
Abstract
Aggrecan is a large proteoglycan that forms giant hydrated aggregates with hyaluronan in the extracellular matrix (ECM). The extraordinary resistance of these aggregates to compression explains their abundance in articular cartilage of joints where they ensure adequate load-bearing. In the brain, they provide mechanical buffering and contribute to formation of perineuronal nets, which regulate synaptic plasticity. Aggrecan is also present in cardiac jelly, developing heart valves, and blood vessels during cardiovascular development. Whereas aggrecan is essential for skeletal development, its function in the developing cardiovascular system remains to be fully elucidated. An excess of aggrecan was demonstrated in cardiovascular tissues in aortic aneurysms, atherosclerosis, vascular re-stenosis after injury, and varicose veins. It is a product of vascular smooth muscle and is likely to be an important component of pericellular matrix, where its levels are regulated by proteases. Aggrecan can contribute to specific biophysical and regulatory properties of cardiovascular ECM via the diverse interactions of its domains, and its accumulation is likely to have a significant role in developmental and disease pathways. Here, the established biological functions of aggrecan, its cardiovascular associations, and potential roles in cardiovascular development and disease are discussed.
Keywords
Introduction
All tissues and organs contain extracellular matrix (ECM) having varying abundance, composition, and specific arrangements with respect to resident cells. 1 Whereas considerable attention is given to cell lineage origins, cell proliferation, differentiation, and apoptosis, the regulatory impact of the ECM in these contexts has received less attention. It is well accepted that ECM provides structural integrity to tissues and organizes tissue-specific architectures. In contrast to these roles as a construction material, it is not as widely appreciated that at the cellular level, pericellular ECM in particular has a strong regulatory impact on cell polarity, adhesion, migration, proliferation, differentiation, and cell death.2–5 ECM can signal directly via specific interactions with cellular ECM receptors such as integrins, 2 or indirectly by binding and regulating several classes of signaling proteins including morphogens, growth factors, and pro-inflammatory cytokines.6,7
Developmental processes are characterized by a dynamic ECM in keeping with rapidly evolving cell numbers and lineages, extensive cell movements, and dramatic shape changes in the embryo. Normal adult ECM is relatively stable, although it likely also undergoes regulated remodeling, albeit at a slower rate than embryonic ECM. However, dysregulated ECM remodeling that occurs in diseases is of profound significance for cell and tissue function. In all settings, the pericellular matrix, which is the ECM juxtaposed to the cell surface, may be especially dynamic, since it is the most accessible and thus most readily modified by cellular synthetic and proteolytic activities. It is well-positioned to influence cell behavior via its content of pro- and anti-adhesive molecules, as well as growth factors, morphogens, and cytokines. Thus, its components and their turnover are correspondingly of great interest. During early embryogenesis, the ECM is of a primordial and provisional nature, whereas adult tissues and organs have an architecturally mature ECM with a specialized and distinctive composition. The broad importance of ECM components such as fibrillar collagens and fibrillin-1, which are widely distributed, is evident from syndromic inherited disorders of connective tissues such as the Ehlers-Danlos syndromes and Marfan syndrome, which result from well-defined gene mutations.8,9 Many ECM components are specialized and have a narrower tissue distribution, reflected in rare organ-specific genetic conditions, such as inherited kidney and skin disorders resulting from ECM gene mutations.10,11 Conversely, in nearly all acquired disorders, ECM undergoes broad changes such as a general increase or decrease in its abundance relative to cell numbers or altered abundance of specific components which leads to inappropriate ECM stoichiometry and imperils cell function. For example, tendons, ligaments, and bone rely for their tensile strength on the specific rope-like arrangement and high abundance of the fibril-forming component collagen I. Yet, an inappropriate excess of collagen I in other organs is disruptive and a hallmark of a severe tissue derangement called fibrosis. 12
The cardiovascular system follows similar developmental principles as other body systems, in that early stages of its development are characterized by a primordial ECM such as cardiac jelly in the ventricular wall and endocardial cushions, whereas later developmental stages are characterized by resorption of this matrix and transition to a mature collagen- or elastin-rich ECM, a process which continues into the neonatal period. Cardiac ECM is altered dramatically in the neonatal period in mice by the expansion of cardiac fibroblasts, 13 which produce fibrillar collagen, resulting in an increase in ECM stiffness which may restrict the proliferative capacity of cardiomyocytes. 5 Elastic lamellae of blood vessels are laid down during late gestation and the neonatal period, 14 and endocardial cushions undergo compaction in late gestation to form mature valve leaflets with a stereotypical three-layered ECM structure. 15 Conversely, dysregulation of adult cardiovascular ECM during disease, including fibrosis or reversion to ECM with embryonic characteristics, may accompany or cause cellular changes with significant impact on disease outcomes and are thus deserving of further research.
Proteoglycans are important, diverse and widely distributed ECM components. They are composite molecules comprising a core protein and covalently attached glycosaminoglycan (GAG) chains of various kinds. Over 40 proteoglycans are known, with several belonging to well-characterized families 16 and their number is growing with the application of improved methods for their identification, such as glycoproteomics. 17 The proteoglycan aggrecan is best known as a quantitatively major, essential, and defining cartilage component. In this review, we examine it in the context of development of the cardiovascular system and its emerging associations with several cardiovascular disorders.
Aggrecan—A Member of the Large Aggregating Proteoglycan Family
The structure and properties of aggrecan are similar to those of other large chondroitin sulfate (CS) proteoglycans collectively known as hyalectans, lecticans, or aggregating proteoglycans, which bind to the GAG hyaluronan (HA) to form large aggregates. 18 The other hyalectans, namely versican, brevican, and neurocan, have fewer CS chains than aggrecan, with versican having the most, yet only one-tenth as many as aggrecan. Brevican and neurocan are thought to be restricted to the nervous system (which also contains aggrecan and versican), whereas versican is widely distributed. 19 It is abundant in the cardiovascular system, especially during development and its accumulation is implicated in cardiovascular development and disease.20,21 Notably, mouse embryos homozygous for a mutation (Vcanhdf) that abolishes production of all versican splice variants die during early development with major cardiac developmental defects.22–24 Versican is upregulated in cardiac intimal hyperplasia following intimal damage or ablation and is associated with atheromatous lesions, where it is thought to promote vascular smooth muscle cell (VSMC) proliferation and migration as well as cytokine and lipoprotein binding. 21 Some of the elucidated roles and mechanisms of versican are likely to be highly relevant to and perhaps even directly applicable to aggrecan given the many structural similarities of the two proteoglycans.
