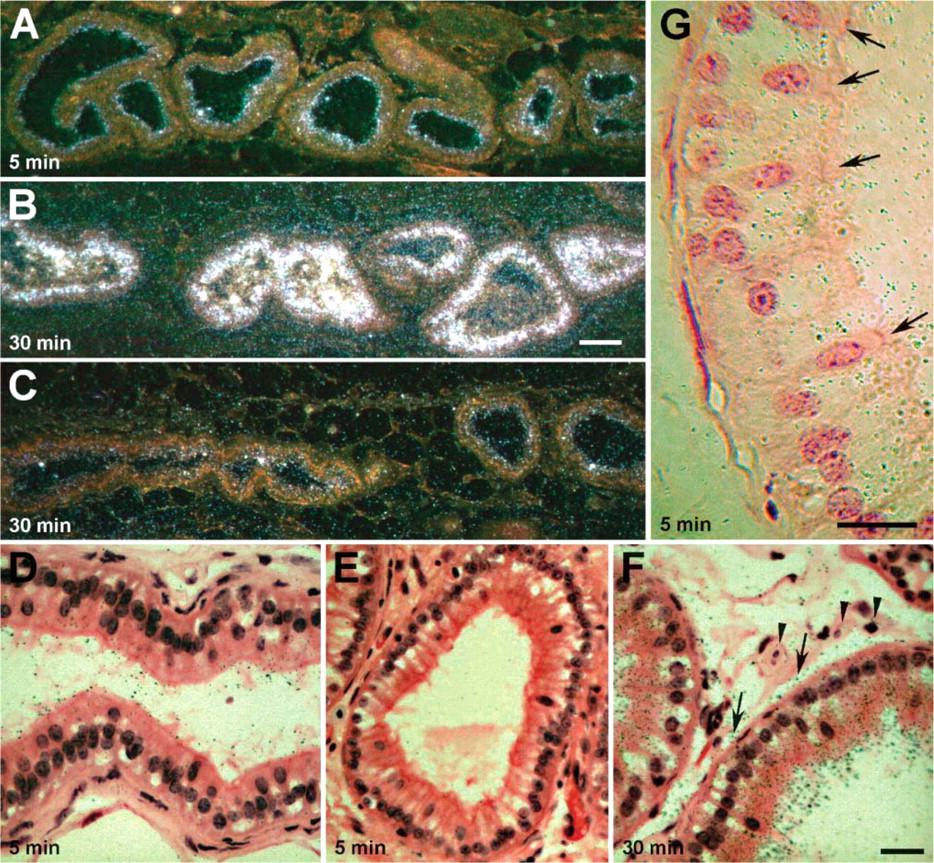
Autoradiographic analysis of non-lipid-modified sonic hedgehog (ShhN) uptake by efferent duct epithelial cells. (
Efferent ducts infused with [32P]-labeled ShhN were also examined using bright-field microscopy. As shown in Figure 1D, at 5-min postinfusion, silver grains could be seen decorating apical membranes and microvilli of the brush border of non-ciliated cells (9 ± 2 SD silver grains per non-ciliated cell; n = 20 cells). No silver grains were seen associated with these structures when [32P]-labeled ShhN was coinfused with RAP (Figure 1E). Few, if any, silver grains were detectable in apical aspects of ciliated cell (1.5 ± 1 SD silver grains per ciliated cell; n = 15 cells) (Figure 1G). Given the relatively large thickness of the emulsion, the possibility existed that the few silver grains found over the ciliated cells were exposed by nuclide decay events occurring in adjacent ciliated cells.
At 30 min following infusion, silver grains were abundant within the non-ciliated epithelial cells (Figure 1F). The highest concentration of silver grains was in the supranuclear regions of non-ciliated cells. Silver grains were also clearly apparent along basal aspects of the epithelial cells as well as on surfaces of smooth muscle cells and in interstitial spaces of the connective tissue surrounding the epithelium (Figure 1F).
The presence of silver grains within the connective tissue raised the possibility that ShhN was transcytosed and released into basolateral extracellular compartments of the efferent duct epithelium. However, the silver grains overlying the connective tissue could correspond to radiolabeled degradation products released following catabolism of the [32P]-ShhN (i.e., occurring within epithelial cell lysosomes). Thus, we performed immunocytochemical labeling and used LM and EM to evaluate the subcellular and tissue distribution of microinjected ShhN protein. Using immunnoperoxidase labeling and LM, we evaluated the distribution of infused ShhN in efferent ducts at different times following infusion. At 5-min postinfusion, anti-ShhN immunostaining was located within different-sized vesicles in the apical region of epithelial cells lining the efferent ducts (Figure 2A). A subset of epithelial cells, the ciliated cells (Figure 2A), which are known to lack megalin expression (Morales et al. 1996; Hermo et al. 1999) (see also Figure 2D), did not take up ShhN. After 30 min, ShhN immunostain was detected deeper within the non-ciliated cells, in the midregion of the cytoplasm as well as near the basal surfaces of some cells (Figure 2B). It is important to note that no endogenous ShhN immunostaining was detectable in efferent duct epithelial cells from rats that were not infused with ShhN (Figure 2C).
