
Abstract
Lymphocyte homing is regulated by a multistep process mediated by sequential adhesive interactions between circulating lymphocytes and high endothelial venules (HEVs). In gut-associated lymphoid tissue (GALT), the initial interactive step, “tethering and rolling,” is partly mediated by integrin α4β7 expressed on GALT-homing lymphocytes and its ligand MAdCAM-1, which is exclusively expressed on HEVs in GALT. To probe functional MAdCAM-1 in tissue sections, we developed a soluble integrin α4β7 heterodimeric IgG chimera by joining the extracellular region of mouse integrin α4 and β7 subunits to a human IgG Fc domain. Western blot analysis revealed that co-transfection of HEK 293T cells with expression vectors encoding integrin α4•IgG and β7•IgG results in the formation of α4β7•IgG heterodimeric chimeras. This complex preferentially binds to CHO cells expressing MAdCAM-1 and, to a lesser extent, to cells expressing VCAM-1, but not to cells expressing ICAM-1. Moreover, α4β7•IgG specifically binds to HEVs in GALT in situ in a divalent cation–dependent fashion and inhibits lymphocyte binding to HEVs in GALT. These findings indicate that α4β7•IgG can be used as a probe for functional MAdCAM-1 expressed on HEVs in GALT and could potentially serve as an anti-inflammatory drug inhibiting GALT-specific lymphocyte migration.
Keywords
The gastrointestinal tract is connected to the external world, bringing “the outside world within.” Thus, the gastrointestinal mucosa is continuously exposed to exogenous antigens, originating from diet and commensal bacteria, as well as to potentially harmful agents such as pathogenic microorganisms. As a result, the gastrointestinal mucosa must be armed with a specialized local immune system for such antigens. Indeed, gut-associated lymphoid tissue (GALT), consisting of isolated and aggregated lymphoid follicles such as Peyer’s patches (PPs) and mesenteric lymph nodes (MLNs), is one of the largest lymphoid organs in the body, containing 70% of the body’s lymphocytes (Corr et al. 2008), and it plays crucial roles in gastrointestinal mucosal immunity.
The ability of lymphocytes to migrate selectively to a particular lymphoid organ is termed “homing.” Homing is regulated by a multistep process mediated by sequential adhesive interactions between circulating lymphocytes and specialized endothelial cells comprising high endothelial venules (HEVs) (Butcher and Picker 1996; von Andrian and Mempel 2003). In peripheral lymph nodes (PLNs) and tonsils, the initial step of the interaction, called “tethering and rolling,” is mediated by L-selectin expressed on lymphocytes and its carbohydrate ligand 6-sulfo sialyl Lewis X–capped glycoproteins, collectively called peripheral lymph node addressin (PNAd), expressed on the luminal surface of HEVs (Rosen 2004). On the other hand, in GALT, the tethering and rolling step is partly mediated by L-selectin–PNAd interactions but also in an L-selectin–PNAd–independent manner by integrin α4β7 expressed on GALT-homing lymphocytes and its ligand mucosal addressin cell adhesion molecule 1 (MAdCAM-1), which is selectively expressed on HEVs in GALT (Streeter et al. 1988). Moreover, after activation by CCL21 chemokine produced by endothelial cells lining PP HEVs (Pachynski et al. 1998; Gunn et al. 1998; Hirose et al. 2001), integrin α4β7 increases its binding affinity to MAdCAM-1 through conformational changes (Hynes 2002) to facilitate subsequent firm attachment or “arrest” of lymphocytes on HEVs in GALT (Berlin et al. 1995). Thus, MAdCAM-1 functions as a molecular “zip code” for integrin α4β7 expressing GALT-homing lymphocytes.