Aggrecan Domain Structure and Established Biological Roles
Aggrecan consists of a large core protein of ~250 kDa with covalently attached GAG chains, specifically ~100 CS chains and ~30 keratan sulfate (KS) chains (KS is present in bovine and human aggrecan, but not rat and mouse) 25 (Fig. 1A). The GAG chains have variable lengths and sulfation and can increase the mass of an average aggrecan molecule to 3 MDa. 25 The core protein has three globular domains, termed G1, G2, and G3, with the GAG chains attached in three main clusters to a long, extended region between the G2 and G3 domains 25 (Fig. 1A). Expression of the aggrecan gene is a defining feature of chondrogenesis during development of much of the bony skeleton in vertebrates. During this process, axial and limb bud mesenchymal cells aggregate to form condensations which initiate aggrecan synthesis in response to specific transcription factors and morphogens. The condensations are subsequently identified as cartilage by the presence of aggrecan and another quantitatively major cartilage ECM component, collagen II. 26 These chondrogenic rudiments form the anlagen (models) of the future bony skeleton, which are initially wholly cartilaginous. Aggrecan forms giant aggregates with HA, which are located both in the pericellular matrix of chondrocytes and interstitial ECM of cartilage 27 (Fig. 1B). The HA-aggrecan complex is stabilized by a link protein structurally homologous to the aggrecan G1 domain called cartilage link protein or HAPLN1, which binds to aggrecan with 1:1 stoichiometry 28 (Fig. 1A and B). Binding of the aggregates to cell-surface HA receptors, as well as CS receptors (Fig. 1A) ensures their proximity to the cell surface. During the subsequent postnatal period, cartilage is present only within well-defined growth plates near the ends of bones and the adjacent joint surfaces. Growth plate cartilage is gradually resorbed by endochondral ossification during the embryonic and juvenile periods, leaving only articular cartilage at the ends of bones upon completion of skeletal growth. In articular cartilage, aggrecan is the major compression load-bearing component, acting via two potential mechanisms: charge repulsion between adjacent aggrecan molecules and/or a Donnan equilibrium effect, in which the net negative charge of the aggrecan GAG chains draws in salt and water, resulting in a swelling pressure (Fig. 1C). Swelling of the aggregates is constrained by the collagen II network to about 10–20% of their free solution volume, 25 resulting in a unique composite structure with resilience against tension, shear and compression. Aggrecan is also enriched in the inner two-thirds of fibrocartilaginous menisci in knee joints, and in regions of tendons subjected to compression loading.29,30
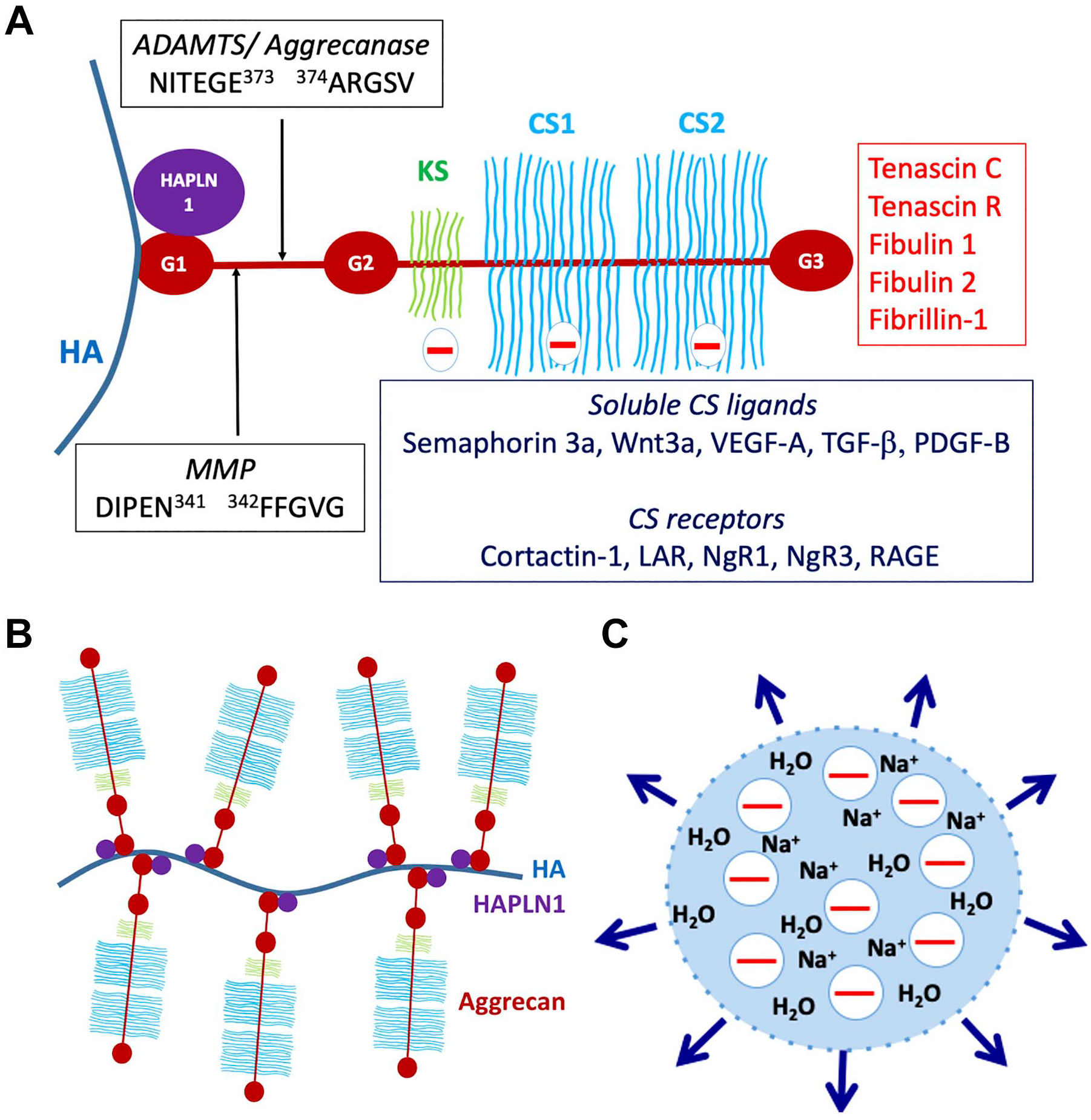
Structure and functional attributes of aggrecan. (A) Aggrecan domains and their intermolecular interactions. G1, G2, and G3 indicate globular domains. The interglobular domain is the region between G1 and G2. HA is hyaluronan, HAPLN1, cartilage link protein. The major MMP and ADAMTS (aggrecanase) cleavage sites in the interglobular domain are shown, as well as the relative locations of the keratan sulfate (KS)—and chondroitin sulfate (CS)—bearing domains. Molecules known to bind to CS chains, including cell-surface receptors and soluble ligands and known ligands of the G3 domain are indicated. (B) The aggrecan-hyaluronan (HA) aggregate can vary enormously in size depending on the length of HA and the number of available aggrecan and HAPLN1 molecules. (C) Representation of the Donnan equilibrium concept. The net negative charge of aggrecan-HA aggregates draws in sodium ions and with them, water, contributing to exertion of a swelling pressure (indicated by the outward pointing arrows) by the aggregates. Abbreviations: MMP, matrix metalloproteases; RAGE, receptor for advanced glycation end-products.
