
Abstract
The brain changes in volume and composition with normal aging. Cellular components of the brain are supported by an extracellular matrix (ECM) comprised largely of hyaluronan (HA) and HA-associated members of the lectican family of chondroitin sulfate proteoglycans (CSPGs). We examined regional differences in microvascular density, neuronal and glial markers, and accumulation of HA and CSPGs in mouse brains during normal aging. The cortex, hippocampus, dentate gyrus, and cerebellum of young (4 months), middle-aged (14 months), and aged (24–26 months) brains were analyzed. Microvascular density decreased in cerebral cortex and cerebellum with age. There were no detectable differences in neuronal density. There was an increase in astrocytes in the hippocampus with aging. HA accumulation was higher in aged brain relative to young brain in the cerebral cortex and cerebellum, but not in other regions examined. In contrast, CSPGs did not change with aging in any of the brain regions examined. HA and CSPGs colocalized with a subset of neuronal cell bodies and astrocytes, and at the microvasculature. Differences in accumulation of ECM in the aging brain, in the setting of decreased microvascular density and/or increased glial activation, might contribute to age-related regional differences in vulnerability to injury and ischemia.
Keywords
Introduction
There are generally decreases in brain volume with normal aging.1–5 Much of the loss of brain size is attributed to reductions in neuronal and microvascular density, which then confer greater vulnerability to dysfunction as a result of injury, neurodegeneration, or poor perfusion. Neurons and microvasculature are supported by the extracellular matrix (ECM), which is comprised largely of hyaluronan (HA) and HA-associated ECM, such as the lectican family of the chondroitin sulfate proteoglycans (CSPGs).6–9 Changes in the ECM with normal brain aging have not been well studied. Consequently, the potential contribution of ECM to age-related changes in brain anatomy and function, in the absence of specific pathology, is not known.
HA is a non-sulfated glycosaminoglycan (GAG) polymer comprised of repeating disaccharides consisting of D-glucuronic acid and N-acetyl-D-glucosamine. 10 HA is synthesized by HA synthases (HASes) 1 and 2, and to a lesser extent HAS 3, as a large polymer of high molecular weight (MW >1000 kDa), but is rapidly degraded to smaller sizes primarily as a result of hyaluronidase (HYAL) activity. In most organs, including the central nervous system, low MW HA (<50 kDa) promotes inflammation.11–13 In the brain, activity of HASes 1–3 accounts for HA accumulation, and HYALs 1–3 are reported to be the main drivers of HA degradation. 14 Multiple cell types express the enzymes responsible for HA synthesis and HA turnover, but it is likely that astrocytes are the predominant source of both HASes and HYALs in the brain. 15
The stability of HA is also determined by the presence of proteoglycans, such as the subset of CSPGs that contains an HA-binding domain. In the brain, the most common CSPGs are the lecticans, which include aggrecan, versican, neurocan, and brevican. 16 Lecticans are characterized by a C-terminal C-type lectin domain, an N-terminal HA-binding domain, and a highly diverse central domain with attached GAG side chains and other poly- and oligosaccharides. The synthesis and breakdown of CSPGs is controlled by more than 15 enzymes and other mediators. 17 Similar to HA, CSPG accumulation is increased in the setting of injury and ischemia and can have both beneficial and detrimental impacts on neuronal tissues. For example, the synthesis of CSPGs can limit glial scar formation, but also creates a barrier that prevents differentiation of oligodendrocyte precursor cells and subsequent remyelination of injured neural tissue. 18
HA and CSPGs are present in both gray and white matter and usually appear in a diffuse pattern or in a localized, pericellular condensed form.6,8 In the diffuse pattern, HA and CSPGs are distributed in multiple brain regions and associate with each other in extracellular spaces. In the condensed form, HA and CSPGs often localize with perineural nets (PNNs), which are specialized structures that surround a subset of neurons that includes inhibitory interneurons. 8 PNNs are responsible for both synaptic stabilization and plasticity,19,20 and their presence can be altered by aging, injury, and neurodegenerative diseases.20,21 HA and CSPGs also accumulate in the microvasculature, which expresses transporters that independently modulate the influx of brain nutrients and the efflux of toxic substances through the blood-brain barrier. The brain vasculature and associated astrocytic end feet that border the perivascular space in the glymphatic system also control convective flow and clearance of cerebrospinal and interstitial fluids, thereby preventing the buildup of deleterious substances in the brain.22,23
We propose that there are changes in brain ECM during normal aging that confer vulnerability to injury and ischemia. In this study, we examined regional differences in microvascular, neuronal, and glial cell density to assess key vascular and cellular components that are responsible for synthesis and turnover of ECM. We utilized young and aged murine brains, an accepted model for brain aging. 24 We then analyzed the accumulation of HA and CSPGs in selected brain regions during normal aging.
Materials and Methods
Animals
Young (4 months, n=15–20), middle-aged (14 months, n=4–9), and aged (24–26 months, n=15–20) male mice of the C57BL/6 strain were obtained from the National Institute of Aging Rodent Colony at Charles River Laboratories (Reno, NV). Mice were acclimated for 5 days before euthanasia. The Office of Animal Welfare at the University of Washington approved the care of mice and all procedures.
Mice were anesthetized, and the heart was perfused with 20 ml of serum-free DMEM (Gibco/ThermoFisher; Waltham, MA) to remove all blood components from the vasculature. After decapitation, the brain was removed and fixed for embedding.
