
Abstract
Nidogen 1 and 2 are ubiquitous basement membrane (BM) components. They show a divergent expression pattern in certain adult tissues with a prominent localization of nidogen 2 in blood vessel BMs. Deletion of either nidogen 1 or 2 in mice had no effect on BM formation, suggesting complementary functions. However, studies in these mice revealed isoform-specific functions with nidogen 1–deficient mice showing neurological abnormalities and wound-healing defects not seen in the absence of nidogen 2. To investigate this further nidogen 1– or 2–deficient mice were intravenously injected with B16 murine melanoma cells, and lung metastasis was analyzed. The authors could show that loss of nidogen 2, but not of nidogen 1, significantly promotes lung metastasis of melanoma cells. Histological and ultrastructural analysis of nidogen 1– and 2–deficient lungs did not reveal differences in morphology and ultrastructure of BMs, including vessel BMs. Furthermore, deposition and distribution of the major BM components were indistinguishable between the two mouse strains. Taken together, these results suggest that absence of nidogen 2 might result in subtle changes of endothelial BMs in the lung, which would allow faster passage of tumor cells through these BMs, leading to a higher metastasis rate and more larger tumors.
Basement membranes (BMs) are specialized extracellular matrices that are instrumental for tissue compartmentalization and maintenance of cell phenotypes, but they also act as a source of morphogenetic stimuli for development and tissue remodeling (Ekblom et al. 1994; Smyth and Paulsson 1998). All BMs contain at least one representative from each of the laminin, collagen IV, nidogen, and proteoglycan (including perlecan) families (Timpl and Brown 1996; Miner and Yurchenco 2004).
In mammals, the nidogen family consists of two members, nidogen 1 and 2. On the basis of a large body of in vitro binding data, nidogens have been proposed to be essential for BM organization by mediating the linkage of the laminin and collagen IV network (Fox et al. 1991; Aumailley et al. 1993; Kohfeldt et al. 1998; Salmivirta et al. 2002). Mice lacking both nidogen isoforms die at birth with abnormalities in lung, heart, and limb development, all directly related to BM defects (Bader et al. 2005; Böse et al. 2006). However, there are also ultrastructurally normal BMs found in certain tissues such as kidney (Bader et al. 2005) and skin (Mokkapati et al. 2008), demonstrating that in vivo BMs can form without nidogen, thus indicating tissue-specific requirements for nidogens.
In mice, nidogen 1 and 2 share overlapping expression patterns during development (Kohfeldt et al. 1998; Miosge et al. 2000; Murshed et al. 2000; Salmivirta et al. 2002; Schymeinsky et al. 2002). In adult mice, however, nidogen 2 deposition is limited and particularly enriched in endothelial BMs, whereas nidogen 1 is evenly distributed in all BMs (Murshed et al. 2000; Miosge et al. 2002). In addition, nidogen 2 shows distinct binding to endostatin and tropoelastin in vitro and colocalizes with both components in elastic fibers of vessel walls (Sasaki et al. 1999; Sasaki et al. 2000).
In contrast to the nidogen double-null mice, most BMs appear ultrastructurally normal in nidogen 1 and 2 single-knockout mice (Murshed et al. 2000; Schymeinsky et al. 2002). The null animals are generally healthy, have a normal life span, and are fertile. In nidogen 1–deficient mice, nidogen 2 shows no alteration upon deletion of nidogen 1. However, in striated muscles, nidogen 2 staining is considerably changed. In normal skeletal muscle, staining for nidogen 2 is weak in the BMs surrounding the myocytes, whereas the endothelial BMs stain strongly. In contrast, in nidogen 1–deficient mice nidogen 2 appeared abundant in both sites with a staining pattern reminiscent of that of nidogen 1 in the control animals. Similar results were obtained for heart tissue (Murshed et al. 2000), supporting the notion that nidogens are capable of compensating for each other. In nidogen 2–deficient mice, no obvious alterations in the expression and distribution pattern of nidogen 1 and the other major BM components could be detected (Schymeinsky et al. 2002). Although there seems to be redundancy within the nidogen family, nidogen 1 null mice show neurological phenotypes (Dong et al. 2002; Köhling et al. 2006; Vasudevan et al. 2010) and wound-healing defects (Baranowsky et al. 2010), not seen in the absence of nidogen 2, suggesting also isoform-specific functions of the two nidogens.
To test this further, lung metastasis was analyzed after tail vein injection of melanoma cells in the absence of nidogen 1 or nidogen 2.
Material and Methods
Animals and Breeding
The mutation in the NID1 gene was generated by deleting exon 3 as described previously (Murshed et al. 2000). The mutation in the NID2 gene was introduced by insertion of a gene trap vector in intron 4 (Skarnes et al. 1995; Schymeinsky et al. 2002). These mouse strains were crossed back into C57BL/6 background for at least 10 generations. All animals used in this study were females between 10 and 12 weeks of age. Littermates carrying one wild-type allele of either NID1 or NID2 were used as controls. Wild-type and mutant alleles were assessed by Southern blot hybridization and/or PCR of DNA isolated from mouse tail biopsies as described previously (Murshed et al. 2000; Schymeinsky et al. 2002). The animals were housed in a specific pathogen-free facility, and all experiments were conducted in compliance with the German Law for Welfare of Laboratory Animals and were approved by the Regierungspräsidium Köln.
Tail Vein Injection of Melanoma Cells and Statistics
The mouse melanoma cell line B16-F1 was routinely cultured in RPMI 1640 medium supplemented with 10% fetal calf serum (FCS), 2 mM glutamine, non-essential amino acids, and 100 U/ml each of penicillin and streptomycin. For tail vein injection, the cells were trypsinized, washed once with sterile phosphate-buffered saline (PBS), and counted. Then, 5 × 105 cells suspended in 100 µl sterile PBS were injected. Twenty days after injection, the mice were killed by cervical dislocation. The lungs were dissected and right and left lungs photographed with a digital camera (Sony Digital Still Camera DSC-F707; Sony, Tokyo, Japan). Three independent experiments were performed with 29 control (NID1+/– and NID2+/–), 20 NID1–/–, and 24 NID2–/– mice in total. The lung metastases were counted and the size of the metastases determined using Image J software (National Institutes of Health; Rockville, MD).