ShhN Undergoes Transcytosis and Lysosomal Degradation
Immunogold labeling and EM were used to characterize the subcellular distribution of ShhN endocytosed by efferent duct epithelial cells. At 5-min postinfusion of ShhN, immunogold staining for ShhN could be seen in 50- to 75-nm-diameter endocytic vesicles and 250-nm-diameter endosomes (Figure 3A) located near the apical surface of the non-ciliated cells. The pattern of gold particle labeling of endosomes indicated that ShhN was associated with the endosomal membrane and to a lesser degree was seen in the endosome interior (Figure 3A). Lysosomes showed no immunogold labeling at 5-min post-ShhN infusion (Figure 3A). When ShhN was coinfused with antagonists of megalin ligand binding (i.e., megalin antibodies or RAP, Figures 3B and 3C, respectively), immunogold particles were not detected in endocytic vesicles and endosomes. Immunogold particles were also absent from intercellular spaces between adjacent cells in the epithelium (Figure 3B). Within 30- and 60-min postinfusion, most of the electron-dense lysosomes within non-ciliated cells contained extensive immunogold labeling, with colloidal gold particles found throughout lysosome luminal compartments (Table 1; Figure 3D). Coinfusion of ShhN with anti-megalin IgG or RAP significantly decreased the ShhN immunogold