IHC and Immunofluorescence (IF)
Brains were fixed in 10% neutral-buffered formalin (NBF), paraffin-embedded, and sectioned at 5 μm. Slide-mounted sections were deparaffinized, and the antigens unmasked by boiling in citrate buffer (Item 3300; Vector Laboratories; Burlingame, CA) for 40 min, followed by cooling for 20 min. For IHC, endogenous peroxidases were inactivated in 3% H2O2 in TBS for 30 min. The sections were then blocked in 2% goat serum and exposed to 2–5 μg of the following primary antibodies: polyclonal rabbit anti-mouse/human platelet endothelial cell adhesion molecule (PECAM)/CD31; Item ab28364; Abcam; San Francisco, CA), monoclonal mouse anti-mouse NeuN (Item MAb377; MilliporeSigma; Burlington, CA), and polyclonal rabbit anti-mammalian glial fibrillary acidic protein (GFAP; Item ab7260; Abcam). Non-antibody primary affinity reagents used were biotinylated HA-binding protein (bHABP; Item 385911; Calbiochem/MilliporeSigma) and biotinylated Wisteria floribunda agglutinin (bWFA), which binds to the N-acetylgalactosamine residues of CSPGs (Item L1516; MilliporeSigma).
Bound bHABP or bWFA, and bound primary antibodies (after exposure to the appropriate biotinylated secondary antibody) were visualized with Vectastain Avidin-Biotin Complex (ABC; Item PK-6105; Vector) in conjunction with 3,3′-diaminobenzidine (DAB; Item SK-4105; Vector). Cell nuclei were stained with hematoxylin. Secondary antibody/ABC staining alone served as negative controls, and human brain samples served as positive controls. The DAB-stained sections were viewed by conventional brightfield imaging using a Leica DM2500 microscope (Leica Microsystems Inc.; Buffalo Grove, IL) equipped with a SPOT Insight 4 megapixel color CCD camera (Diagnostic Instruments; Sterling Heights, MI).
To verify the specificity of HABP and WFA staining, selected sections were pretreated with HYAL or chondroitinase, respectively. To pretreat with HYAL, sections were exposed to 20 Units/ml of Streptomyces hyalurolyticus HYAL (Item H1136; MilliporeSigma) in 50 mM NaOAc, 0.15 M NaCl, pH 6.0 for 1 hr at 37C and then exposed to bHABP to detect HA, as described above. To pretreat with chondroitinase, sections were exposed to 0.2 Units/ml of Proteus vulgaris chondroitinase ABC (Item C3667; MilliporeSigma) in Tris buffer with 1 mg/ml BSA for 1 hr at 37C, followed by exposure to biotinylated WFA to detect CSPGs, as described above.
For double IF, sections were exposed to bHABP or bWFA in conjunction with one of the primary antibodies to NeuN, GFAP, or CD31 described above. Bound bHABP, bWFA, and primary antibodies were visualized by exposure of the sections to Alexa-Fluor 488-strepavidin (for bHABP and bWFA), Alexa-Fluor 594-goat anti-rabbit IgG for GFAP and PECAM, and Alexa-Fluor 594 goat anti-mouse IgG for NeuN (all Alexa-Fluor conjugates were obtained from ThermoFisher). Images were recorded using an epifluorescence microscope (Leica model DMR) equipped with a SPOT RT 1.4 megapixel color/monochrome CCD camera (Diagnostic Instruments). Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI).
Image Analysis
For quantification of neuronal, glial, and vessel density, the stained area was expressed as an area fraction (area of the section stained with PECAM, NeuN, and GFAP/area of the standard field) × 100% using ImageJ (National Institutes of Health image analysis freeware, http://imagej.nih.gov/ij/). Analysis of vessel density was based on appropriate morphology and PECAM staining. Deposition of HA and CSPGs in DAB-stained sections was graded independently by three separate observers and then confirmed by ImageJ (for these analyses, the sections were not counterstained with hematoxylin). All staining and image acquisitions for each antibody were performed in parallel, using identical acquisition settings and exposure times. To express DAB staining as intensity, there was modification of the threshold in the blue channel until the DAB-stained areas showed optimal contrast in the red channel, and at least four digital images per section were obtained at 10× magnification. The images were opened in ImageJ and the brightness and contrast adjusted before conversion to red-green-blue (RGB)-stacked images and measurement of staining intensity.
Statistical Analysis
Initial comparisons for significant differences among aged, middle-aged, and young mice were made by ANOVA. Significant differences between aged and young mice were then determined using a paired Student’s t-test with unequal variance. Statistical significance was defined as p<0.05.
Results
There were no apparent differences between young and middle-aged brains or middle-aged and aged brains of the mice in any of the regional parameters examined. This indicated that brain aging occurs at a steady rate with no abrupt acceleration of changes at middle age. Accordingly, further comparisons focused on differences between the young and the aged groups of brains. The regions selected were based on their relevance to aging: cerebral cortex, cerebellum, hippocampus, and dentate gyrus (Fig. 1).25–27 Studies in the cortex focused on gray matter, which comprises the majority of the cortex in the mouse.28,29 All layers of the cerebellum, hippocampus, and dentate gyrus were assessed.