Cell Adhesion Assays
For cell adhesion assays, 96-well tissue culture plates were coated with recombinant his-tagged nidogen 1 (0.25, 0.5, 1 µg), nidogen 2 (0.25, 0.5, 1 µg), His6 peptides (~70 nM), and human plasma fibronectin (1 µg/well) with 1 µg nidogen 1 or 2 corresponding to ~70 nM. In addition, human collagen IV (1 or 2 µg, with 2 µg corresponding to ~70 nM; Sigma-Aldrich, Munich, Germany), a mixture (1:1 molar ratio) of collagen IV and nidogen 1 or 2, Matrigel (1 or 2 µg; BD Bioscience, Heidelberg, Germany), or a mixture of Matrigel and nidogen 1 or 2 was used as immobilized substrates. The protein mixtures were preincubated at 37C for 1 hr before coating at 4C overnight. BSA coating and blocking of nonspecific binding sites were performed with heat-denatured BSA (1% in Ca2+/Mg2+-free PBS) for 1 hr at room temperature. After washing the wells twice, B16-F1 melanoma cells (2 × 104 cells/well) were seeded and incubated for 1 hr at 37C. Non-adherent cells were removed by washing twice with PBS. Adherent cells were fixed with 2% paraformaldehyde in PBS, pH 7.6, and stained with 0.5% crystal violet in 20% (v/v) methanol. The dye was released from the cells by the addition of 0.1 M sodium citrate in 50% (v/v) ethanol. The optical density of the released dye solution was determined at 595 nm. Adhesion was expressed relative to fibronectin, which was set arbitrarily as 1. The means and standard deviations in comparison with fibronection were calculated according to Bishop et al. (1975). Cell adhesions assays were also performed using unfixed cryosections of the vena cava caudalis as a substrate. The vena cava caudalis was dissected from two wild-type, nidogen 1– and nidogen 2–deficient adult male mice, cut open and frozen unfixed in optimal cutting temperature compound (O.C.T.; Sakura, Torrance, CA). The cryosections (inner surface up) were placed in the middle of a coverslip and incubated with B16-F1 melanoma cells suspended in RPMI medium (1 × 105/100 µl) at 37C for 3 hr. Non-adherent cells were removed by washing with PBS. Then the sections were fixed with 4% paraformaldehyde for 10 min at room temperature, mounted in aqueous mounting medium, and photographed (Leica DM 4000B microscope [Leica, Wetzlar, Germany] and DISKUS software [Carl H. Hilgers–Technisches Buro, Konigswinter, Germany]). Adherent cells per tissue section (750 µm2) were counted using Image J software. Two independent experiments were performed using each time three sections per genotype.
Histological and Immunofluorescence Analysis
The lungs were either fixed for 2 hr in 4% paraformaldehyde on ice before paraffin embedding or frozen unfixed in O.C.T. For histology, sections from paraffin-embedded lungs were stained with hematoxylin and eosin following standard protocols. Immunofluorescence was performed on paraffin sections after antigen retrieval or cryosections fixed for 10 min with ice-cold ethanol. Rabbit polyclonal antibodies raised against the following antigens were used: nidogen 1 and nidogen 2 (kindly provided by the late R. Timpl, Max-Planck-Institute for Biochemistry, Martinsried, Germany), laminin α4 and α5 chains (kindly provided by L. Sorokin, Institute of Physiological Chemistry and Pathobiochemistry, University of Münster, Germany), α–smooth muscle actin (α-sma; Abcam, Cambridge, UK), and collagen IV (Biozol; Eching, Germany). As a control for the specificity of the primary antibodies, lung sections were incubated with normal rabbit serum or IgG before adding the secondary antibodies (data not shown). The appropriate secondary antibodies conjugated to Alexa 488 (green) or Alexa 594 (red) (Molecular Probes; Eugene, OR) were applied together with propidium iodide for counterstaining the nuclei. Sections were embedded in mounting media.
Electron Microscopy
Lungs of control and mutant mice were excised and immediately fixed in 2.5% glutaraldehyde followed by 2.5% OsO4, stained en bloc with uranyl acetate, and processed for embedding in Epon 812-equivalent (Serva Bioproducts; Heidelberg, Germany) as described previously (Miosge et al. 1999). Ultrathin sections were counterstained with uranyl acetate and lead citrate.
Statistical Analysis
All statistical analyses were conducted using GraphPad Prism (GraphPad Software, La Jolla, CA). For each group, means and standard deviations were calculated. Statistical analysis was performed using one-way analysis of variance (ANOVA) with Tukey’s multiple comparison test and independent sample t-tests as appropriate. Differences were considered significant at p<0.05 (*).
Results
In the absence of nidogen 2 but not nidogen 1, lung metastasis is significantly increased
To study whether the deletion of the ubiquitous BM components nidogen 1 or nidogen 2 could contribute to tumor metastasis, we performed intravenous inoculation of murine B16-F1 melanoma cells into the tail vein of nidogen 1– and nidogen 2–deficient mice. The presence of lung metastases was examined 20 days later (Fig. 1a). As shown in Fig. 1b, the number of lung metastatic colonies was statistically significantly higher in nidogen 2–defcient mice in comparison with their control littermates. In contrast, the number of lung metastases in nidogen 1–deficient mice was comparable to that determined in control littermates. Furthermore, the number of larger individual metastatic colonies was higher in nidogen 2–deficient mice when compared with numbers obtained in nidogen 1–deficient and control mice (Fig. 1c). More important, 12 of 24 nidogen 2–deficient mice showed tumors larger than 3 mm2, whereas only 2 of 20 nidogen 1–deficient and 3 of 29 control mice had tumors of this size. These results suggest that deletion of nidogen 2, but not of nidogen 1, facilitates lung metastasis of melanoma cells.