Although its etiopathogenesis has not yet been fully elucidated, pathological lymphocyte recruitment to the gut characterizes inflammatory bowel disease (IBD), consisting of ulcerative colitis (UC) and Crohn’s disease (CD), and plays a central role in initiation and progression of the disease. To block such lymphocyte accumulation in the gut, therapeutic monoclonal antibodies that target the interaction between integrin α4β7 and MAdCAM-1, such as Natalizumab and Vedolizumab (otherwise known as MLN-02 or LDP-02), have been developed and shown to be effective in treating IBD (Gordon et al. 2002; Ghosh et al. 2003; Feagan et al. 2005; Soler et al. 2009). However, because these antibodies contain elements foreign to the patient, that is, mouse peptide sequences in the complementarity determining region (CDR) (Rutgeerts et al. 2009), administration of these proteins occasionally elicits an immune response (Aarden et al. 2008), namely increased serum levels of human anti-human antibodies (HAHA), which may cause severe hypersensitivity-type reactions, decrease treatment efficacy, or induce autoimmunity (Nechansky 2010). Thus, an alternative strategy to block integrin α4β7–MAdCAM-1 interactions is required. Thus far, researchers have developed chimeric proteins consisting of integrins and immunoglobulin G (IgG) and analyzed their activity (Stephens et al. 2000; Strauch et al. 2001; Coe et al. 2001). However, to date, there are no reports of development of functional integrin α4β7 heterodimeric IgG chimeras, which could be useful as molecular targeting therapy for IBD.
In the present study, we developed a soluble integrin α4β7 heterodimeric IgG chimera (α4β7•IgG) by joining the extracellular region of mouse integrin α4 and β7 subunits to a human IgG Fc domain, utilizing the hinge region to drive heterodimer formation (Stephens et al. 2000) (Fig. 1). This antibody-like heterodimeric molecule preferentially binds to MAdCAM-1 and can be used as an immunohistochemical reagent to stain functional MAdCAM-1 expressed on HEVs in GALT. α4β7•IgG inhibits binding of lymphocytes to HEVs in GALT, making it a candidate as an effective anti-inflammatory drug to inhibit GALT-specific lymphocyte migration.
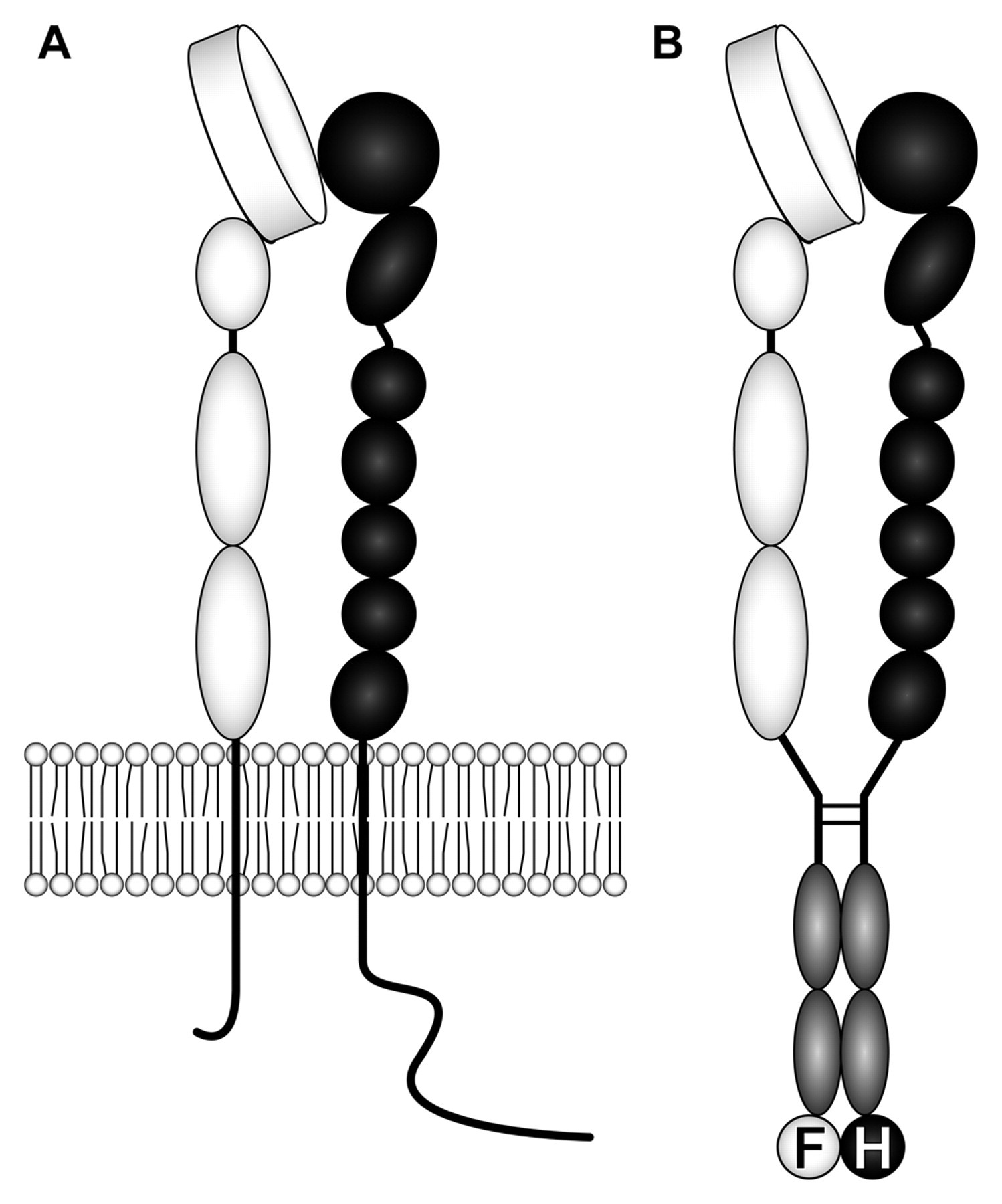
Schematic of native integrin α4β7 expressed on lymphocytes (A) and recombinant integrin α4β7 heterodimeric IgG chimera (α4β7•IgG) (B). The extracellular domain of integrin α4 (white) and β7 (black) subunits is fused to IgG Fc (gray), and the IgG hinge is utilized to drive heterodimer formation. FLAG (F) and HA (H) tags are attached to the C-termini of α4•IgG and β7•IgG, respectively. Putative molecular weights of α4•IgG-FLAG and β7•IgG-HA are 170 kDa and 160 kDa, respectively.