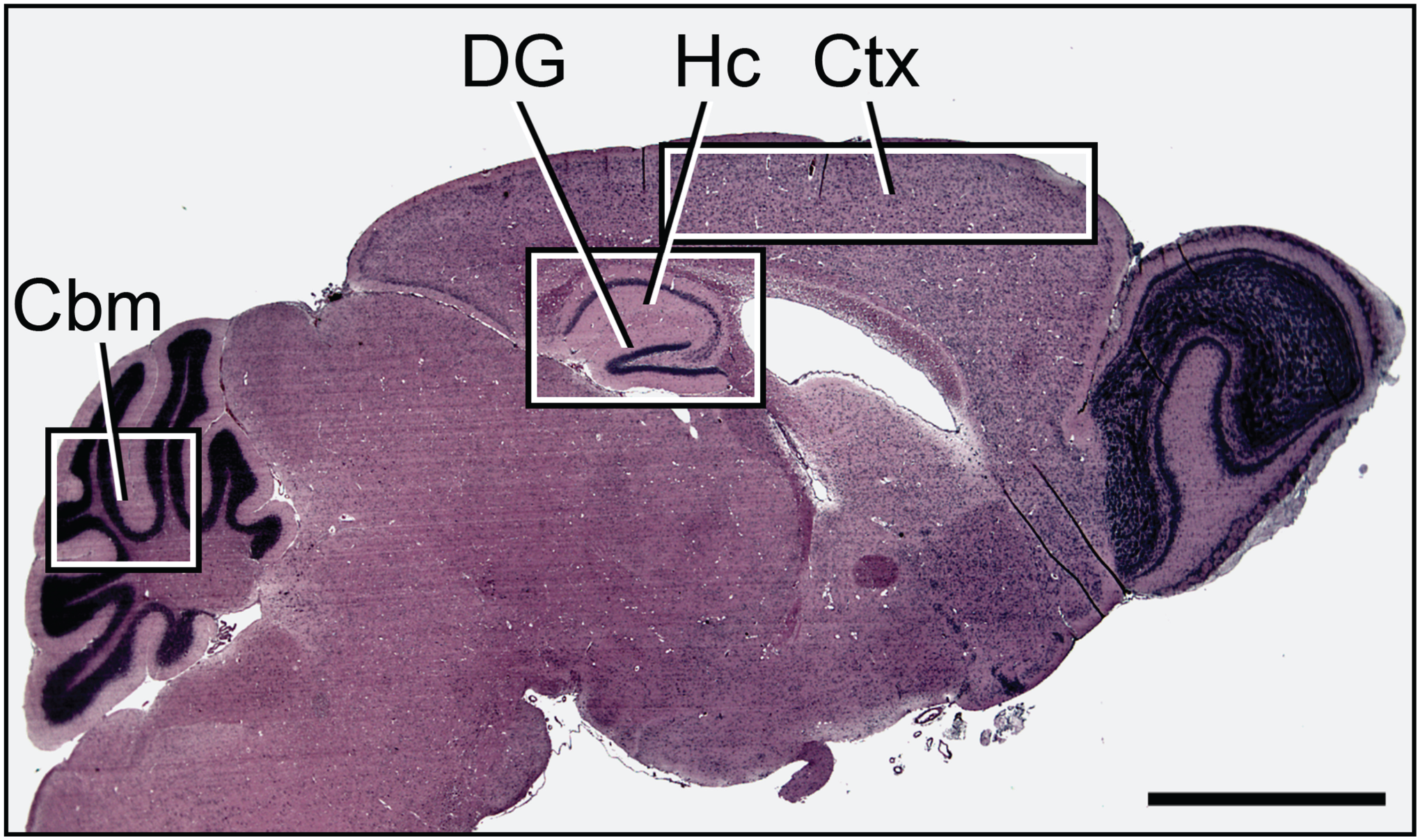
Overview of the brain regions examined. Shown is cresyl violet stain of regions of interest in the mouse brain. Scale bar = 2 mm. Abbreviations: Ctx, Cortex; Hc, hippocampus; DG, dentate gyrus; Cbm, cerebellum.
We first focused on key cellular components (microvasculature, neurons, and astrocytes), which contribute to brain volume and biosynthesis of ECM (Fig. 2). As expected, based on prior studies by our group and others, there was a significant decrease in microvascular density (as defined by vessel morphology and PECAM staining) in the cerebral cortex and cerebellum of aged brains relative to young brains30–33 (Fig. 2A–E). In contrast, we were not able to find differences in the density of neuronal bodies, as detected by NeuN, in aged brains relative to young brains (Fig. 2F–J). There was a significant increase in GFAP-positive cells, indicating activated astrocytes, in the hippocampus of aged brains relative to young brains, but not in the other regions examined (Fig. 2K–O). The cortex had very few GFAP-positive cells, which limited the detection of differences in this area (Fig. 2L–M).
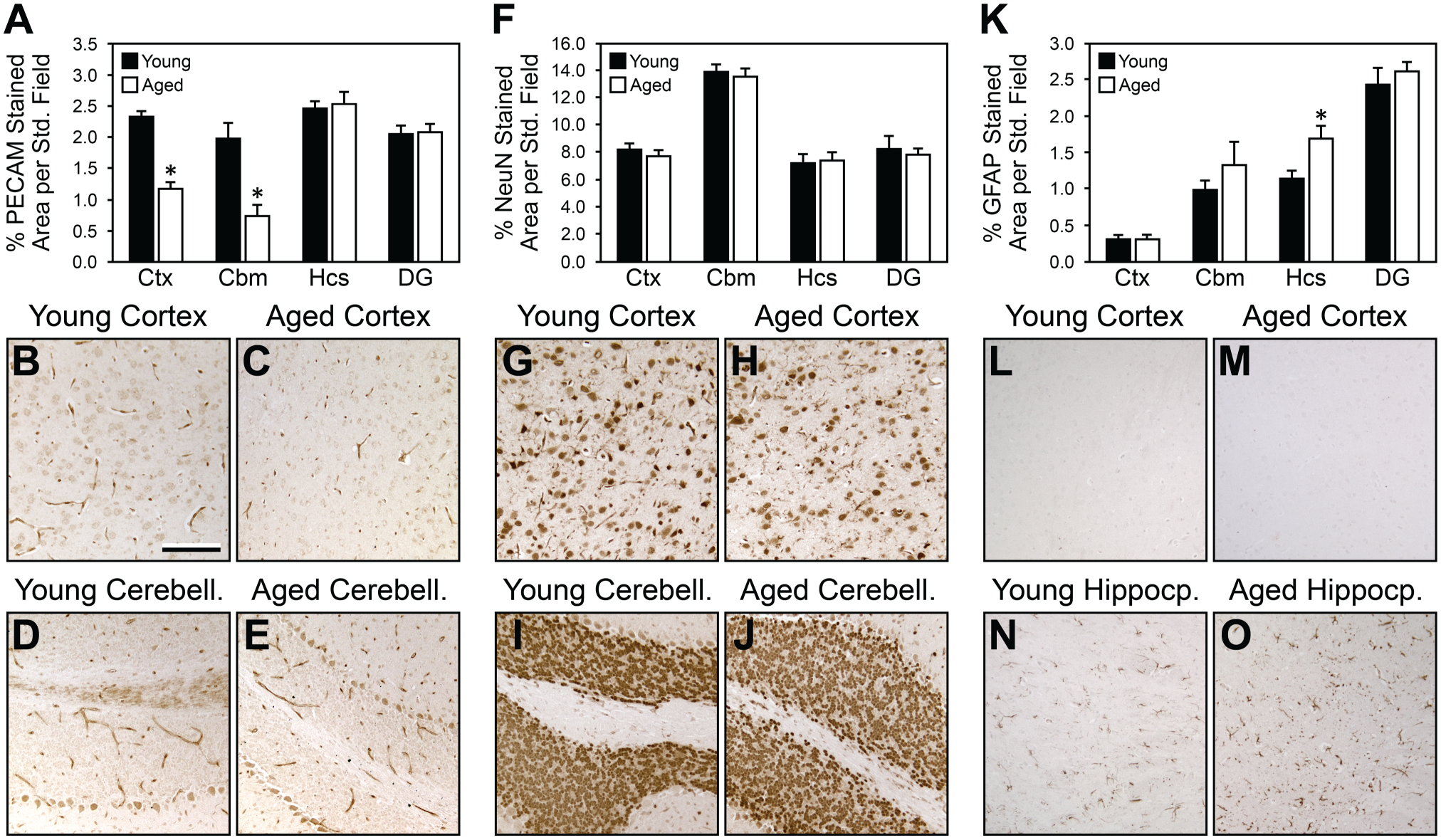
Measurement of the density of microvessels, neurons, and glial cells in young versus aged brains by IHC (brown DAB staining). (A) Histogram shows decreased density of microvessels (as defined by morphology and PECAM staining) in aged brain versus young brain in Ctx and Cbm, but not in Hcs or DG. (B–E) show representative PECAM staining in young and aged Ctx and Cbm. Non-vascular structures in the Cbm that cross-reacted with the PECAM antibody were not included in the analysis. (F) Histogram of NeuN staining shows no difference in neuronal density in young