In addition to cartilage, aggrecan has been investigated in the central nervous system, especially in the context of perineuronal nets (PNNs), a pericellular matrix present around the soma (body) of parvalbumin-positive interneurons. 31 PNNs are assembled on a scaffold of cell-surface attached HA and also contain other aggregating proteoglycans. 31 They appear toward the end of the critical period of neuronal plasticity and are thought to insulate the neurons from further synaptic remodeling, 32 a possibility supported by the recently reported effect of conditional in vivo aggrecan deletion in these cells. 33 PNNs are implicated in the loss of neuronal plasticity occurring after the end of the critical period, which happens around the age of 5 years in humans and they have been considered as the basis of long-term memory. 34
Aggrecan Interactions
The aggrecan G1 domain is divided into three sub-domains, A (an immunoglobulin repeat), B, and B′ (each comprising a proteoglycan tandem repeat). Binding to HAPLN1 occurs through the N-terminal A sub-domain, whereas the B- B′ subdomains bind HA and are not known to have additional interactions. 35 Whereas the G1 and G3 domains are present in all the aggregating proteoglycans, the G2 domain is unique to aggrecan, homologous to the proteoglycan tandem repeat, and lacks known interactions. The G3 domain has a variety of known ligands including tenascin-C, tenascin-R, fibulin-1 and -2, fibrillin-1, and cell-surface glycolipids and carbohydrates and connects the HA-aggrecan pericellular matrix to an extensive ECM network. 36 Like G1, the G3 domain also has distinct sub-domains, namely two epidermal growth factor (EGF)–like repeats, a C-type lectin domain (CLD) and a complement regulatory protein (CRP, also known as a sushi domain) repeat. 36 Unlike G1, these G3 sub-domains can undergo alternative splicing, which affects the intermolecular interactions of aggrecan. 37 The KS and CS chains attached to the aggrecan core protein are believed to be of heterogenous number, length and charge density, with further modifications occurring during development and disease. CS chains have numerous known ligands including the growth factors VEGF-A, TGF-β, semaphorin-3a, Wnt3a, and platelet-derived growth factor B (PDGF-B), and bind to diverse receptors on the cell surface, such as cortactin-1, the receptor for advanced glycation end-products (RAGE), leukocyte common antigen-related phosphatase and nogo receptors NgR1 and NgR338,39 (Fig. 1A). The interglobular domain (IGD) between G1 and G2 is susceptible to cleavages that separate the GAG-bearing region from the G1 domain and HA 40 (Fig. 1A). These cleavages are considered critical for aggrecan loss from articular cartilage in osteoarthritis (OA), since they release that part of aggrecan which carries the compression-load bearing element (the GAG chains), and disrupt the network linking the cell-surface to the interstitial matrix. 40 Two cleavage sites identified in the IGD have attracted considerable interest. One is attributed to matrix metalloproteases (MMPs) (occurring at IDIPEN341↓F342FGVG) and the other to A disintegrin-like and metalloprotease domain with thrombospondin type 1 motifs (ADAMTS) proteases (also referred to as the aggrecanase site, occurring at NITEGE373↓A374RGSVIL) (Fig. 1A). 41 They are among numerous cleavage sites in the core protein targeted by proteases (only the IGD sites are shown in Fig. 1A). Among them, sites of ADAMTS protease activity are well-characterized, since these proteases were implicated as major contributors to aggrecan proteolysis in OA.41,42 Although ADAMTSs are secreted, they have a propensity for binding cell-surfaces and/or pericellular ECM, and can be regarded as operational cell-surface proteases. 24 ADAMTS cleavage of versican and aggrecan is known to release bioactive matrikines.24,43,44 The 32-mer aggrecan peptide resulting from cleavages at the major MMP and ADAMTS cleavage sites (Fig. 1A) has been shown to interact with toll-like receptors, potentially acting as a damage associate molecular pattern (DAMP) component, and also to function in stimulation of pain in the arthritic joint.45,46
Aggrecan Genetics
That aggrecan is indispensable for skeletal development and function is evident from the severe impact of mutations in several species (Table 1). Naturally occurring recessive ACAN mutants in chicks (the nanomelia [nm] mutant) 47 and the bulldog ACAN mutation in Dexter cattle 48 have an embryonically lethal effect. Two naturally occurring mouse null mutants, one termed cartilage matrix deficiency (Acancmd) 49 resulting from a 7-bp N-terminal deletion affecting the G1 domain, and the Acancmd-Bc mutation, a near-complete Acan deletion, 50 lead to a severe, lethal recessive chondrodysplasia and have been extensively characterized. Mutant mice die soon after birth with respiratory insufficiency likely resulting from soft rib and tracheal cartilage. Mice with Acan hemizygosity are apparently normal at birth, but develop mild dwarfism after 4 weeks of age and abnormal spinal curvature is noticeable in 1-year old mice. 51 In another mouse mutant, Acanb2183Clo, biventricular cardiac hypertrophy was reported in addition to the expected skeletal and craniofacial defects, but few details of this phenotype are presently available. 52 Complete loss of function in humans has not been reported and is likely also lethal at birth. ACAN mutations in humans were identified in spondyloepimetaphyseal dysplasia (Online Mendelian Inheritance in Man [OMIM] 612813), 53 osteochondritis dissecans with short stature and early OA (OMIM 165800), 54 or spondylometaphyseal dysplasia, Kimberly type (OMIM608361). 55 No cardiovascular manifestations are reported in these genetic disorders. Hapln1 knockout in mice leads to a milder chondrodysplasia than aggrecan deficiency, but is nevertheless lethal at birth in the majority of Hapln1-null mice,28,56 emphasizing the important supporting role that link protein plays in forming stable HA-aggrecan and HA-versican aggregates. Hapln−/− mice have a cardiac developmental phenotype comprising atrioventricular (AV) septal and myocardial defects similar to that seen in Vcan haploinsufficient mice. 57 Since versican linkage to HA is also stabilized by HAPLN1, 58 and because cardiac anomalies have not been reported in Acan mouse mutants other than the Acanb2183Clo allele, it is possible that the cardiac roles of HAPLN1 are primarily related to loss of ability to form stable versican-HA complexes.
Naturally Occurring Mutations in the Aggrecan Gene.
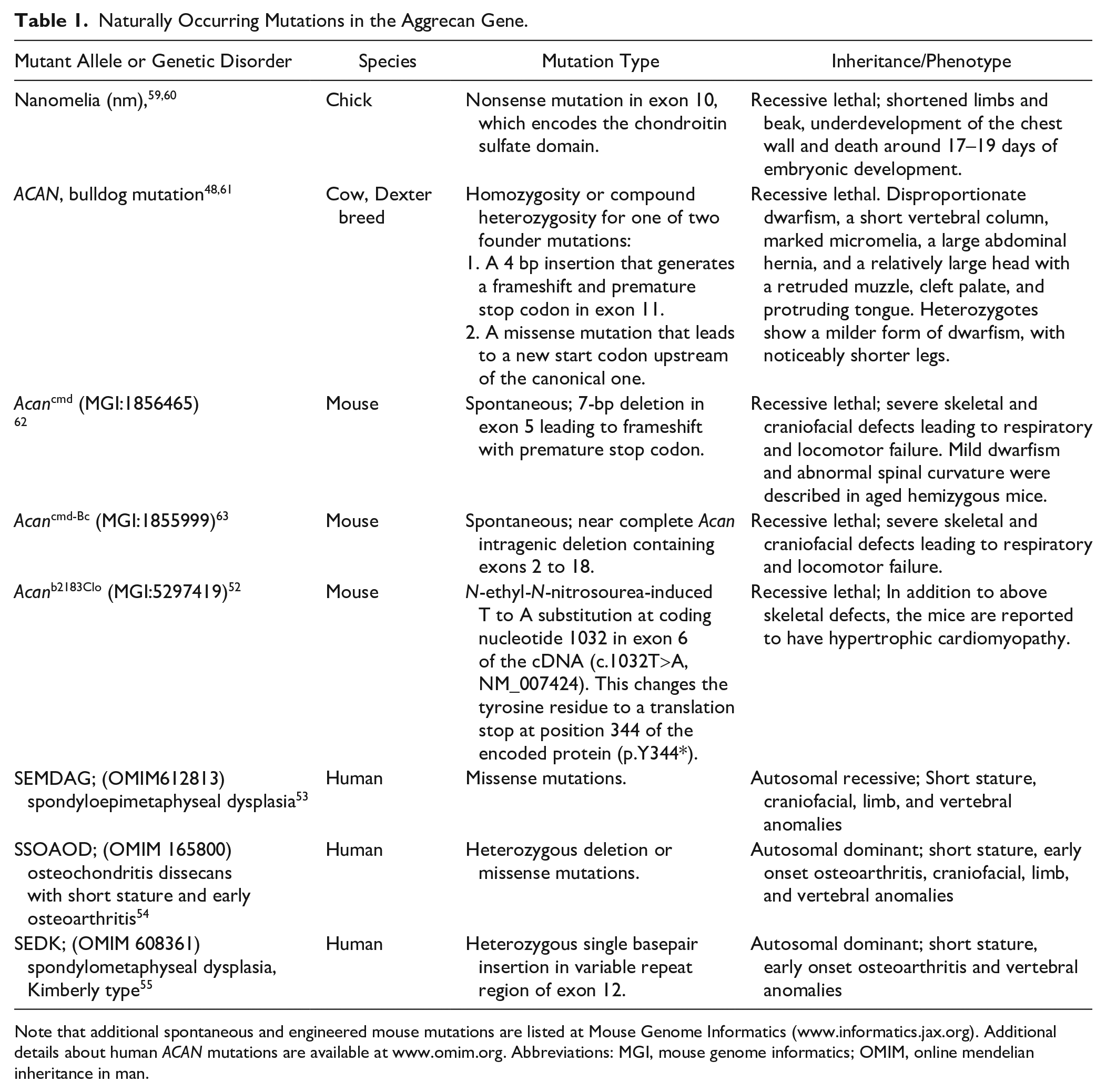
Note that additional spontaneous and engineered mouse mutations are listed at Mouse Genome Informatics (www.informatics.jax.org). Additional details about human ACAN mutations are available at www.omim.org. Abbreviations: MGI, mouse genome informatics; OMIM, online mendelian inheritance in man.