label detectable within lysosomes of non-ciliated cells at 60-min postinfusion (Table 1; Figures 3E and 3F). When efferent duct tissue sections from animals injected with PBS alone were incubated with ShhN antibody and anti-IgG-gold conjugate and examined by EM, no immunogold labeling was detectable in any endocytic vesicle (Table 1).
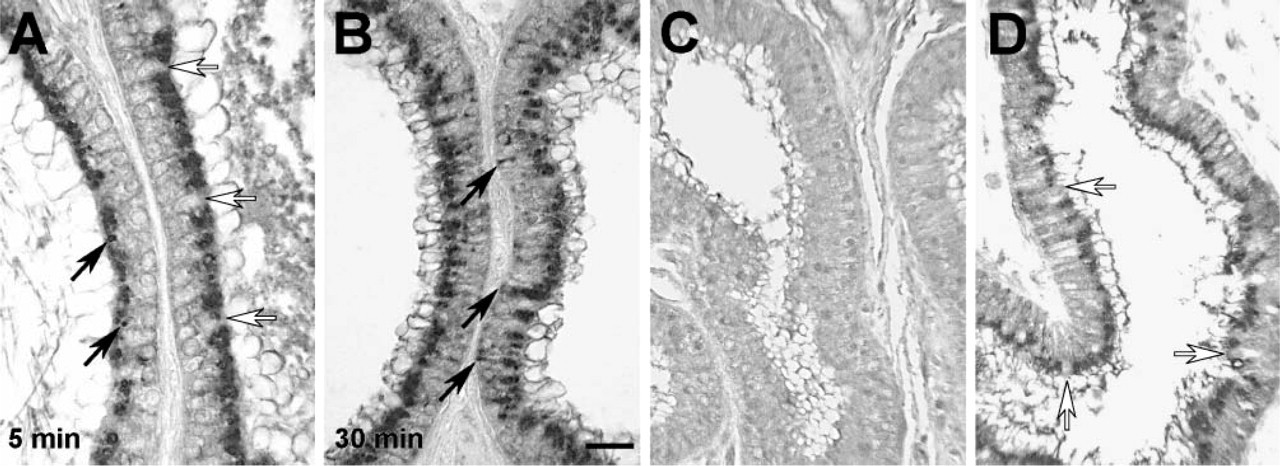
Efferent duct epithelial cell distribution of infused ShhN analyzed by immunnoperoxidase detection. Efferent ducts were infused with ShhN and after 5-min (
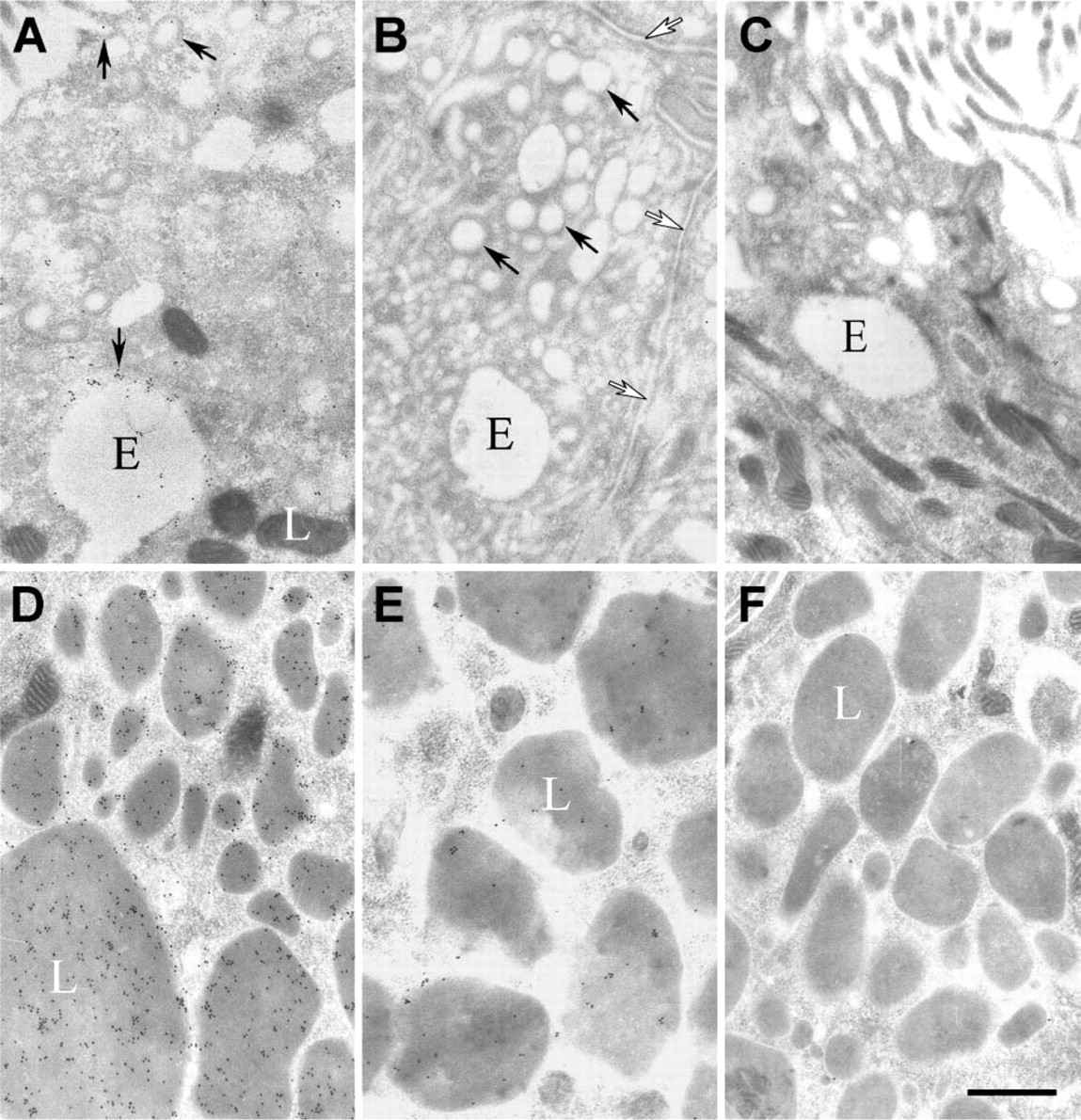
Immunogold electron microscopy analysis of the intracellular trafficking of ShhN. (
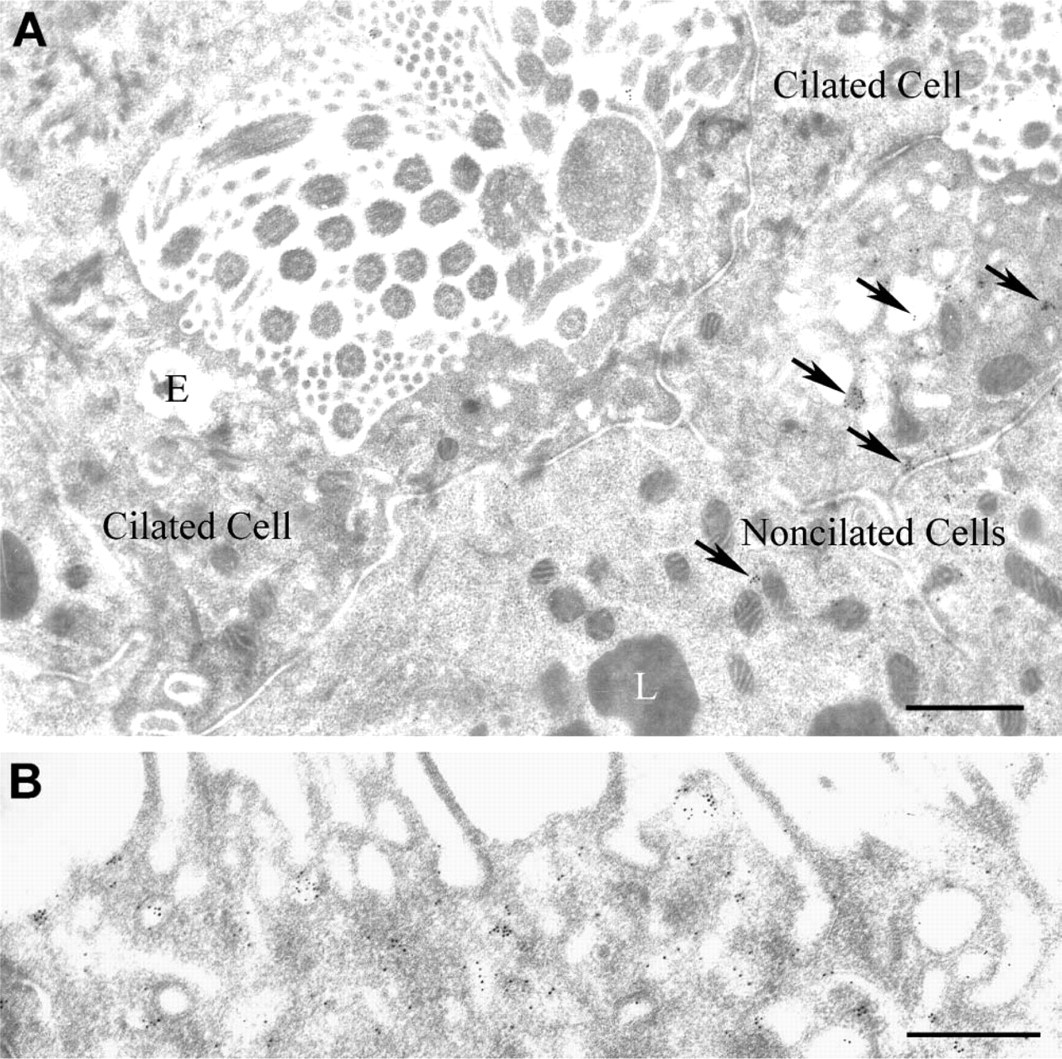
Whereas apical membrane regions of non-ciliated cells showed large numbers of gold particles within coated pits and vesicles at 5 min after ShhN infusion (Figures 3A and 4B), apical membrane regions of ciliated cells showed few, if any, gold particles at 5-min (Figure 4A) or at 30-min post-ShhN infusion (data not shown). These findings were consistent with those showing that few, if any, silver grains were detectable in the apical regions of ciliated cells at 5- and 30-min postinfusion of [32P]-labeled ShhN. The apparent lack of apical uptake of ShhN by ciliated cells correlates with the fact that they do not express megalin.