versus aged brain regions. (G–J) show representative NeuN-positive cells in young and aged Ctx and Cbm. (K) Histogram of GFAP staining shows an increase in astrocytes in the aged versus young Hcs. This difference was not detected in the other regions examined. Panels L–O show stain for representative GFAP-positive cells in young and aged Ctx and Hcs. Scale bar in B = 100 μm. Abbreviations: DAB, 3,3’-diaminobenzidine; Ctx, cortex; Cbm, cerebellum; Hcs, hippocampus; DG, dentate gyrus; GFAP, glial fibrillary acidic protein; PECAM, platelet endothelial cell adhesion molecule.
We then examined HA accumulation, as detected by HABP (Fig. 3A–F). HA was present in both diffuse and localized patterns in all brain regions examined. HA was significantly increased in aged brains relative to young brains in the cortex and cerebellum. This increase was primarily due to greater amounts of diffuse HA accumulation in aged relative to young cortex and cerebellum. There were no significant differences in HA accumulation with aging in the hippocampus or dentate gyrus. However, there was more diffuse and localized HA in the CA2 and CA3 regions relative to other hippocampal regions and the dentate gyrus. There was also less HA accumulation in the area of the hippocampus that was adjacent to the dentate gyrus. In the dentate gyrus, there was very little accumulation of diffuse HA and rare instances of localized HA (usually in the granular layer), irrespective of age. Brain sections either pretreated with HYAL (Fig. 3G) or exposed to secondary labeling agents only (Fig. 3I) were not stained, indicating the specificity of the HABP label for HA. In contrast, pretreatment of sections with chondroitinase (Fig. 3H) did not eliminate HABP staining for HA.