Aggrecan in Cardiovascular Development
Aggrecan localization has been described previously in various regions of the cardiovascular system, and there is a new interest in its distribution and biological roles because of numerous recent associations with cardiovascular disease. Moreover, the observation of cardiac hypertrophy in embryos recessive for the Acanb2183Clo allele may have uncovered a hitherto cryptic role for aggrecan in cardiovascular development. 52 The cellular impact of excess aggrecan has not been sufficiently investigated. Here, we review the literature on the cardiovascular developmental and disease associations of aggrecan to identify possible directions for future research.
Aggrecan in Zebrafish Cardiovascular Development
Zebrafish have two paralogous genes encoding aggrecan, aggrecan a and aggrecan b (symbols: acana and acanb), but only acana is expressed in cardiac development, specifically in the outflow tract (OFT), and its expression is dependent on normal hemodynamic forces. 64 Knockdown of acana using morpholinos and knockout using CRISPR-Cas9 impaired OFT development and led to a larger atrium and associated edema at 3 days post-fertilization. 64 acana morphants had reduced cardiac output and stroke volume, and AV regurgitation, which were attributed to failed OFT development. 64 Adult zebrafish valves, unlike those of humans and mice, lack distinct stratified layers of elastin, collagen and proteoglycan, and the valve leaflets comprise two major regions along their proximal-distal axis, a base to mid-region zone containing large polygonal cells and a tip region of small cells with high cellularity. A morphologic analysis of valvular interstitial cells in zebrafish and comparison with mouse valves noted chondrocyte-like morphology of cells in the mid-region, which expressed Sox9 (a transcription factor driving aggrecan and collagen II expression) and a surrounding ECM containing aggrecan and collagen II. 65
Aggrecan in Avian Cardiovascular Development
Zanin et al. 66 studied aggrecan and versican in chick heart development using immunostaining, western blot, RT-PCR, and RNA in situ hybridization, concluding that aggrecan and versican were expressed in distinct regions. VCAN expression precedes that of ACAN, which is primarily present between Hamburger-Hamilton (HH) stages 16–21 in the developing chick heart, with highest expression around HH stage 20. 66 At HH stage 18/19, ACAN mRNA is strongly expressed within the endocardial cushions of the OFT and the AV junction, albeit at higher levels in the inferior cushion of the AV endocardial cushion tissue than the superior side. 66 ACAN mRNA was also present in the sino-atrial junction as well as in a population of migrating cells in the dorsal mesocardium of the inflow tract. ACAN mRNA expression was not detected in HH stage 45 hearts or adult hearts. 66
By immunohistochemistry, aggrecan core protein was detected in multiple groups of migrating mesenchymal cells in the chick heart. HH stage 16/17 is marked by cells migrating into the thickened basement membrane to form the cardiac jelly, separating the myocardium, and endocardium and differentiating into cushion tissue. At this stage, aggrecan was present in the epicardium and in mesenchymal cells within the cardiac jelly of the OFT. 66 However, no aggrecan was seen in the AV cushions, except at the sino-atrial junctions. 66 By HH stage 18/19, AV cushions were moderately infiltrated with cells, locations of which correlated with aggrecan staining. By HH stage 20/21, AV cushions were fully infiltrated, showing strong aggrecan staining. 66
Zanin et al. 66 concluded that ACAN was expressed where mesenchymal cells contributed to the future septation of the primary heart tube, such as the ventricles, common atrium, and sinus venosus. In contrast, they showed VCAN expression to be associated with myocardium and cardiac jelly in the OFT and ventricle, and to the border between the myocardium and OFT in the atrium and AV region. 66 Weaker ACAN expression was observed in the septal conal cushion, consistent with the presence of fewer mesenchymal cells. 66 Chicken ACAN seems to be under the regulation of T-box transcription factor 20 (TBX20) in the endocardial cushion, since TBX20 overexpression reduced aggrecan expression, whereas siRNA-knockdown of TBX20 in primary chicken endocardial cushion cells increased ACAN expression. 67 In this system, Shelton and Yutzey also found that TWIST1 acted upstream of TBX20 to repress ACAN mRNA expression. 68
Another study noted the expression of ACAN during avian cardiac cushion elongation and cusp remodeling, and persistence of expression in the juvenile valve cusps (8 weeks). 69 During cushion elongation, ACAN is expressed throughout the valve cusp, with increased expression in the fibrosa along with type III collagen and tenascin. 69 During cusp remodeling, ACAN is strongly expressed in the annulus, while at the juvenile stage, aggrecan is absent in the annulus and is only present in the fibrosa and spongiosa. 69 In the pre-fused endocardial cushion, BMP2 and FGF4 regulate the diverging differentiation of valve precursor cells into valves or the valve-supporting structures, such as chordae tendineae. 70 ACAN is co-expressed with transcription factors SOX8 and SOX9, the latter being strongly associated with chondrogenesis, in the cushion tissue and differentiating valve leaflets. 71 Increased BMP2 or decreased FGF4 signaling induces ACAN and SOX9, mediated by the phosphorylation of SMAD1/5/8. Conversely, decreased BMP2 or increased FGF4 signaling induces tenascin and scleraxis through phosphorylated MAPK. 70 Consistent with this, at HH stage 25, ACAN was not expressed in the unfused AV endocardial cushions but was expressed after cushion fusion at HH stage 29, where ACAN and SOX9 mRNA were co-expressed in the primitive mural and septal tricuspid valve, septal mitral valve primordia in the AV canal, and in the atrial sub-septal area of the central cushion. 70 At HH stage 36, ACAN and SOX9 are only expressed in the septal and parietal mitral valve leaflets extending to the tips and in the trans-septal fibrous continuity across the ventricular septum, but not in the chordae tendineae. 70 Treatment of cultured valve precursor cells from the pre-fused endocardial cushions with BMP2 or FGF inhibitor SU5402 increased the percentage of cells expressing aggrecan by 20–30% and led to a 62-fold increase in the ACAN transcriptional level compared to untreated controls. 70 These findings suggested the involvement of aggrecan in the cell lineage determination of bipotent valve precursor cells.