Materials and Methods
Construction of α4•IgG-FLAG and β7•IgG-HA Expression Vectors
DNA fragments encoding human IgG hinge and constant regions (CH2 and CH3) connected to either FLAG (IgG-FLAG) or hemagglutinin (HA) epitope tag (IgG-HA) were amplified by polymerase chain reaction (PCR) using pcDNA3-IgG (Aruffo et al. 1990) as template. PCR products were digested with BamHI and XbaI and inserted into the BamHI/XbaI sites of pcDNA3.1/Hygro (Invitrogen, Carlsbad, CA), resulting in pcDNA3.1/Hygro-IgG-FLAG and pcDNA3.1/Hygro-IgG-HA. DNA fragments encoding the extracellular region of mouse integrin α4 and β7 subunits, corresponding to amino acid residues 1−977 and 1−724, respectively, were amplified by PCR. Template cDNA was prepared from RNA isolated from mouse PPs using SuperScript III reverse transcriptase (Invitrogen), according to the manufacturer’s instruction. In the PCR reaction, 1772G>T (R591L) in the α4 subunit was introduced to eliminate a posttranslational endoproteolytic cleavage site (Teixido et al. 1992; Bergeron et al. 2003), and three silent mutations—108T>C, 132C>T, and 384C>T—in the β7 subunit were introduced to eliminate BamHI sites for subsequent subcloning by site-directed mutagenesis, as described previously (Higuchi et al. 1988; Ho et al. 1989). PCR products for α4 and β7 subunits were digested with NheI and BamHI and inserted into the NheI/BamHI sites of pcDNA3.1/Hygro-IgG-FLAG and pcDNA3.1/Hygro-IgG-HA, respectively, resulting in pcDNA3.1/Hygro-α4•IgG-FLAG and pcDNA3.1/Hygro-β7•IgG-HA. All PCR was carried out using PrimeSTAR Max DNA polymerase (Takara, Kyoto, Japan), according to the manufacturer’s instruction, and PCR products were inserted into pCR-Blunt (Invitrogen) and sequenced. Primers used are listed in Table 1.
Primers Used in This Study

Expression of α4β7•IgG Heterodimeric Chimeras
Human embryonic kidney (HEK) 293T cells were transiently transfected with pcDNA3.1/Hygro-α4•IgG-FLAG and pcDNA3.1/Hygro-β7•IgG-HA at a 1:1 ratio using Lipofectamine Plus (Invitrogen), according to the manufacturer’s instruction. Transfected cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen) supplemented with 10% fetal bovine serum (FBS) (HyClone, South Logan, UT). For subsequent Western blot analysis, DMEM was substituted with HyClone SFM4Transfx-293 serum-free medium (Thermo Fisher Scientific, Waltham, MA) supplemented with 4 mM L-glutamine (Invitrogen). Conditioned medium was recovered 72 hours later and concentrated approximately 10-fold using Centriprep YM-30 (Millipore, Billerica, MA).