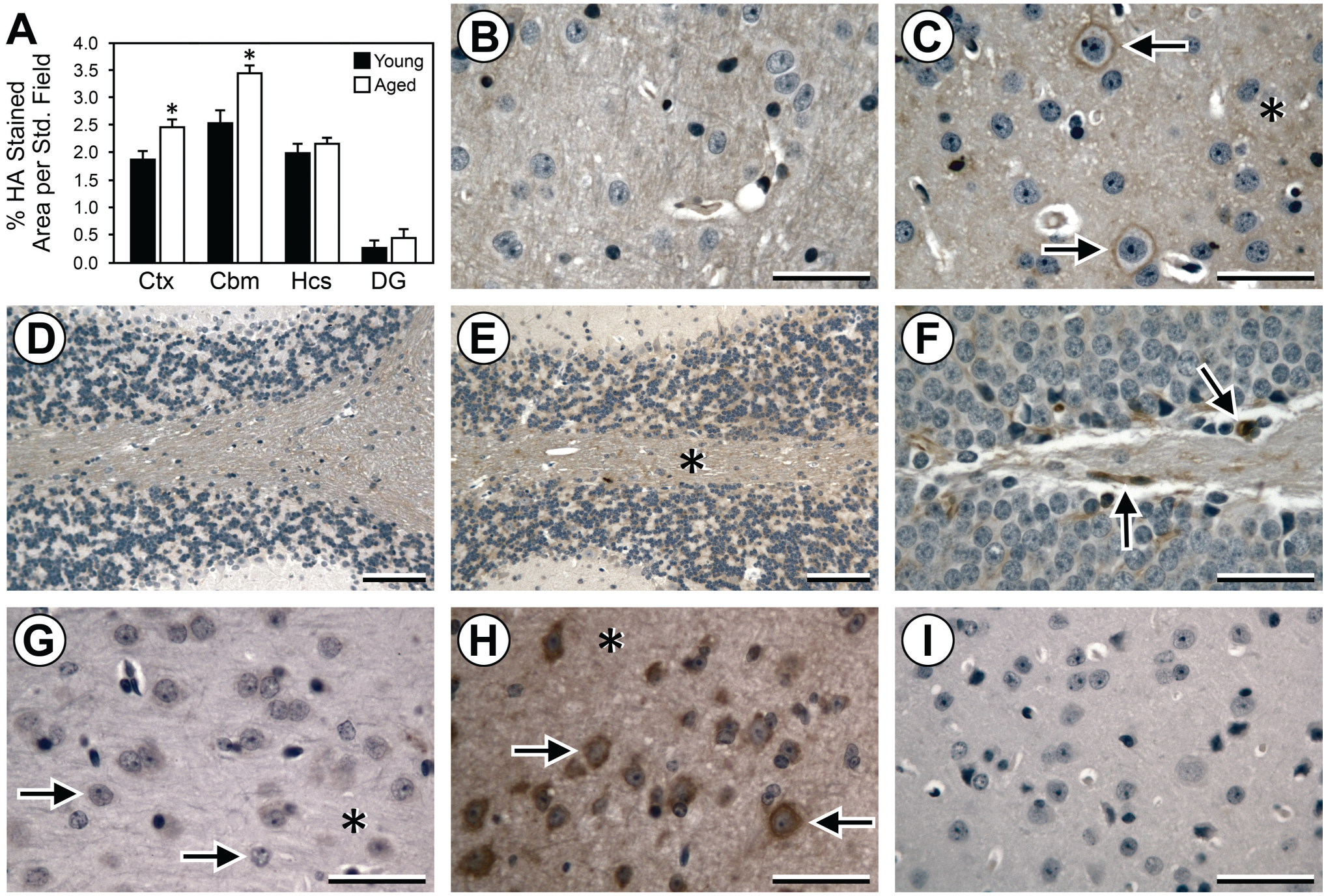
Measurement of the accumulation of HA in young versus aged brains by IHC. (A) Histogram shows increased accumulation of HA (HABP staining) in aged brain versus young brain in Ctx and Cbm, but not in Hcs or DG. (B–E) Representative IHC (brown DAB staining) for HA in young Ctx (B), aged Ctx (C), young Cbm (D), and aged Cbm (E). Increased HA accumulation in aged Ctx and Cbm is largely due to increases in diffuse staining (C, E; asterisks) with additional increases in localized perineural staining in Ctx (C; arrows). Panel F shows HA accumulation in a subset of the cells (arrows) of the granular layer of the DG of young brain, which is not demonstrably different from aged brain. (G–I) Specificity of IHC for HA in sections of aged Ctx. Hyaluronidase pretreatment (G) before IHC eliminates perineural staining (arrows) and diffuse staining (asterisk) for HA, whereas chondroitinase pretreatment (H) does not affect HA staining (arrows, asterisk). Exposure to secondary Vectastain agents and DAB in the absence of bHABP results in no HA staining (I). B–I are counterstained with hematoxylin. Scale bars = 50 μm in B, C, and G–I and 100 μm in D–F. Abbreviations: HA, hyaluronan; HABP, HA-binding protein; Ctx, cortex; Cbm, cerebellum; Hcs, hippocampus; DG, dentate gyrus; DAB, 3,3′-diaminobenzidine; bHABP, biotinylated HA-binding protein.
HA in the ECM of the brain and other tissues is highly associated with CSPGs, which confer stability to the HA polymer. CSPGs were also present in both diffuse and localized patterns in all regions examined, and were especially prominent in a subset of PNNs in the cortex, hippocampus, and cerebellum. In contrast to the increase in HA with aging, there was similar accumulation of CSPGs in all brain regions in the young and aged brains (Fig. 4A–F). In the hippocampus, there were relatively small amounts of CSPGs overall, but there were notably higher levels of CSPGs present as localized accumulations in PNNs in the CA2/CA3 region and as diffuse areas in the CA4/hilus region. Brain sections pretreated with chondroitinase showed complete loss of both localized and diffuse CSPG staining (Fig. 4G), and sections exposed to secondary labeling agents only were not stained (Fig. 4I), indicating the specificity of the WFA label for CSPGs. In contrast, pretreatment of sections with HYAL (Fig. 4H) did not eliminate WFA staining for CSPGs.