In the developing chick aorta, aggrecan is mostly localized in the outer region of the aortic wall. In embryos, little or no aggrecan was present on day 7 (HH stage 31), but weak staining was detected by day 14 (HH stage 40) around the cells organized into lamellae, which continued to day 21 (HH stage 46). 72 By immunoblotting, aggrecan was detected at low levels at day 14 (HH stage 40). These observations contrasted to versican, which was present throughout the aortic wall and at higher levels. 72
Aggrecan in Mammalian Cardiovascular Development
The expression and localization of aggrecan in the mammalian cardiovascular system has not been investigated in the same detail as in chick embryos. Although aggrecan staining was reported in mouse cardiac jelly during cardiomyogenesis, 73 there are no reports of aggrecan expression during mouse cardiac valve development. In gestational day 18.5 (E18.5) embryonic mouse aorta, Acan mRNA is significantly higher than in 12-week-old adult aorta (delta Ct values 3.88 ± 0.58 versus 0.28 ± 0.10, p=0.001). 74 At postnatal day 16 (P16), aggrecan is immunolocalized to the inner third of the aortic tunica media and limited to half of the aortic circumference, but disappears by P30 and is absent at P45, P60, and P90. 75 On the other hand, ADAMTS-cleaved aggrecan increases from P16 to P60, but is absent at P45 and slightly reduced at P90. 75 Acan is also upregulated by 3.67-fold in the ductus arteriosus (DA) of Brown-Norway rats, which are used as an in vivo animal model for patent DA and display dysregulated DA elastogenesis. 76 No cardiovascular anomalies have been described in mouse Acan mutants, other than in Acanb2183Clo which was identified in a chemical mutagenesis screen that specifically sought cardiac developmental anomalies, probably because the severe phenotypes readily evident in the skeleton have attracted greater attention. The observed cardiomyopathy could plausibly result from a primary defect in myocardial development, since aggrecan is present in cardiac jelly. 73 Alternatively, it could be secondary to an obstructive defect in the aorta and/or heart valves.
Adamts5−/− mice, which lack the major ADAMTS protease involved in aggrecan cleavage in OA, have aortic thickening with aggrecan accumulation. 77 Aggrecan immunostaining in the ascending aorta of mutants was more intense around E14.5, that is, at the beginning of elastogenesis and following septation of the cardiac OFT, and was seen up to 1 month postnatally, preferentially on the aortic arch lesser curvature. Aggrecan co-localized with smooth muscle actin positive cells, suggesting that differentiated VSMCs accumulated aggrecan in their pericellular matrix in the absence of ADAMTS5. 77 This possibility is consistent with RNA in situ hybridization and immunostaining which demonstrated Acan expression by mouse and human aortic VSMC. 75 Aggrecan staining in Adamts5−/− aorta was polarized, with stronger staining on the adventitial aspect of VSMCs, accompanying widened interlamellar distance and elastic fiber breakage. The findings suggest that ADAMTS5 participates in physiological aggrecan turnover to maintain optimal levels during mouse aortic development, but reduced cleavage of other ADAMTS5 substrates could potentially also occur.
It was observed that inhibition of the MEK pathway by the chemical U0126 in the wild-type E13.5 heart in vitro led to 2.9 ± 0.4-fold induction of Acan and 56 ± 4% increase in Sox9 expression. 78 MEK/ERK are upstream regulators of Ets-1, which is an effector transcription factor for FGF signaling. 78 Ets1−/− mice exhibit perinatal lethality and develop an abnormal focus of intra-cardiac cartilage as well as a membranous ventricular septal defect due to anomalous neural crest cell migration. 78
Knowledge of ACAN expression in human embryonic cardiovascular tissues is limited. Rambeau et al. 64 determined relative ACAN mRNA abundance in developing and adult human aortic valves by RT-qPCR and detected low expression at 9 weeks and 10 weeks of gestation, robust expression at 13 weeks of gestation, and higher mRNA levels in adult human valves. They compared relative ACAN mRNA abundance between tricuspid aortic valves and bicuspid type 0 valves and found reduced ACAN mRNA (16-fold difference) in the latter, although the number of valves analyzed was far too small (4 tricuspid valves, 2 bicuspid) to be conclusive. 64 Low levels of ACAN mRNA were identified in VSMCs of adult non-diseased human ascending aorta using RNA in situ hybridization. 75
Aggrecan in Cardiovascular Disease
Although ACAN mRNA is expressed constitutively in normal arteries75,79 aggrecan accumulation has been identified in several pathologic conditions affecting the vasculature (see Table 2).
Major Cardiovascular Disease Associations of Aggrecan.
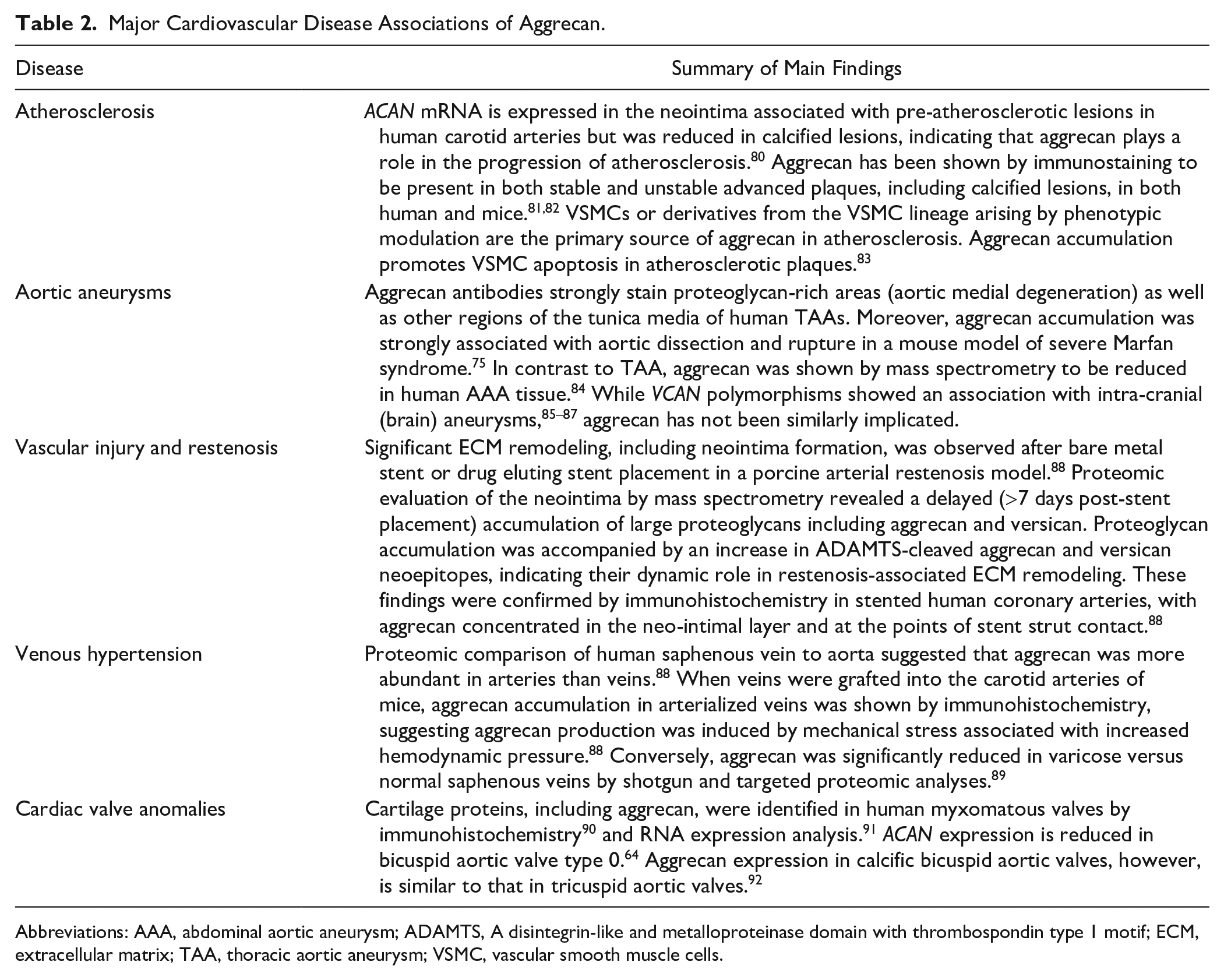
Abbreviations: AAA, abdominal aortic aneurysm; ADAMTS, A disintegrin-like and metalloproteinase domain with thrombospondin type 1 motif; ECM, extracellular matrix; TAA, thoracic aortic aneurysm; VSMC, vascular smooth muscle cells.