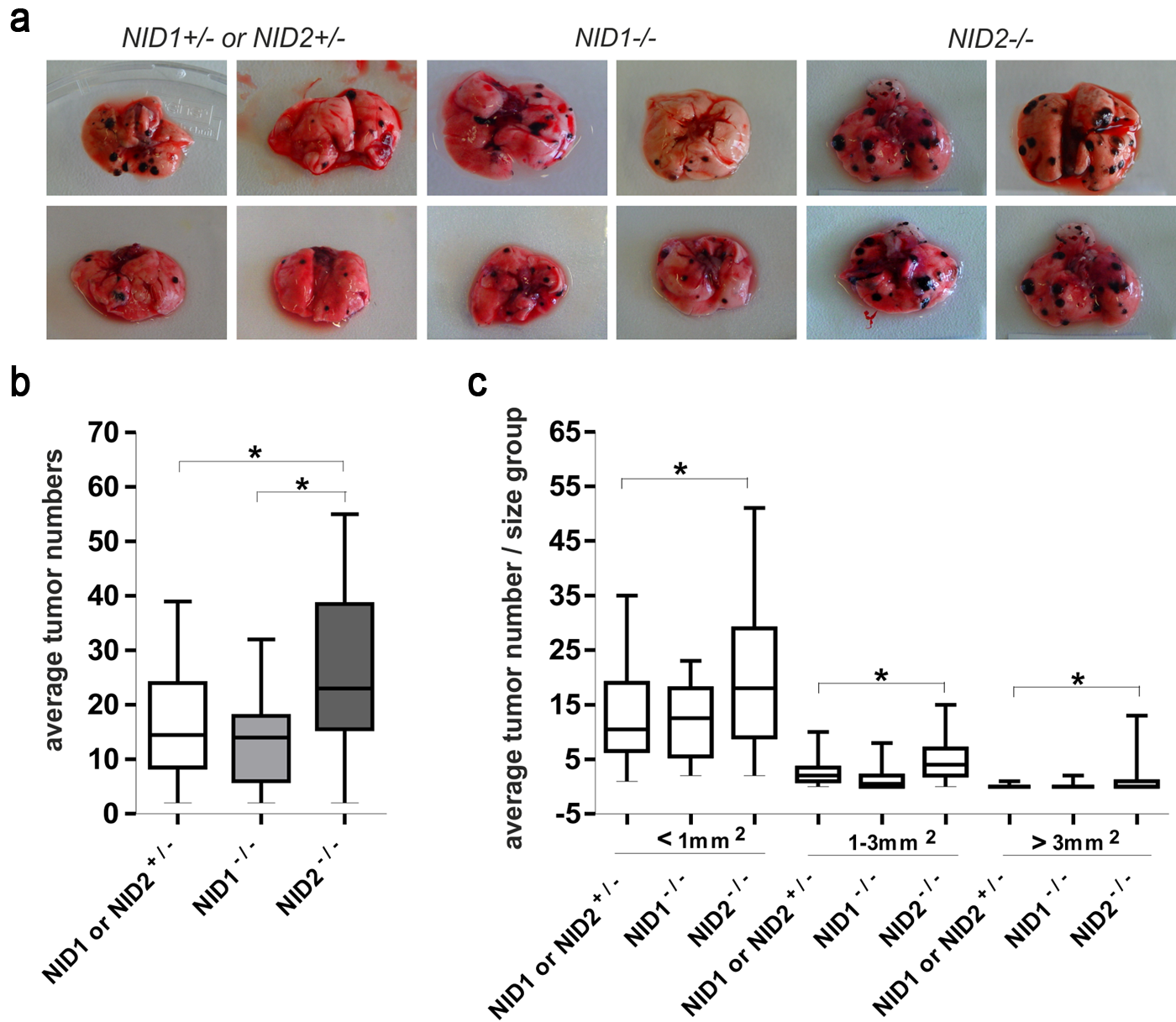
Tumor metastasis in nidogen-deficient mice. (a) Representative lung metastasis in NID1–/–, NID2–/–, and control (NID1+/– and NID2+/–) mice 20 days after injection of B16-F1 melanoma cells into the tail vein. (b) Numbers of lung surface metastatic colonies in NID1–/–, NID2–/–, and control (NID1+/– and NID2+/–) mice 20 days after injection of B16-F1 melanoma cells into the tail vein. The total number of lung metastatic colonies was counted on both sides of the lung, and the mean values were calculated. Three independent experiments were performed with 29 control (NID1+/– and NID2+/–), 20 NID1–/– mice, and 24 NID2–/– mice. The p-value (*) was different between control and NID2–/– mice (p<0.05). (c) Tumors were graded into three types based on the area covered by individual tumors (tumors with areas less than 1 mm2, between 1 and 3 mm2, larger than 3 mm2). In all three categories, NID2–/– mice showed statistically significantly higher tumor numbers (*). Statistical analysis was performed using one-way ANOVA.
Next we analyzed adhesion of B16-F1 melanoma cells to different BM components. B16 melanoma cells adhere equally well to nidogen 1 or nidogen 2. The differences seen in Fig. 2a are not statistically significant. There were also no significant differences observed with collagen IV and Matrigel alone or enriched with nidogen 1 or 2 (Fig. 2b). This indicates that differences in adhesion do not account for the higher tumor rate in the absence of nidogen 2. To test whether adhesion to vessel walls might account for the differences seen between nidogen 1– and nidogen 2–deficient mice, we used unfixed cryosections of the vena cava caudalis as adhesive substrate. However, as shown in Fig. 2c, adhesion of B16 melanoma cells was not statistically significantly different between tissue sections prepared from wild type, NID1–/–, or NID2–/– male mice.