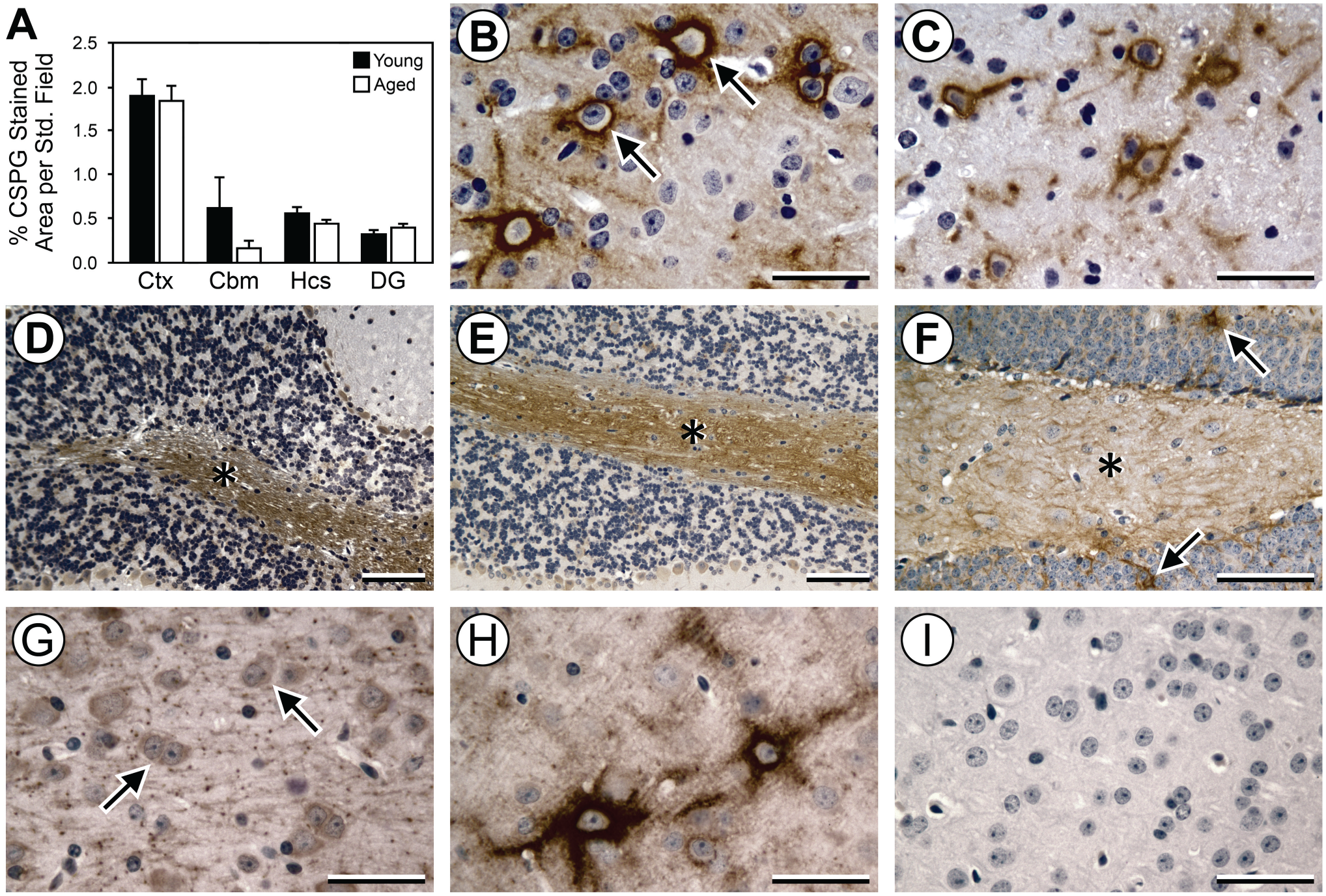
Measurement of CSPGs in young versus aged brains by IHC. (A) Histogram shows that levels of CSPGs are similar in aged brain versus young brain in Ctx, Cbm, Hcs, and DG. Representative IHC images (B–F) show similar accumulation of CSPGs (brown DAB staining) in the young Ctx and Cbm (B, D, respectively) relative to the aged Ctx and Cbm (C, E, respectively). Accumulation of CSPGs was localized primarily to PNNs in the Ctx (B; arrows) and white matter in the Cbm (D, E; asterisks) with no differences with aging. Panel F shows representative diffuse CSPG accumulation (asterisk) and rare CSPG-positive PNNs (arrows) in young brain in the hilus (CA4) region of the Hcs, which is not demonstrably different from the aged brain. (G–I) Specificity of IHC for CSPG in sections of aged Ctx. Chondroitinase pretreatment (G) before IHC for CSPG eliminates PNN staining (arrows), whereas hyaluronidase pretreatment (H) does not affect CSPG staining of PNNs. Exposure to secondary Vectastain agents and DAB in the absence of WFA results in no CSPG staining (I). B–I are counterstained with hematoxylin. Scale bars = 50 μm in B, C, and F–I and 100 μm in D and E. Abbreviations: CSPGs, chondroitin sulfate proteoglycans; Ctx, Cortex; Cbm, cerebellum; Hcs, hippocampus; DG, dentate gyrus; DAB, 3,3′-diaminobenzidine; PNNs, perineural nets; WFA, Wisteria floribunda agglutinin.
We have previously shown that there is increased HA in microvessels isolated from the cortex of aged mice. 31 Examination of HA and CSPG accumulation in the young and aged mouse brains demonstrated that both were prominent in PNNs and colocalized with a small subset of cells that were positive for NeuN (Fig. 5A and D). There was rare, but definite, colocalization of HA and CSPGs with astrocytes (Fig. 5B and E). As expected, both HA and CSPGs were associated with brain microvasculature (Fig. 5C1, C2, F1, F2).