Atherosclerosis
The presence of aggrecan throughout the temporal development of atherosclerotic lesions has been described in mice, rats, and humans. Because versican, biglycan, decorin, and perlecan have long been known to be the predominant proteoglycans of the arterial wall, more attention was given to their interactions with low density lipoproteins (LDL) in mechanisms of atherosclerosis.93–96 Despite containing up to 10-fold more constitutive CS attachments sites than versican, 97 aggrecan’s role in GAG-mediated LDL retention has not been investigated. In Apo−/− mice, loss of ADAMTS5 was associated with proteoglycan accumulation and ADAMTS5 was shown to reduce the proteoglycan content of atherosclerotic lesions by release of cleaved core protein fragments, primarily arising from versican, but also biglycan and aggrecan, from the arterial wall; ADAMTS5 activity also contributed to release of proteoglycan bound lipoproteins. 93 ACAN mRNA is expressed in the neointima associated with pre-atherosclerotic lesions in human carotid arteries but was reduced in calcified lesions, suggesting that aggrecan plays a role in the progression of atherosclerosis. 80 Aggrecan has been shown by immunostaining to be present in both stable and unstable advanced plaques, including calcified lesions, in both human and mice.81,82
Several studies have attempted to identify the cells responsible for aggrecan production in atherosclerotic lesions as well as the underlying mechanisms driving its expression. VSMCs, specifically pericytes, can display chondrocyte-like biosynthesis in response to BMP-2 and Wnts and produce aggrecan as part of an intermediate phenotype.98–100 A growing body of evidence suggests that VSMCs or derivatives from the VSMC lineage arising by phenotypic modulation are the primary source of aggrecan in atherosclerosis. Indeed, a number of studies have described VSMC trans-differentiation to a chondrocyte-like phenotype in vascular pathologies. In a uremic rat model for chronic kidney disease, chondrogenic VSMCs led to calcification in the tunica media of the aorta. 101 In mouse and human atherosclerotic lesions, smooth muscle cells with transitional phenotypes, described as myochondrocytes, were identified in which chondrocyte markers such as SOX9 were increased while SMC markers were present but reduced. While the exact mechanism of chondrocyte-like phenotype transition has not been determined, several studies have provided potential insights. In response to vascular injury, typical SMC markers such as SM α-actin (SMA) and myosin heavy chain (MYH11) are downregulated concomitantly with the upregulation of chondrogenic markers including collagen II, bone morphogenetic protein-2 (BMP-2), and aggrecan. 102 In mouse models of atherosclerosis, VSMC transition to a chondrocytic phenotype can be driven by BMP-2. 103 Disruption of matrix GLA protein, which inhibits BMP-2 activity, is known to result in arterial calcification. 104 Inhibition of BMP-2 in Ldlr−/− mice reduced both atheroma development and arterial calcification, although aggrecan was not evaluated. 105 While these studies suggest that BMP-2 is a potential driver of a chondrocyte-like phenotype transition in VSMCs, its effect on aggrecan production has not yet been directly tested. It is also unclear what events trigger VSMCs transition to an aggrecan-producing phenotype. Aggrecan accumulation was enhanced in Tagln−/− mice in response to endothelial injury. 102 VSMCs of the rat aorta express aggrecan and other chondrogenic markers when cultured on calcified elastin or hydroxyapatite and revert to their original phenotype when removed from calcific conditions, 106 suggesting SMC phenotype modulation/ trans-differentiation can also be induced through interactions with the surrounding ECM in which they reside.
While its production by VSMCs appears to be part of an injury response mechanism, the accumulation of aggrecan promotes apoptosis of VSMCs in atherosclerotic plaques. 83 Conversely, cleavage of aggrecan by the macrophage-derived proteases MMP9, ADAMTS4, and ADAMTS8 is increased in unstable plaques. 107 It is unclear, however, if aggrecan cleavage itself causes plaque instability and whether it is contributory to the severity and progression of atherosclerosis. Other ADAMTS proteases implicated in versican turnover and expressed in the vascular wall, such as ADAMTS1108–110 and ADAMTS9,111–114 which can both cleave aggrecan, may also be involved in vascular aggrecan turnover.
Aneurysms
Proteoglycan accumulation is a defining feature of medial degeneration, a histopathologic hallmark of thoracic aortic aneurysms (TAAs). 115 The swelling pressure thus exerted was postulated to contribute to dissection risk by creating stress risers in the aortic wall. 116 Aggrecan antibodies strongly stain these proteoglycan-rich areas as well as other regions of the tunica media of human TAAs. Moreover, aggrecan and versican accumulation are strongly associated with aortic dissection and rupture. 75 While TAA can arise from several genetic defects and thus unique disease pathways, the consistent presence of medial degeneration and the associated aggrecan accumulation in diseased aortic tunica media suggests a common final pathogenic sequence leading to vessel wall failure, with aggrecan accumulation making an important contribution75,117 (Fig. 2). Aggrecan accumulation is asymmetric within the ascending aortic wall and has been shown to favor the aorto-pulmonary septum. 118 Its production has been proposed to be influenced by both VSMC cell lineage and vessel hemodynamics. 118 Consistent with this hypothesis, aggrecan expression in the normal mouse aorta decreases as the vessel progresses from the aortic arch, an area that subjected to high shear stress, toward the abdominal aorta, an area with relatively lower shear stress. 119 This heterogeneous distribution of mural proteoglycans could reflect regional vascular wall adaptations to hemodynamic stresses. 