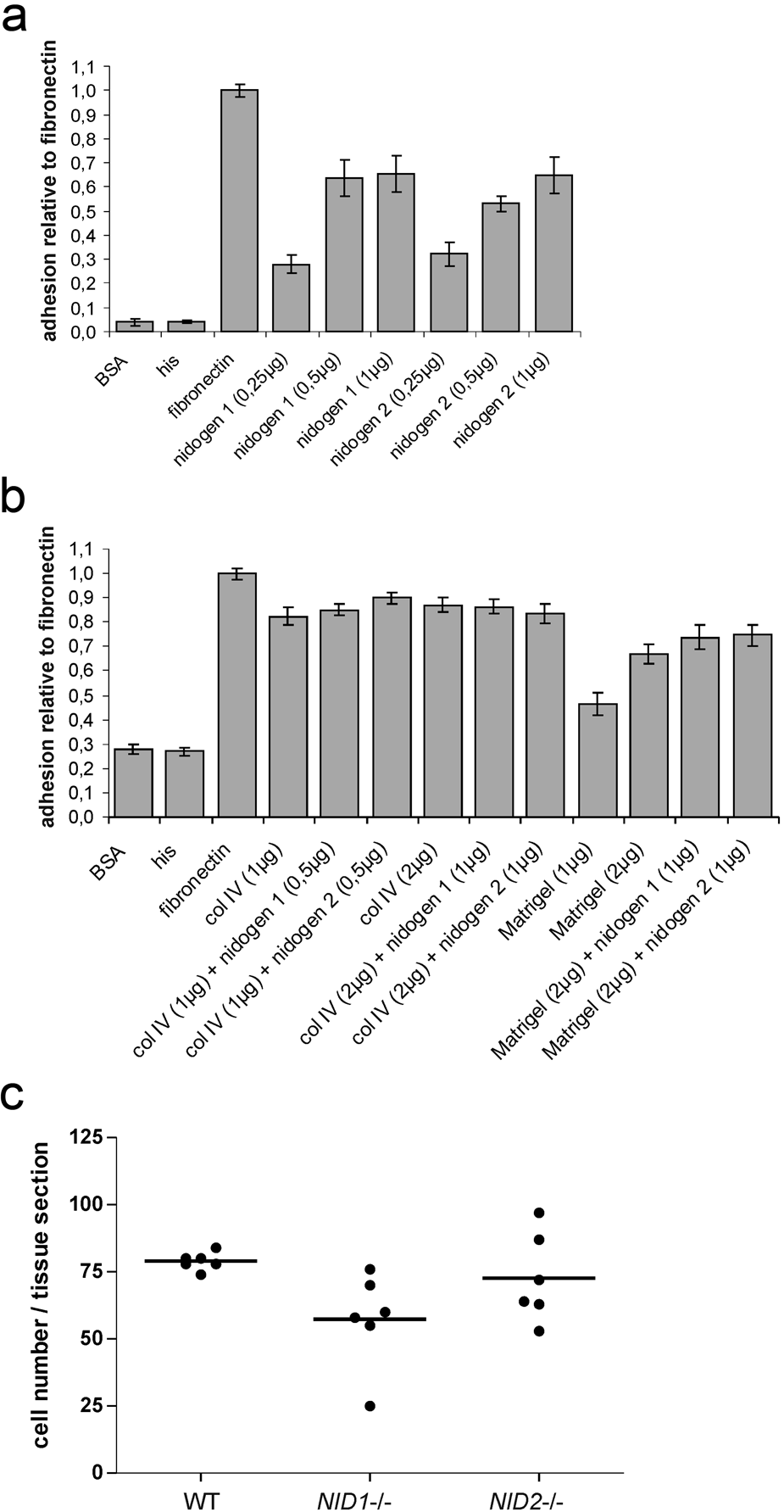
B16-F1 melanoma cells were seeded on microtiter plates coated with increasing concentrations of his-tagged recombinant nidogen 1 and nidogen 2 (a), different concentrations of collagen IV and Matrigel alone or enriched with nidogen 1 or 2 as described in Materials and Methods (b), and His peptides (a, b) as indicated. Coating with BSA and fibronectin was used as negative and positive controls, respectively. Adhesion is expressed relative to the adhesion to fibronectin, which was arbitrarily set as 1. Results are shown as mean ± SD from three independent experiments performed in triplicates. B16-F1 melanoma cells were also seeded on unfixed cryosection of the vena cava caudalis prepared from wild-type (WT), NID1–/–, and NID2–/– mice (c). Shown are the results obtained using two different preparations of the vena cava (three tissue sections of each genotype per adhesion assay). Statistical analysis using one-way ANOVA did not reveal significant differences.
BM composition in the lung is not altered in the absence of nidogen 1 or 2
To understand the potential mechanisms contributing to enhanced metastasis and tumor growth in nidogen 2–deficient mice, we first analyzed lung morphology and BM composition in nidogen-deficient lungs in comparison with control lungs. As shown in Fig. 3, in the absence of nidogen 1 or 2, lung morphology appeared normal with well-formed, thin-walled respiratory bronchioles, respiratory ducts, and alveoli when compared with lung sections from control littermates.