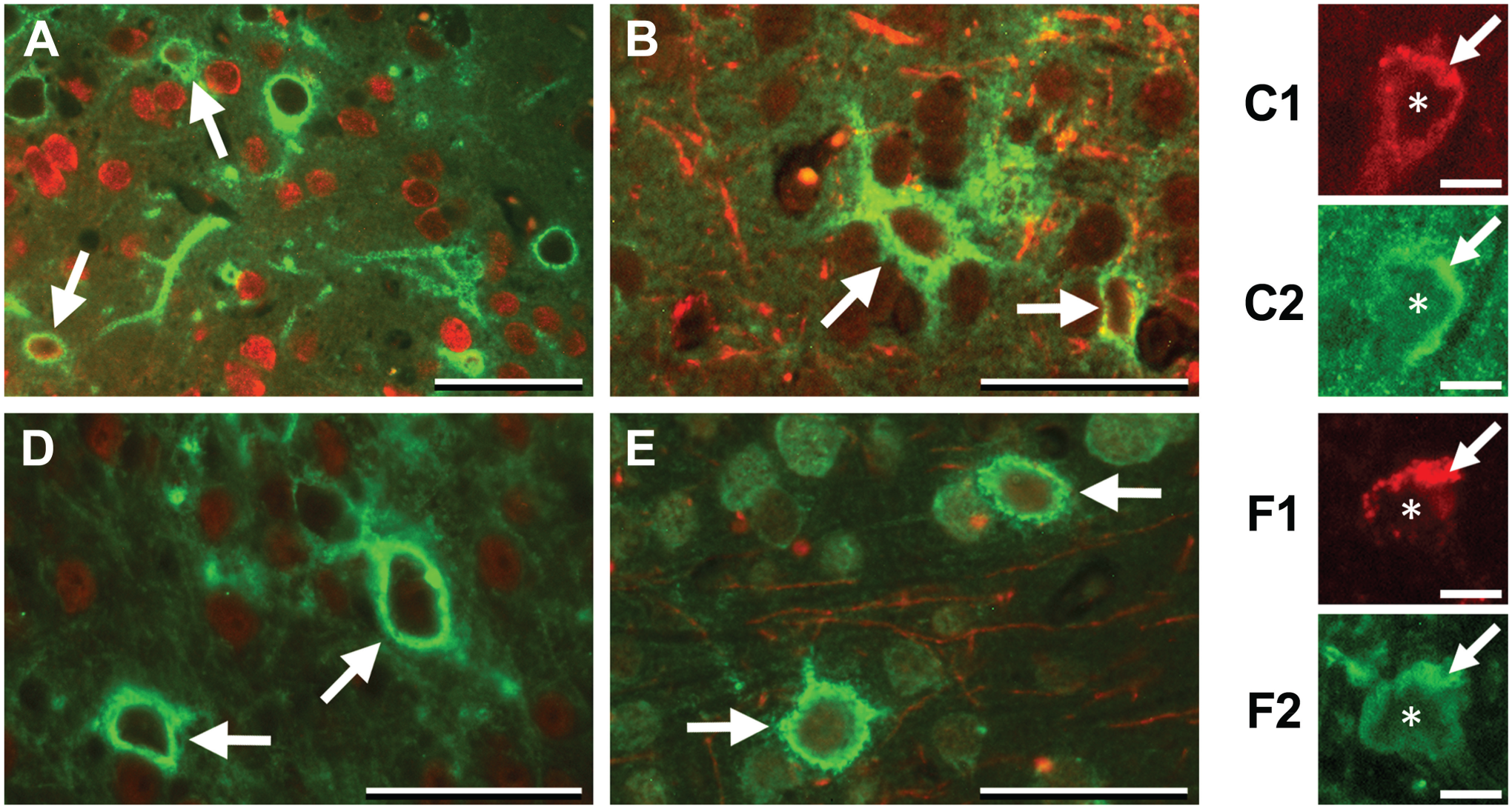
Accumulation of HA and CSPGs in the cortex. (A–C) HA staining (green). HA accumulates in a subset of NeuN-positive (red) cells (A; arrows) and, rarely, in GFAP-positive (red) astrocytes (B; arrows). A PECAM-positive (red) microvessel seen in cross-section (C1; arrow) has an accumulation of HA (C2; arrow). Asterisks in C1/C2 indicate the lumen of the vessel. (D–F) CSPG staining (green). CSPG accumulates in a subset of NeuN-positive (red) cells (D; arrows) and GFAP-positive (red) astrocytes (E, arrows). Like HA, a PECAM-positive (red) microvessel (F1, arrow) is associated with CSPG (F2; arrow). Asterisks in F1/F2 indicate the lumen of the vessel. Scale bars = 50 μm in A, B, D, and E; and 10 μm in C1, C2, F1, and F2. Abbreviations: HA, hyaluronan; CSPGs, chondroitin sulfate proteoglycans; GFAP, glial fibrillary acidic protein; PECAM, platelet endothelial cell adhesion molecule.
Discussion
There is increasing interest in how neurodegeneration, stroke, and other injury processes affect the ECM of the brain.34–36 The ECM has always been ascribed a supporting role for brain cells and microvasculature, but it is now appreciated that ECM has regulatory functions during development and disease. In the brain, the ECM modulates cellular plasticity and can promote or inhibit immune and inflammatory responses.35,36 More recently, it has been found that the glymphatic system, which supports the convective flux of cerebrospinal and interstitial fluids and serves as one mechanism to eliminate solute waste from the brain, utilizes the ECM for flow along perivascular pathways. 37 However, little is known about the influence of normal aging on brain ECM. Indeed, although overall brain volume and weight are consistently shown to decrease with aging in humans, the data on mice are variable depending on strain and size.1–5 Using a murine model of normal brain aging, we measured microvascular, neuronal, and glial cell content, and accumulation of HA and CSPGs. 24 HA and CSPGs are key elements of the brain ECM that are distributed in diffuse networks as well as in localized, pericellular areas, such as PNNs and the microvasculature. We focused on cortex, cerebellum, hippocampus, and dentate gyrus as regions of interest in brain aging.25,27 We found that in certain brain regions, microvascular density decreases and glial cells increase with aging. Whereas HA accumulation increases in some regions of the brain, the accumulation of CSPGs does not change during normal aging.