We next evaluated ShhN gold distributions in basolateral regions of the efferent duct epithelial cells. At 5-min postinfusion of ShhN, no immunogold staining for ShhN was present in the basolateral regions of the epithelial cells (not shown). However, at 30 min (Figure 5A) and to a greater extent at 60-min post-ShhN infusion (Figures 5C-5E), colloidal gold particles were present in association with extracellular surfaces of basolateral plasma membranes (Table 1; Figures 5A-5E). In areas where plasma membranes of adjacent non-ciliated cells were interdigitated, the density of ShhN-colloidal gold particles was highest along the extracellular surface of the plasma membrane (Figures 5A-5E). At 60-min postinfusion, ShhN-colloidal gold particles could be detected within vesicles at the basal surfaces of nonciliated cells (Figure 5D). Furthermore, ShhN-colloidal gold particles were also sparsely distributed in the extracellular space beneath basal regions of the efferent duct epithelial cells (Figure 5D).
Quantification of the number of anti-ShhN-colloidal gold particles in lysosomes and intercellular spaces of non-ciliated cellsa
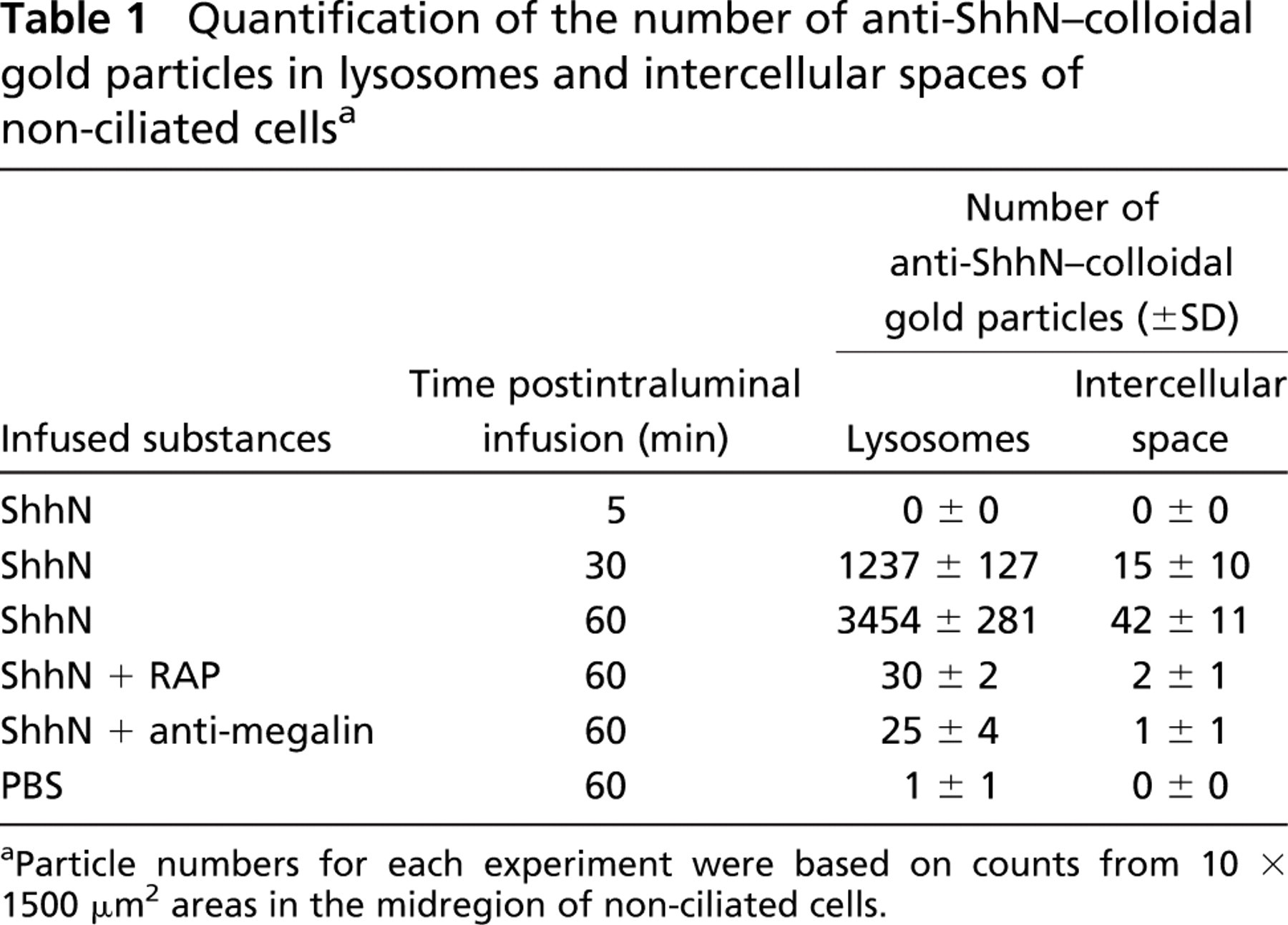
Particle numbers for each experiment were based on counts from 10 × 1500 μm2 areas in the midregion of non-ciliated cells.