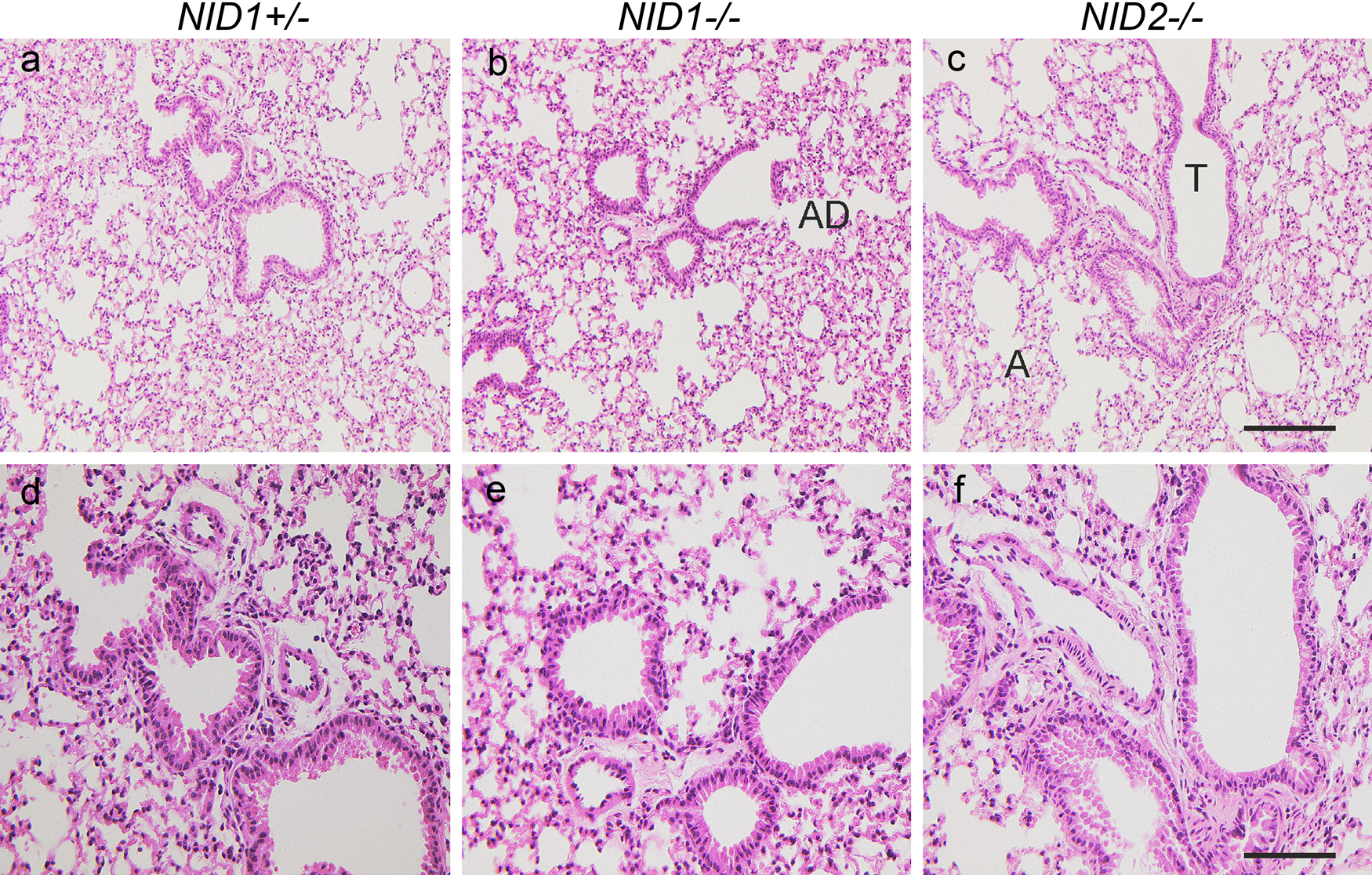
Lung histology in nidogen-deficient mice. Hematoxylin and eosin staining of lung sections from NID1+/– (a, d), NID1–/– (b, e), and NID2–/– (c, f) shows a comparable tissue architecture in all three genotypes. T, terminal bronchiole; A, alveoli; AD, alveolar duct. Scale bars = 180 µm (a–c); = 90 µm (d–e).
BM composition was analyzed by indirect immunofluorescence on paraffin-embedded or cryostat lung sections. In control sections, nidogen 1 staining was detected in the alveolar and capillary BMs as well as the BMs of large vessels and airways (Fig. 4a). As shown in Fig. 4d, the nidogen 2 staining pattern is comparable to that seen with nidogen 1 antibodies. However, immunostainings for nidogen 2 in lung sections from nidogen 1–deficient mice (Fig. 4e) and for nidogen 1 in lung sections from nidogen 2–deficient mice (Fig. 4c) were unchanged with respect to intensity and distribution when compared with control sections (Fig. 4a,d). Immunofluorescence analysis applying antibodies raised against collagen IV (Fig. 4g–i) and the laminin α5 chain (laminin 511) (Fig. 4j–1) also did not reveal differences between control and nidogen-deficient mice. Comparable results were obtained for α-sma, a marker for fully mature medium-sized and large vessels (Jostarndt-Fogen et al. 1998) (Fig. 5a–c). Immunofluorescence applying antibodies against CD 31 (Fig. 5d–f), an endothelial cell marker, and the laminin α4 chain (laminin 411; Fig. 5g–i), a marker for endothelial BMs (Hallmann et al. 2005), also did not display obvious alterations in the distribution patterns of these markers between the three genotypes.