The finding that microvascular density decreases in the brain with aging has been previously shown by our group and others.30–33 Whether this occurs in every brain region is not known, as we were only able to detect significant differences in the cortex and cerebellum. Decreases in microvasculature with aging have been primarily studied in tissues with high turnover (such as the skin) and are attributed to loss of existing vessels as well as decreased capacity to undergo angiogenesis. In the absence of pathology, there should not be a stimulus for making new microvasculature in the brain; however, the mechanism by which vessels are lost in the brain with aging is unknown. Apoptosis has been considered, but not proven, which has led to use of the term “rarefaction” in some studies.38,39
We found no significant differences in numbers of NeuN-positive cells in the brain during normal aging. Although neuronal loss is ubiquitous during ischemic injury or neurodegeneration, and some decreases in neuron density contribute to lower brain weight and volume in the aged, we did not expect to find a significant change with age alone in healthy mice.2,4,40 In contrast, where there was measurable GFAP staining, such as in the hippocampus, there was an increase in reactive astrocytes in the aged brains relative to the young brains. These results are consistent with prior studies showing higher levels of inflammatory cells in healthy aged brains.6,41–47 This increase in a reactive glial cell phenotype is thought to confer susceptibility of the aging brain to neurodegeneration and disease.
An increase in HA was found in both the cerebral cortex and the cerebellum of the aged brains relative to the young brains. It is possible that other regions had age-related increases in HA, but we were not able to detect significant differences. Others have shown increased HA in the gray matter of the cortex with aging in rats. 15 We have demonstrated increased HA in microvessels isolated from the aged mouse brain. 31 In the present study, we found that diffuse and localized (e.g., PNN-associated) accumulations of HA were both increased in aged versus young cortex and cerebellum; however, diffuse HA made the greater contribution to this increase. It is possible that brain volume loss with aging increases the density of the ECM, thereby resulting in higher intensity of HA staining. Our previous finding that HAS 2 increases with aging in isolated brain microvessels, 31 and our present observation that staining intensity for CSPGs does not change with age, indicates that ECM compaction alone does not explain an age-related increase in HA accumulation.
HA accumulation is associated with many biological processes and has both beneficial and detrimental effects. Increased levels of HA during wound repair is viewed as a source of low MW HA, which can promote immune responses and inflammation that influence healing.48,49 We showed in a previous study that HA synthesized by isolated mouse brain microvessels has an intermediate (200 kDa range) and is produced in greater quantity by the aged versus the young microvessels. 31 However, we were not able to determine if the increased level of this intermediate MW HA was correlated with an elevated level of low MW HA (although it is reasonable to suspect that this might be so). The origin of age-associated increases in HA quantity in the brain is unknown, although we found increased HAS 2 when specifically examining the microvasculature. 31
In contrast to the increased HA accumulation in the aged cerebral cortex and cerebellum, we did not find any differences in CSPGs with aging in any of the brain regions examined. This could reflect the high basal accumulation of CSPGs, which would make any changes with age alone difficult to detect. Indeed, CSPGs, as a class, are highly diverse in structure and in distribution such that it would take significant injury or degeneration to show a change in their accumulation.50,51 It is possible that an examination of specific CSPGs, such as neurocan or aggrecan, would show that there are differences in specific forms of CSPGs during normal brain aging. Moreover, recent data suggest that CSPG sulfation, which we did not measure, increases with aging and promotes an ECM environment that inhibits PNN plasticity. 21
Although it is appreciated that there is great variation in the ECM composition of the layers of the hippocampus and dentate gyrus, little is known about specific changes in the ECM in these areas with normal aging. 52 Using a proteomic approach, Vegh et al. reported increased ECM in the hippocampus with aging in mice ranging from 20 to 100 weeks of age. 53 We found specific areas had differences in ECM, for example, greater amounts of diffuse HA in the hippocampus, such as in the CA2 and CA3 regions, relative to CSPGs. In contrast, there was more localized accumulation of CSPGs, but not HA, in the CA2, CA3, CA4/hilus regions and the dentate gyrus. Of note, we were not able to detect significant differences in these patterns between young and aged mice. The hippocampus mediates many processes necessary for memory and cognition, and alteration of this region (either due to atrophy or deposition of extracellular products) is a hallmark of neurodegenerative diseases in humans such as Alzheimer’s disease.53–56 Consequently, additional studies are needed to define the role of HA and CSPG accumulation in hippocampal structure and function with aging.
Although most ECM components of the brain are present in a diffuse pattern in the interstitial spaces between cells, HA and CSPGs are notable for their localized pericellular accumulation. We examined cellular localization patterns to determine if either HA or CSPGs associate with neuronal or glial cell markers. Both HA and CSPGs colocalized with a subset of NeuN-positive cells in a classic PNN-like pattern. Rare astrocytes also demonstrated accumulation of HA and CSPGs. We did not find that colocalization of HA and CSPGs with astrocytes was significantly more common in regions that had a high density of PNNs. HA and CSPGs are major components of the glycocalyx of the brain microvasculature.57–59 Correspondingly, in the present study, we found that HA and CSPGs were associated with microvessels.
In summary, we find that with normal brain aging, there are region-specific decreases in microvasculature and that there is a greater accumulation of HA, but not CSPGs, with age. These changes with normal aging likely confer regional variation in vulnerability to injury, inflammation, and neurodegeneration.
Footnotes
Acknowledgements
The authors thank Thomas N. Wight, PhD, for many helpful discussions and Virginia M. Green, PhD, for her assistance with the manuscript.
Competing Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
All authors have contributed to this article as follows: MJR supervised all experiments and data analyses and drafted the manuscript; MD performed the IHC and the data analysis; JLP performed the IHC and assisted with data analysis; MAE assisted in the design and implementation of the study and the drafting of the manuscript; WAB assisted in the design and implementation of the study and the drafting of the manuscript; RBV assisted in data analysis, creating the final images, and the drafting of the manuscript; and all authors have read and approved the final manuscript as submitted.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health grants (R03AG051071, R21AG056883 to M.J.R., R01AG046619 to W.A.B., and R01EB012558 to R.B.V.), the Klorfine Foundation (R.B.V.), and the VA Puget Sound Health Care System (W.A.B.).