At 60-min postinfusion, ShhN-colloidal gold particles could also be seen on the basolateral plasma membrane of ciliated cells in coated pit-like vesicles near endocytic vesicles (Figure 6A, inset). The apparent basolateral endocytosis of ShhN by ciliated cells raised questions as to the potential receptor that could be mediating this uptake, given that ciliated cells lack megalin. Because Ptc-1 has been demonstrated to play a role in Shh endocytosis (Incardona et al. 2000), we investigated the possibility that ciliated cells expressed Ptc-1. As shown in Figure 6B, immunohistochemical staining of the efferent ducts revealed that ciliated cells, but not non-ciliated cells, expressed Ptc-1. Furthermore, subcellular distribution of Ptc-1 in ciliated cells extended throughout the cell including basolateral regions (Figures 6B and 6C), as opposed to the apical distribution of megalin found in non-ciliated cells (Figure 2D). The cilia of ciliated cells did not show anti-Ptc-1 immunolabeling. The immunohistological detection of Ptc-1 in ciliated cells was consistent with findings from RT-PCR analysis, which showed Ptc-1 transcripts in RNA isolated from efferent ducts (Figure 6D). Furthermore, immunoblot analysis of extracts from efferent duct using Ptc-1 antibodies detected an ~170-kDa polypeptide as well as immunoreactive polypeptides of ~100, 90, and 75 kDa. It is not known whether these later polypeptides are the result of proteolytic cleavage or alternative splicing.
Double-immunogold labeling was performed to determine the extent to which Shh and megalin colocalize within endocytic compartments. Anti-mouse IgG conjugated to 15-nm gold was used to detect bound anti-Shh antibody, and anti-rabbit IgG conjugated to 10-nm gold was used to detect bound anti-megalin antibody. Labeling with gold particles of both sizes was observed in pale endocytic vesicles and endosomes (Figures 7A and 7B). On the other hand, only large-size gold particles decorated the electron-dense lysosomes (Figure 7B). In endocytic vesicles and endosomes the small-size particles were seen associated with the membrane of these structures (Figures 7A and 7B). None of these antibodies labeled ciliated cells (data not shown).
Discussion
Although genetic and biochemical evidence support a role for megalin in regulation of Shh signaling in vertebrates (McCarthy et al. 2002; Spoelgen et al. 2005), the mechanism by which this occurs is not clear. Here we present evidence supporting a role for megalin in mediating both in vivo lysosomal targeting and transcytosis of Shh infused into the lumen of a vertebrate tubular epithelium.
Immunogold electron microscopy analysis of ShhN within apical regions of ciliated epithelial cells.
Endocytosis of Shh leading to lysosomal degradation can be a means by which Shh gradients are shaped in vivo. Based on our immunogold EM, the bulk of the ShhN endocytosed by the efferent duct epithelial cells was targeted to lysosomes, thus suggesting efficient degradation. This is in contrast to what has been observed in endoderm-like cells where the majority of ShhN internalized by megalin is not degraded in lysosomes (McCarthy et al. 2002; Spoelgen et al. 2005). Therefore, trafficking of ShhN internalized via megalin is differentially regulated. The proportional targeting of ShhN to the lysosome by megalin should now be considered as a factor that can influence Shh gradient formation.
The importance of endocytosis in the context of planar movement of Shh through an epithelium is a topic receiving considerable scrutiny (Zhu and Scott 2004). Findings of Han et al. (2004) showed that the planar movement of hedgehog during Drosophila wing development is not dependent on dynamin and therefore not dependent on clathrin-mediated endocytosis. By contrast, our findings indicate that endocytosis of ShhN by megalin, a receptor associated with clathrincoated pits and vesicles (Czekay et al. 1997), can lead to ShhN being trafficked to basolateral regions of non-ciliated epithelial cells. This conclusion was based on quantitative analyses showing that ~1.2% of the ShhN endocytosed in efferent duct epithelial cells was detected in basolateral spaces. No labeling of basolateral spaces was observed when megalin function was blocked with RAP or megalin antibody. Therefore, presence of ShhN in the basolateral space is not due to rupture of tight junctions during the infusion of the protein but is a consequence of its transport to the basolateral compartment. The observation that ShhN can also be found on basolateral membranes of ciliated cells in coated pits and endocytic vesicles raises the possibility that ShhN endocytosed via megalin is basolaterally transported from non-ciliated cells to ciliated cells where it is endocytosed by a receptor other than megalin. The findings provide evidence that megalin-mediated endocytosis of ShhN can contribute to planar movement of the morphogen in certain epithelia.