120 Aortic stiffness is associated with a number of cardiovascular diseases and also occurs with aging as loss of elastic fibers reduces the ability of the aortic wall to store elastic energy. 121 Reduced aggrecan has been observed in aging, and postulated to contribute to increased vessel stiffness. 122 Taken together, these studies support an adaptive mechanism in which vascular proteoglycans with high load-bearing capabilities, like aggrecan, assist elastic lamellae in resisting hemodynamic forces. 88 This process appears to be dynamic and controlled not only by the production of aggrecan but also by its proteolysis. 123 Evidence for this comes from mouse models lacking the aggrecanase ADAMTS5 in which aggrecan accumulation and aneurysmal disease progression is exacerbated.77,119 ADAMTS5 mRNA was significantly reduced in human TAA tissue as was Adamts5 mRNA in a mouse model of Marfan syndrome, the Fbn1 mutant Fbn1mgR/mgR. 75
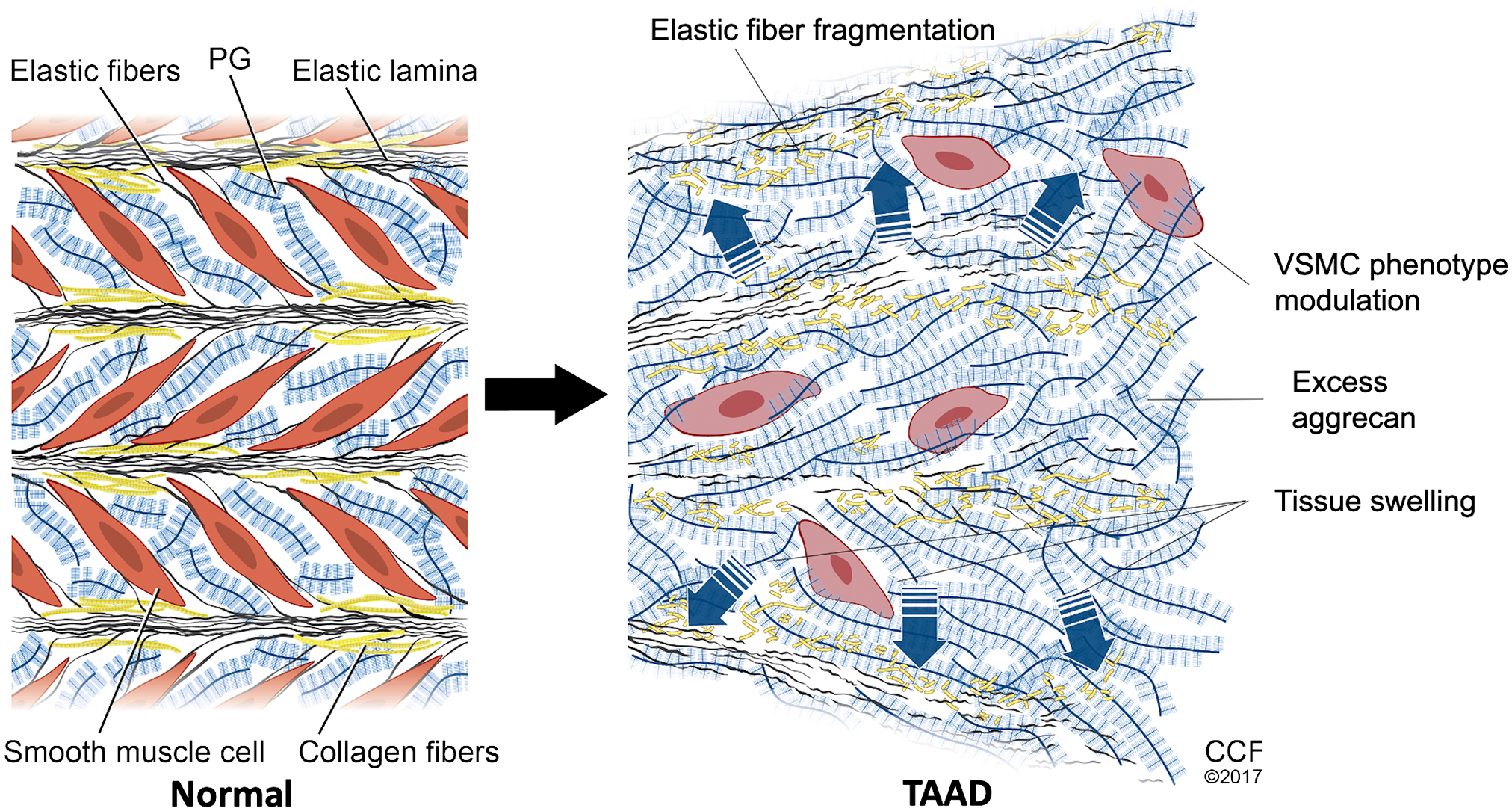
Schematic depicting the impact of aggrecan accumulation in ascending thoracic aortic aneurysm and dissection (TAAD). In a normal aortic wall (left), aggregates of hyaluronan and the proteoglycans (PG) aggrecan and versican provide compressive stiffness to the elastic lamellar units and place elastic fiber/microfibril-cell connections into tension, aiding mechano-sensing by vascular smooth muscle cells (VSMC). Pathologic accumulation of aggrecan and versican in TAAD (right), occurring together with elastic fiber fragmentation disrupts VSMC-extracellular matrix connections. This loss of adhesive and mechanical input to VSMC may result in phenotypic modulation, depicted here as loss of the typical spindle shape of VSMC and assumption of a more rounded appearance. The figure is modified from Cikach et al. 75
In contrast to TAA, aggrecan accumulation is not implicated in the pathogenesis of other types of aneurysms. Abdominal aortic aneurysm (AAA) VSMCs have increased production of the proteoglycans versican, biglycan, and perlecan but an increase in aggrecan production was not observed. 124 Indeed, aggrecan was shown by mass spectrometry to be reduced in human AAA tissue. 84 While VCAN polymorphisms showed an association with intra-cranial (brain) aneurysms,85 –87 ACAN has not been similarly implicated.
Vascular Injury and Restenosis
Neointima formation is a common complication following stenting for coronary artery disease. 125 Significant ECM remodeling, including neointima formation, was observed after bare metal stent (BMS) or drug eluting stent (DES) placement in a porcine arterial restenosis model. 88 Proteomic evaluation of the neo-intima by mass spectrometry revealed a delayed (>7 days post-stent placement) accumulation of large proteoglycans including aggrecan and versican. Proteoglycan accumulation was accompanied by an increase in ADAMTS-mediated aggrecan and versican neoepitopes, indicating a dynamic role in restenosis-associated ECM remodeling. These findings were confirmed by immunohistochemistry in stented human coronary arteries, with aggrecan concentrated in the neo-intimal layer and at the points of stent strut contact. 88
Venous Hypertension
Proteomic comparison of human saphenous vein to aorta suggested that aggrecan was more abundant in arteries than veins. 88 When veins were grafted into the carotid arteries of mice, aggrecan accumulation in arterialized veins was shown by immunohistochemistry, suggesting aggrecan production was induced by mechanical stress associated with increased hemodynamic pressure. 88 Conversely, aggrecan was significantly reduced in varicose versus normal saphenous veins by shotgun and targeted proteomic analyses. 89 Although varicose veins arise from venous hypertension, venous pressures are lower than arterial, and other biological differences between arteries and veins may be contributory to the distinct responses. Taken together, the findings suggest that vascular aggrecan remodeling is a major adaptation to altered hemodynamic pressure, and that aggrecan levels could additionally be altered by a structural failure of vessels.
Cardiac Valve Disorders
Cartilage proteins, including aggrecan, were identified in human myxomatous valves by immunohistochemistry 90 and RNA expression analysis. 91 Mice deficient in Axin2, an inhibitor of Wnt/β-catenin signaling, develop myxomatous valve disease (MVD) 126 and the same signaling pathway is activated in human MVD. 127 Adult aortic valves of Axin2 KO mice had both increased thickness and aggrecan immunofluorescence. 128 The role of Wnt-β-catenin signaling remains unclear as it has also been shown to reduce nuclear localization of Sox9, preventing aggrecan expression. 126 Increased aggrecan expression in valves has also been shown by PCR and in situ hybridization in mice lacking periostin, a matricellular protein thought to play a major role in valve development and homeostasis. 129 Conversely, ACAN expression is reduced in bicuspid aortic valve type 0. 64 Aggrecan expression in calcific bicuspid aortic valves, however, is similar to that in tricuspid aortic valves. 92
Myocardial Overload
Proteoglycans have an important role in cardiac remodeling and fibrosis. 130 In a study designed to evaluate the contribution of ADAMTS proteases to myocardial ECM remodeling, Vcan and Acan expression were found elevated in a rat model of cardiac overloading induced by aortic banding. 131 Acan mRNA expression was increased 4- to 6-fold over sham-operated rats. Concomitant increases of both mRNA and protein levels of the aggrecanases ADAMTS 1, 4, and 8 were observed. Other major aggrecanases, such as ADAMTS5 were not evaluated. Pharmacological inhibition of ADAMTS4 reduced versican cleavage at the ADAMTS cleavage-site and mitigated the decline in cardiac contractility, suggesting an association between versican cleavage and deterioration of systolic function. 131 The role of aggrecan and its proteolytic turnover on cardiac biomechanics requires further exploration.