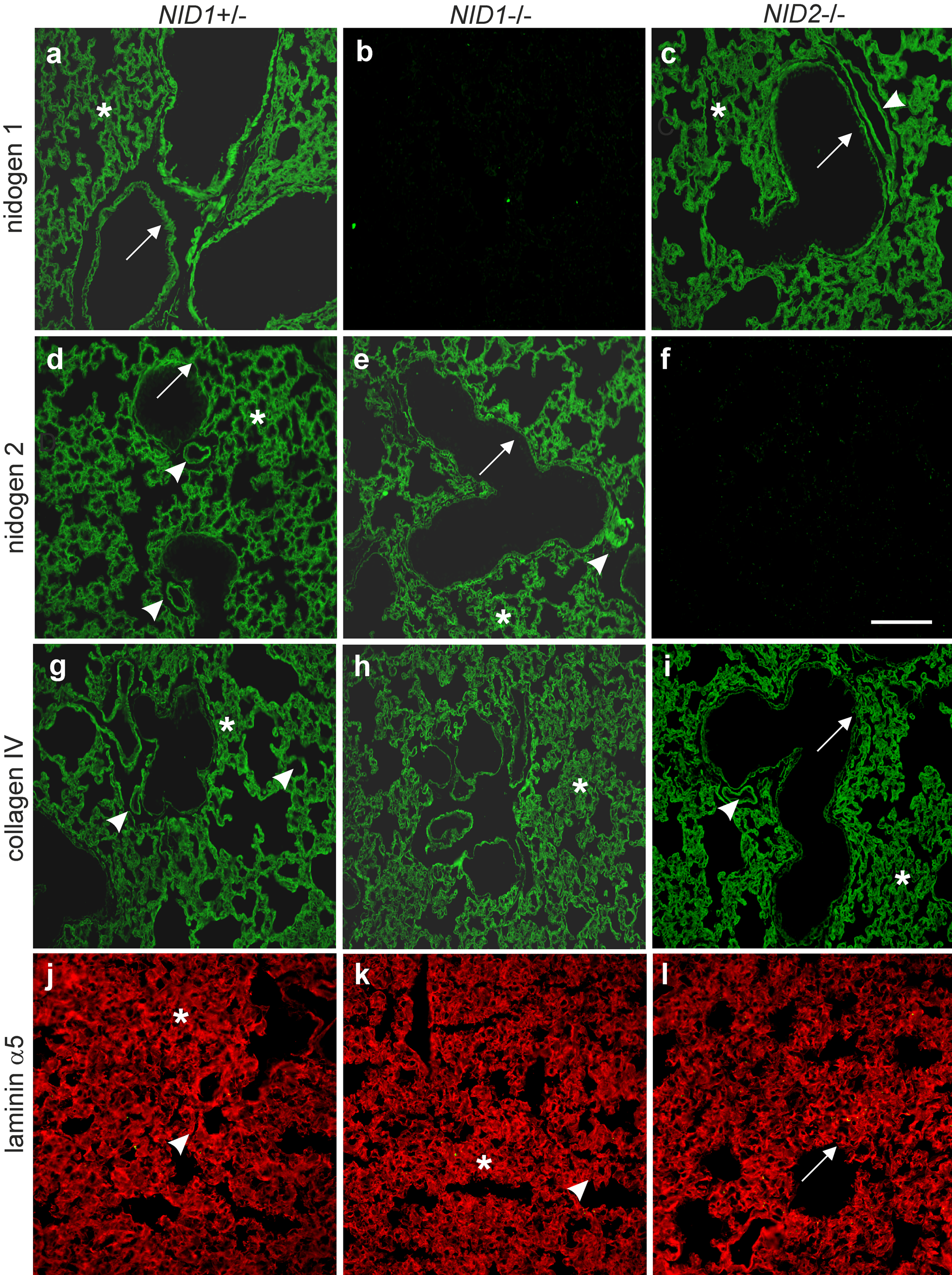
Indirect immunofluorescence staining was performed on lung sections from control (NID1+/–; a, d), NID1–/– (b, e), and NID2–/– (c, f) mice. Paraffin-embedded lung sections were incubated after antigen retrieval with antibodies directed against nidogen 1 (a–c), nidogen 2 (d–f), and collagen IV (g–i). Cryosections were used for staining with antibodies directed against the laminin α5 chain (j–1). Secondary antibodies conjugated to Alexa 488 (green) and Alexa 594 (red) were applied to detect the primary antibodies. Arrows, basement membranes (BMs) of large airways; asterisks, alveoli and BM of alveoli; arrowheads, vessel BMs. Scale bar = 90 µm.
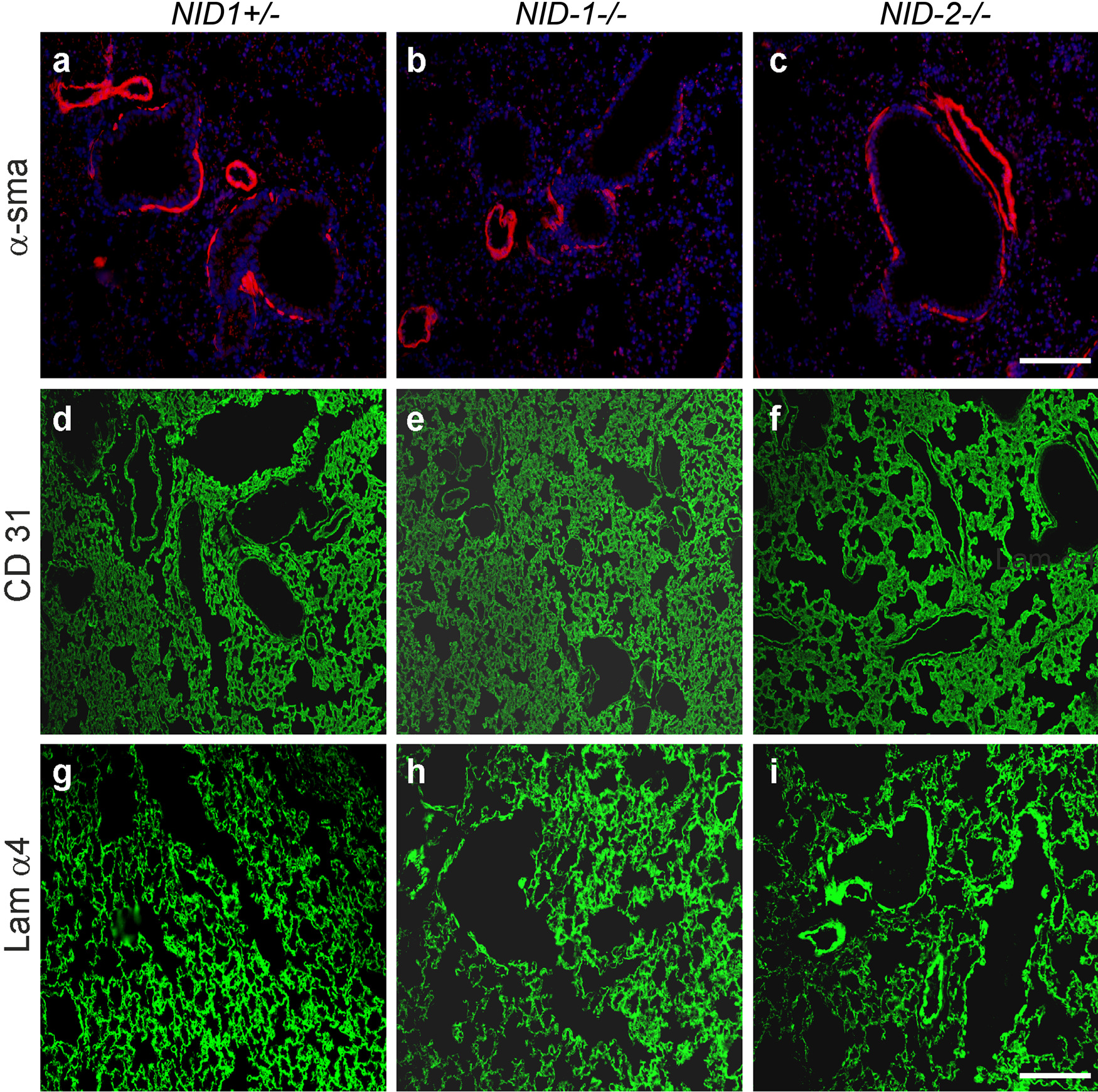
Indirect immunofluorescence staining was performed on lung sections from control (NID1+/–; a, d, g), NID1–/– (b, e, h), and NID2–/– (c, f, i) mice applying antibodies raised against α–smooth muscle actin (α-sma; a–c), CD31 (d–f), and the laminin α4 chain (Lam α4; g–i). α–Smooth muscle actin and laminin α4 stainings were performed on paraffin-embedded lung sections and CD31 staining on cryosections. Secondary antibodies conjugated to Alexa 488 (green) and Alexa 594 (red) were used to detect the primary antibodies. In a–c, the nuclei were counterstained with DAPI. Scale bars = 90 µm (a–c); = 180 µm (d–i).
Ultrastructure of vessel BMs is not altered in the absence of nidogens
In addition to histological and immunofluorescence analysis, electron microscopy was performed. Lung sections from control and nidogen-deficient mice were compared in the hilus region (Fig. 6a–c), where medium-sized and larger blood vessel are found, and in the lung periphery (Fig. 6d–f), where alveolar/capillary BMs are located. Electron micrographs of the hilus region of the lung show a medium-sized artery with smooth muscle and endothelial cells being separated by the lamina elastica interna in the control sections (Fig. 6a). The periphery of the lung displays in control sections capillaries with erythrocytes in their lumen and thin, elongated endothelial cells separated from pneumocytes type I by the alveolar/capillary BM (Fig. 6d). In nidogen 1– and nidogen 2–deficient lung sections, the ultrastructure of the hilus and peripheral regions is comparable to that in control lungs (Fig. 6b,c,e, f). At higher magnification, the alveolar BM in control mice is seen as a continuous and homogeneous layer between the epithelial lining of the alveoli and the endothelium of capillaries in the interalveolar space (Fig. 6g). In the absence of either nidogen 1 or nidogen 2, the ultrastructural appearance of the alveolar BM is indistinguishable from that seen in lung sections of control mice (Fig. 6h,i). In summary, no obvious differences in the ultrastructural appearance of BMs of blood vessels could be detected in the absence of either nidogen 1 or nidogen 2.