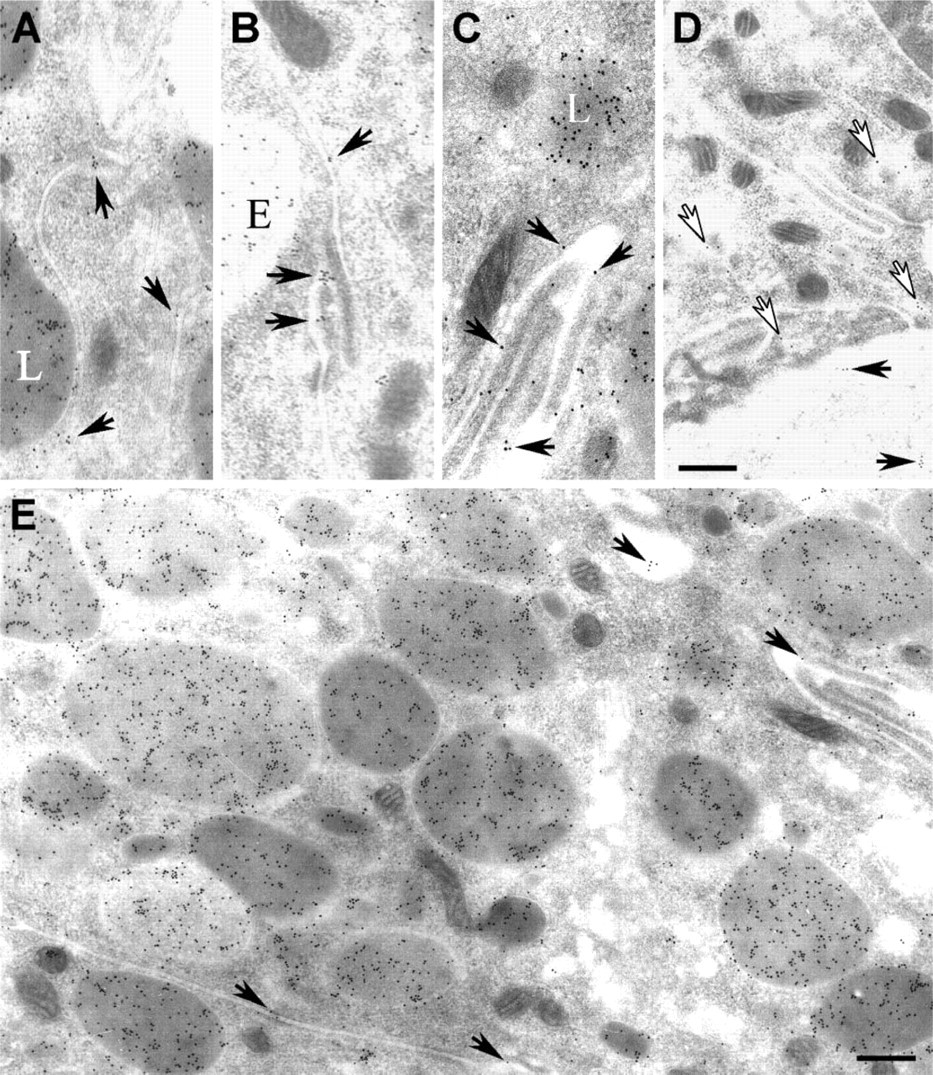
Immunogold electron microscopy analysis of the trafficking of ShhN to basolateral regions of epithelial cells. Shown are electron micrographs of anti-ShhN-gold labeling of basolateral regions of efferent duct non-ciliated epithelial cells from rats 30 min