Future Research Needs
This literature review leads us to conclude that aggrecan is likely to participate in cardiovascular development, with definitive data from different vertebrates (zebrafish, chick, mouse, and human) each showing aggrecan gene expression and localization. However, its roles are likely to be more subtle than those of versican. Aggrecan accumulation around mouse aortic VSMC in the absence of ADAMTS5, 77 suggests that the aggrecan produced by these cells is normally rapidly turned over by the action of this protease (Fig. 3), and possibly, related proteases such as ADAMTS9 113 and ADAMTS1. 132 Further work is needed to understand the factors that regulate aggrecan production by VSMCs and to better define the dynamics of aggrecan turnover by these proteases. Eliciting the potential developmental cardiovascular impact of aggrecan requires conditional Acan deletion, for example, in mouse VSMCs, which are major aggrecan-producing cells in blood vessels. Aggrecan, like versican, also has a potential role in myocardiogenesis, but it is a quantitatively minor component of cardiac jelly compared to versican. Precisely which cells in the heart express aggrecan at different developmental stages or during cardiac failure and repair needs to be carefully determined before conditional deletion strategies can be effectively employed.
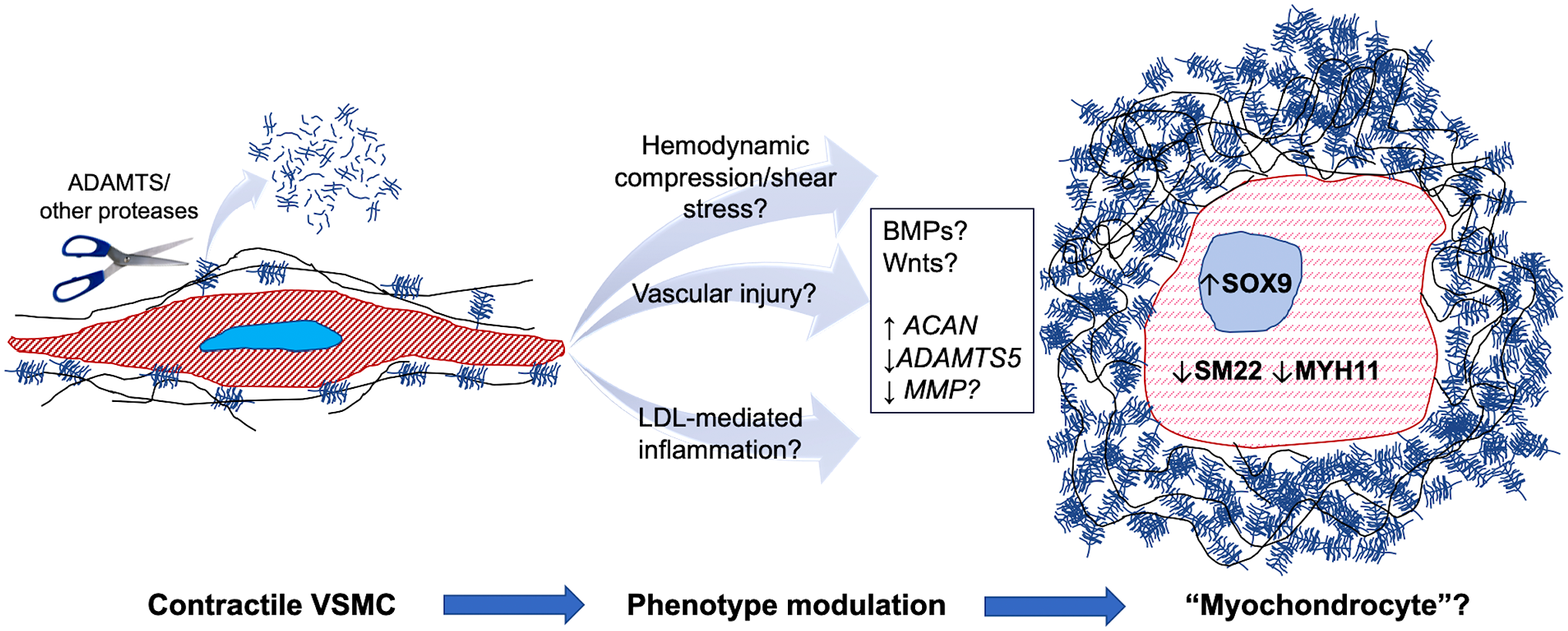
Cellular impact of aggrecan accumulation in vascular smooth muscle cells (VSMC). The cartoon shows that normal VSMCs (left) have a low level of pericellular aggrecan, because of continuous trimming away by ADAMTS5 and potentially other proteases. This may optimize adhesion receptor access to pro-adhesive ECM, promoting formation of a robust cytoskeleton (red hatching) and maintaining the contractile phenotype. Altered hemodynamics, injury, or inflammation may lead to increased aggrecan production or reduced proteolytic turnover as an adaptive response, resulting in net pericellular aggrecan accumulation, which may impair focal adhesion formation and lead to VSMC phenotype modulation (light red broken hatching) toward a poorly characterized phenotype designated here as a “myochondrocyte.” Abbreviations: ADAMTS, A disintegrin-like and metalloproteinase domain with thrombospondin type 1 motif; ECM, extracellular matrix; LDL, low density lipoproteins; MMP, matrix metalloproteases.
We have summarized considerable evidence for aggrecan accumulation in diverse vascular and valve anomalies, as well as in the cardiac response to pressure overload. Because aggrecan is likely to be highly disruptive in excess, its roles in diseases may be more dramatic than in development. Mechanisms of aggrecan accumulation in disease are not yet fully understood, although it is clear that reduced turnover by proteases such as ADAMTS5 may be important in specific disease settings such as TAAD and atherosclerosis (Fig. 3). Among the approaches that could be considered in future are conditional Acan deletion in VSMCs applied to mouse models of aortic aneurysms and atherosclerosis. In particular, the potential mechanistic role of aggrecan in atherosclerosis, a relatively common vascular disorder, deserves further analysis. Rapid aggrecan turnover by ADAMTS proteases may be an important physiological requirement if excess pericellular aggrecan is deleterious to VSMCs and/or vascular wall biomechanics. This possibility could be tested by generating transgenic mice with targeted VSMC over-expression of aggrecan which would also be useful for further investigation of its roles in aneurysms and atherosclerosis in vivo. For example, it was previously shown that excess aggrecan, link protein and HA impaired fibroblast adhesion to the substratum in vitro, 133 and similar approaches could be applied to cultured VSMCs. In this regard, versican accumulation in pericellular ECM of uterine smooth muscle cells impaired the formation of focal adhesions and led to their dedifferentiation. 4 Recent work from our group shows that aggrecan accumulation in the umbilical artery, which does not occur in the umbilical vein, is developmentally and evolutionarily ensured by robust arterial ACAN transcription and limited aggrecan degradation. 134 Computational analysis suggested that abundant aggrecan in the inner umbilical artery tunica media could promote rapid closure of this vessel at birth. 134 Furthermore, both Acancmd-bc/cmd-bc embryos and mice lacking ADAMTS1, the most highly expressed proteoglycan-degrading enzyme in the umbilical vein, have a severely altered VSMC phenotype. 134 There is thus a definite possibility that aggrecan accumulation around VSMCs may lead to phenotype modulation in disease (Fig. 3) and perhaps even compromise their survival in the setting of TAAD, such as via induction of anoikis. For these reasons, exploring the cardiovascular roles of aggrecan at the organ and cellular level is a pressing research need in the cardiovascular disease arena.
Footnotes
Acknowledgements
We thank David Shumick for the artwork that is the basis for Figure 2.
Competing Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the NIH-NHLBI awards HL107147 and HL141130 (to S.S.A.), and by the Allen Distinguished Investigator Program, through support made by The Paul G. Allen Frontiers Group and the American Heart Association (to S.S.A).