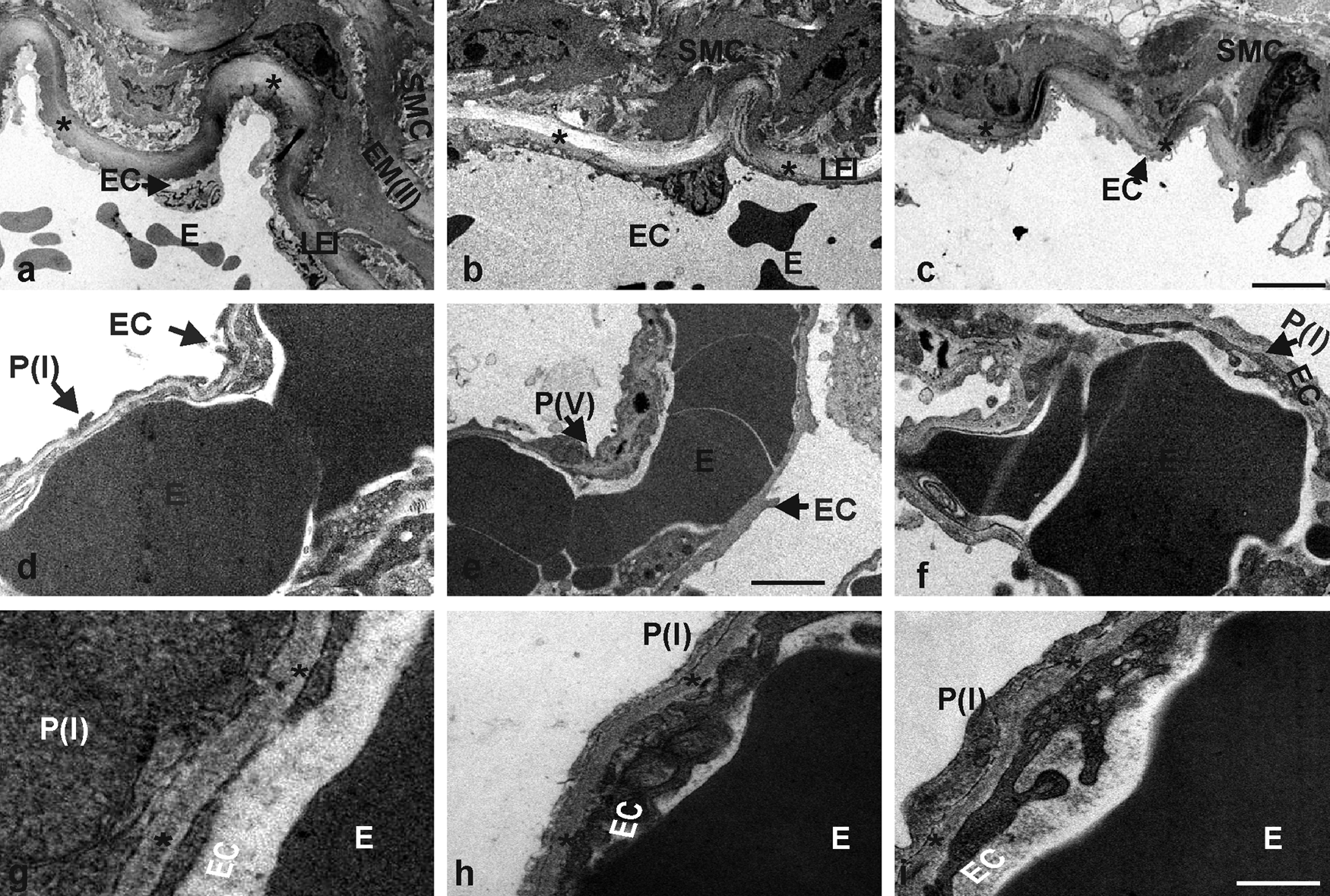
Electron micrographs of ultrathin sections of the hilus region (a–c) and the lung periphery (d–f) from control (a, d, g), NID1–/– (b, e, h), and NID2–/– (c, f, i) mice. The lamina elastica interna (LEI) is marked by asterisks (a–c). The alveolar BM is recognized as a homogeneous layer of extracellular matrix (indicated by asterisks) between the epithelial lining of the alveoli and the endothelia cells of the capillaries of the interalveolar septa (g–i). The arrows mark the different cell types; E, erythrocyte; EC, endothelial cell; EM(II), second elastic membrane; P(I), pneumocyte type I; P(V), pneumocyte type V; SMC, smooth muscle cell. Scale bars = 5 µm (a–c); = 1.75 µm (d–f); = 0.4 µm (g–i).
Discussion
Tumor metastasis is a multistage process that requires complex tumor cell–matrix interactions. Those interactions include adhesion of tumor cells to BM components of vessels, localized proteolysis of the BM, and migration into the connective tissue of distant organs (Sahai 2007; Row and Weiss 2008). However, it has been also demonstrated that tumor cells can extravasate from blood or lymph vessels in a protease-independent manner via amoeboid or bleb-associated motility (Friedl and Wolf 2003; Hotary et al. 2006; Sahai 2007; Fackler and Grosse 2008).
Nidogen 1 and 2 are ubiquitous BM proteins. Analysis of nidogen 1 and nidogen 2 null mice revealed overlapping functions of the two isoforms showing no ultrastructural BM deformities in the absence of either nidogen 1 or 2 (Murshed et al. 2000; Schymeinsky et al. 2002).
Here we report that after tail vein injection of B16 melanoma cells, the number of lung metastases was significantly enhanced in nidogen 2 null mice compared with nidogen 1 null and wild-type mice. In addition, the size of individual metastatic colonies was increased, showing only in nidogen 2 null mice significant numbers of large tumors.
Although nidogen 1 and 2 show in vitro a similar binding repertoire (Fox et al. 1991; Kohfeld et al. 1998; Salmivirta et al. 2002), the binding behavior of nidogen 1 and 2 seems to differ in vivo. Analysis in mice with deletion of the nidogen binding module on the laminin γ1 chain revealed that in vivo, only nidogen 1 deposition is critically dependent on this nidogen binding module, whereas nidogen 2 is independently recruited most likely via an alternative binding site on the laminin molecule. Although the molecular composition and interactions