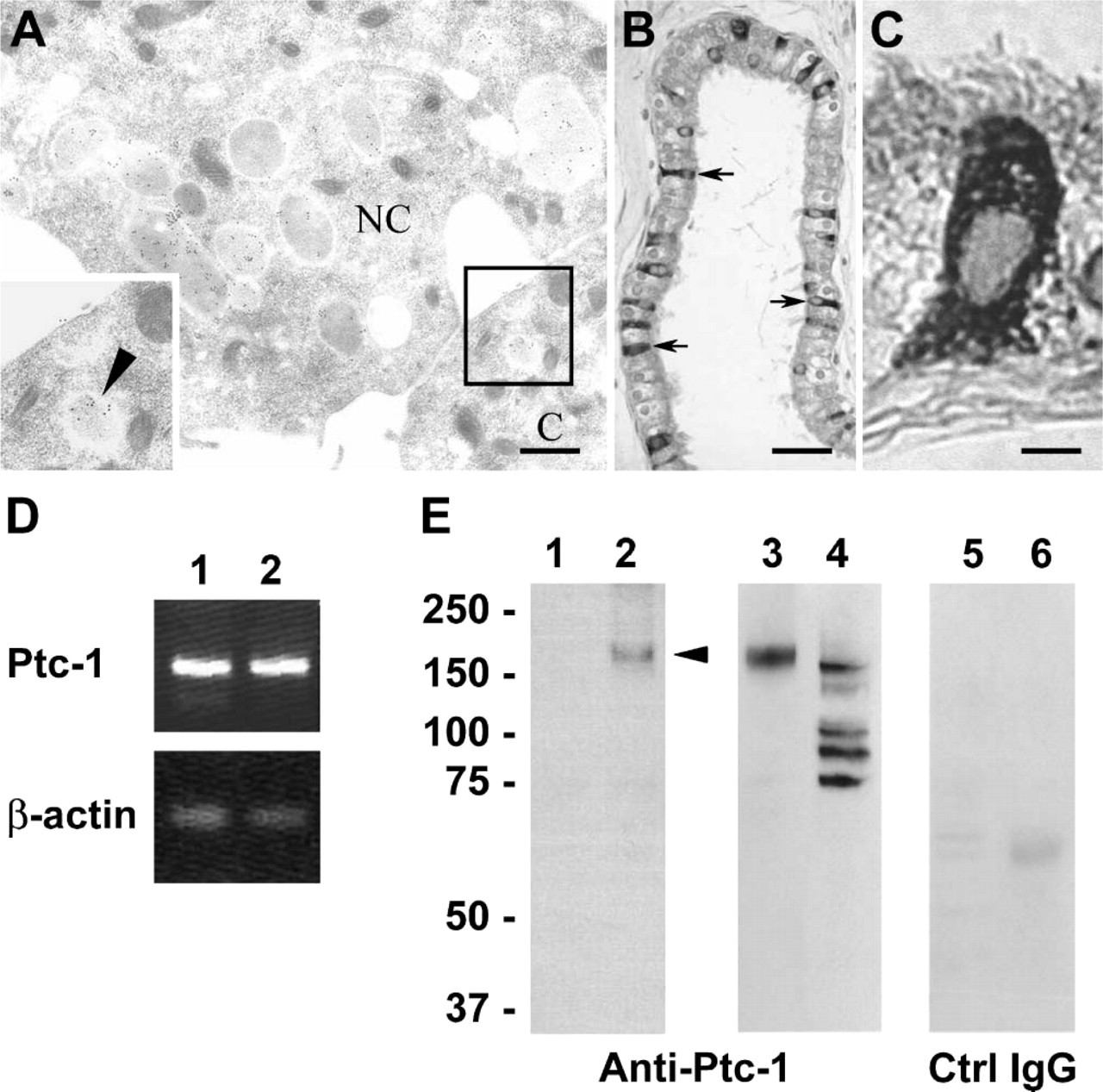
Immunogold labeling of ShhN in basolateral regions of epithelial cells lining the efferent ducts and detection of patched-1 (Ptc-1) expression in ciliated cells.
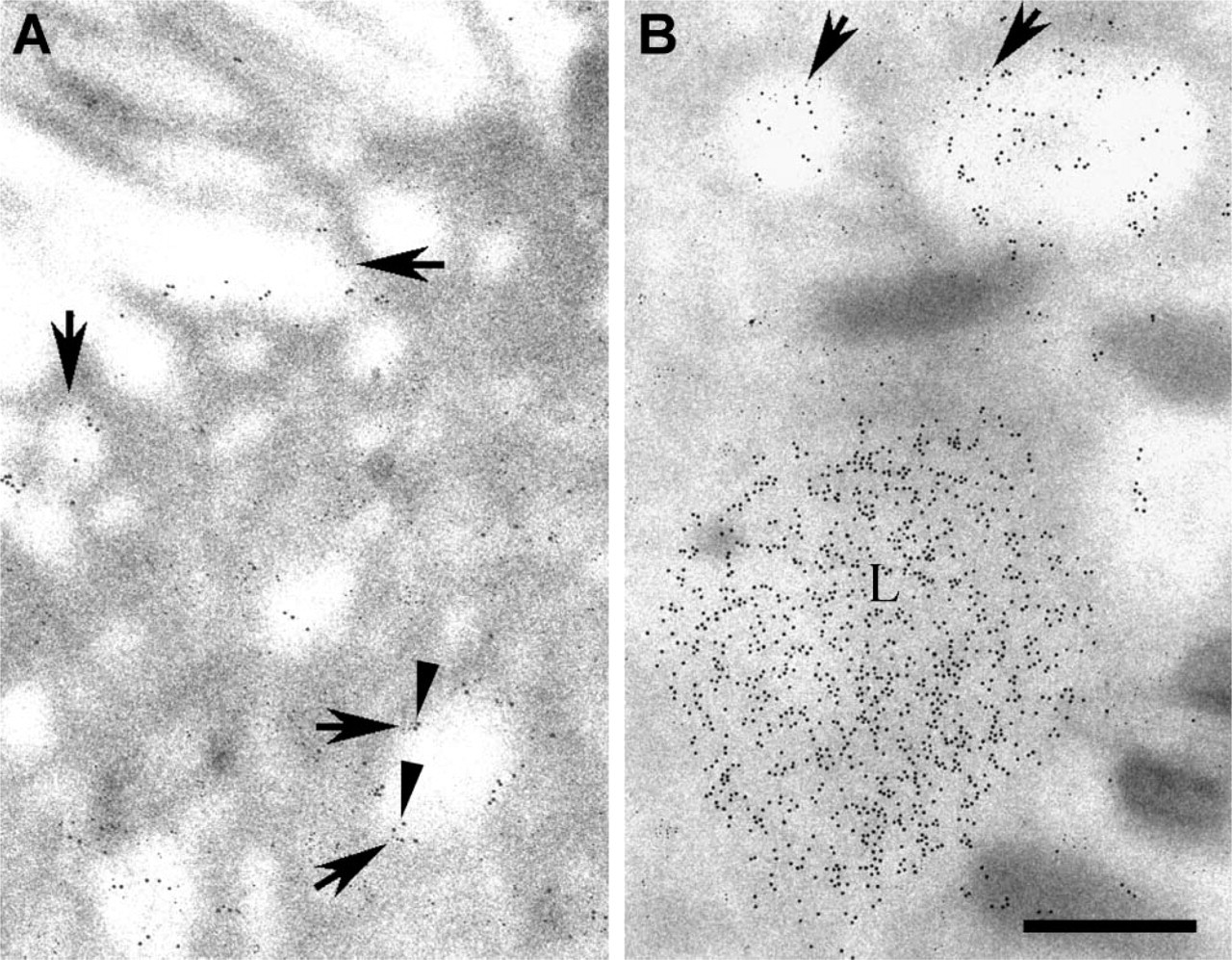
Double-immunogold labeling of megalin and ShhN in a non-ciliated cell.