within the BMs have been changed in these mutant mice, only certain BMs showed ultrastructural alteration (Miosge et al. 2000; Willem et al. 2002; Mokkapati et al. 2011). Based on these observations and the fact that nidogen 2 is particularly enriched in vascular BMs (Kohfeldt et al. 1998; Murshed et al. 2000; Schymeinsky et al. 2002), it is tempting to speculate that in the absence of nidogen 2, the molecular organization of the vascular BM is modified in such a way that transmigration of the melanoma cells is facilitated. Facilitated transmigration would allow more tumor cells to form metastatic colonies in the given time frame of 21 days and would also provide more time for the melanoma cells to grow, resulting in larger metastases. This is supported by the observation that 50% of nidogen 2–deficient mice and only 10% or less of nidogen 1–deficient and control mice, respectively, had tumors larger than 3 mm2. In this process, adhesion of B16 melanoma cells does not appear to play a role as we could show that B16 melanoma cells adhere equally well to recombinant nidogen 1 or 2, collagen IV, and Matrigel, a BM matrix, alone or enriched with nidogen 1 or 2. In addition, even adhesion to sections of the vena cava used as an in vivo substrate was indistinguishable between the three genotypes.
Analysis of lungs from nidogen 1– or 2–deficient mice did not reveal differences in morphology, BM composition of the vascular endothelium, and ultrastructural appearance of BMs, including endothelial BMs.
As indicated by the results obtained in mice with deletion of the nidogen binding module on the laminin γ1 chain (Willem et al. 2002; Mokkapati et al. 2011), changes in molecular composition and interactions of BMs do not necessarily result in detectable ultrastructural alterations. Therefore, the lack of the major vascular BM component nidogen 2 could render this BM or individual BM components more protease sensitive, thus facilitating the passage of melanoma cells. Alternatively, alteration in the intermolecular spacing of BM components by the lack of nidogen 2 could generate gaps allowing the melanoma cells to squeeze through in a protease-independent manner.
Footnotes
Acknowledgements
We gratefully acknowledge the excellent technical assistance of Jan Zamek (Department of Dermatology, University Hospital of Cologne).
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research and/or authorship of this article: This work was supported by the Deutsche Forschungsgemeinschaft through the Sonderforschungsbereich 829 at the University of Cologne and grant 304/11–1 (to R.N.). This work was also supported by the Maria Pesch Stiftung (to R.N.).