Our findings also indicate that megalin-mediated endocytosis can lead to transport of ShhN to basal regions of epithelial cells and release into the subepithelial compartment. Several recent studies have shown that megalin acts in the process of transepithelial transport of various macromolecules (Marino et al. 2000,2001; Marino and McCluskey 2000; Nielsen et al. 2001; Pan et al. 2004). The importance of transepithelial transport of Shh has not been extensively investigated. However, both megalin and Shh localize to apical surfaces of neuroepithelial cells (Willnow et al. 1996; Gritli-Linde et al. 2001; McCarthy et al. 2002), both in floor-plate cells that function as a major Shh source during patterning of the neural tube and in Shh-responsive cells at a considerable distance dorsal to the floor plate. It is likely that Shh is released apically from the floor plate, and our findings support a plausible role for megalin-mediated transcytosis of luminally derived Shh during neural tube patterning.
The mechanism by which certain megalin ligands can bypass lysosomal degradation is not known. One possibility is that the interaction between megalin and these ligands might not be readily dissociated by acidic pH such as occurs in acidic endocytic vesicles and in the compartment for uncoupling of receptor and ligand. As a consequence, ligands might traffic together with the receptor and be delivered to apical or basolateral regions of the cell (McCarthy et al. 2002). The interaction between megalin and ShhN has been shown to be resistant to dissociation by low pH (McCarthy et al. 2002). However, the findings presented here indicate that most of the ShhN endocytosed by non-ciliated cells is targeted to lysosomes. Furthermore, double labeling indicates that megalin is not trafficked to the lysosomal compartment, indicating that the megalin-Shh complex is indeed dissociable. These findings do not preclude the possibility that some fraction of the megalin-Shh complex are transcytosed or that Shh becomes dissociated from megalin and is segregated into cargo vesicles destined to the basolateral space. In support of the chaperon hypothesis are findings that megalin is detected in the basolateral regions of absorptive epithelial cells in vivo (Drake et al. 2004), and ectodomain components of megalin have been found in complex with thyroglobulin transcytosed by cultured megalin-expressing cells as well as in the blood of rats and humans (Marino et al. 2000). However, the fact that we could not detect megalin on the basolateral plasma membranes of non-ciliated cells tends to weaken the possibility that megalin is chaperoning Shh in these cells, although detection sensitivity of the immunogold labeling technique must be considered.
Finally, we show that non-ciliated cells that express megalin, but not Ptc-1, efficiently endocytose ShhN, whereas ciliated cells that express Ptc-1, but not megalin, do not efficiently internalize ShhN (via apical entry). These findings highlight potentially important differences in functions of megalin and Ptc-1 in polarized epithelia with respect to endocytosis. In non-ciliated cells, megalin was apically localized as it is in many absorptive epithelial cells. By contrast, Ptc-1 expressed by ciliated cells was not apically localized but was distributed throughout the cells, including basolateral regions. One explanation for this staining is that the antibody is not only detecting Ptc-1 in intracellular compartments such as endoplasmic reticulum, Golgi, and secretory vesicles but also detecting proteolytic fragments of Ptc-1 released into the cytoplasm. Indeed, the intracellular carboxy terminal domain of Ptc-1 is proteolytically cleaved by caspase-3 (Thibert et al. 2003).
The arrangement of megalin-positive, Ptc-1-negative cells lying adjacent to Ptc-1-positive cells as seen in the efferent ducts is similar to the arrangement of neural epithelial cells in which floor plate cells are negative for Ptc-1 (Concordet et al. 1996) but positive for megalin (McCarthy et al. 2002), and epithelial cells adjacent to floor plate cells are Ptc positive (Concordet et al. 1996). It is therefore possible that Shh expressed in the floor plate is apically endocytosed and transcytosed via megalin and then delivered basolaterally to Ptc-1-positive cells.
In conclusion, our investigation provides evidence for the lysosomal targeting and transcytosis of Shh in mammalian epithelial cells in vivo and highlights a potential mechanism by which megalin may influence Shh morphogen gradients.
Footnotes
Acknowledgements
This work was supported by National Institutes of Health Grant DE-14347 (to WSA).
The authors thank Dr. Phillip Beachy for providing the pRK5mptcNEN Ptc1 expression construct.