Western Blot Analysis
Conditioned serum-free medium was purified by Protein A-Sepharose 4B Fast Flow from Staphylococcus aureus (Sigma-Aldrich, St. Louis, MO) and lysed in sample buffer, with or without reduction (2-mercaptoethanol). After incubation at 65C for 15 minutes, the sample was subjected to 7% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a polyvinylidene difluoride (PVDF) membrane (Millipore). After blocking in Tris-buffered saline (TBS) (pH 7.6) containing 5% nonfat dry milk for 60 minutes, the membrane was incubated with mouse anti-FLAG monoclonal antibody (Sigma-Aldrich) and rabbit anti-HA polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA), followed by horseradish peroxidase (HRP)–conjugated goat anti-mouse IgG and anti-rabbit IgG (Jackson ImmunoResearch, West Grove, PA), respectively. To detect the human IgG Fc region, the membrane was also incubated with HRP-conjugated goat anti-human IgG (Jackson ImmunoResearch). The membrane was developed using the ECL system (Amersham Pharmacia Biotech, Piscataway, NJ).
Subcloning of Mouse MAdCAM-1, VCAM-1, and ICAM-1
cDNAs encoding mouse MAdCAM-1, vascular cell adhesion molecule 1 (VCAM-1), and intercellular adhesion molecule 1 (ICAM-1) were amplified by PCR. As template, cDNAs prepared from mouse PPs were used for MAdCAM-1, and cDNAs from mouse hearts were used for VCAM-1 and ICAM-1. After sequencing, PCR products were digested with BamHI and XhoI for MAdCAM-1, EcoRI and XhoI for VCAM-1, and BamHI and EcoRI for ICAM-1 and inserted into corresponding sites of pcDNA1.1 (Invitrogen), resulting in pcDNA1.1-mMAdCAM-1, pcDNA1.1-mVCAM-1, and pcDNA1.1-mICAM-1, respectively. Primers used are shown in Table 1.
α4β7•IgG Binding Assay on CHO Cells Expressing MAdCAM-1, VCAM-1, and ICAM-1
Chinese hamster ovary (CHO) cells grown on coverslips were transiently transfected with pcDNA1.1-mMAdCAM-1, pcDNA1.1-mVCAM-1, pcDNA1.1-mICAM-1, or pcDNA1.1 (mock) using Lipofectamine Plus (Invitrogen). Thirty-six hours later, cells were fixed with 20% neutral buffered formalin for 15 minutes, followed by blocking with 1% bovine serum albumin (BSA) (Sigma-Aldrich) in TBS for 30 minutes. Cells were incubated at room temperature (RT) for 60 minutes with α4β7•IgG in DMEM supplemented with 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (pH 7.0) and 1 mM MnCl2. In our preliminary experiment, we determined optimal concentration of Mn2+ by using titrated Mn2+, ranging from 0.1 mM to 10 mM. The α4β7•IgG bound to MAdCAM-1 with 0.1 mM Mn2+; however, the binding was stronger when we administered 1 mM Mn2+. This finding is in agreement with a previous report by Stephens et al. (2000), showing α4β1 integrin IgG chimera binding to VCAM-1. To show α4β7•IgG binding clearly, we used 1 mM Mn2+ throughout the study. After washing with DMEM, cells were incubated with Alexa Fluor 488–labeled goat anti-human IgG (Invitrogen) for 15 minutes. Cells were mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, CA) and observed under an AX-80 fluorescence microscope (Olympus, Tokyo, Japan). For negative controls, DMEM was supplemented with 5 mM ethylenediaminetetraacetic acid (EDTA). To quantify the α4β7•IgG binding, cell enzyme-linked immunosorbent assay (ELISA) was performed basically as described previously (Mitoma et al. 2009).
α4β7•IgG Binding Assay on Mouse Tissue Sections
PPs, MLNs, and PLNs were dissected from C57BL/6 mice (Japan Charles River, Kanagawa, Japan), embedded in Tissue-Tek OCT compound (Sakura Finetek, Tokyo, Japan), and frozen at −80C. Frozen tissues were sectioned at 6 µm and fixed with acetone for 5 minutes, followed by air drying. α4β7•IgG binding assays on sections were performed as described above. Animals were treated in accordance with the institutional guidelines of Shinshu University School of Medicine, and the experimental protocol was approved by the Animal Research Committee of Shinshu University.
Lymphocyte Adhesion Inhibition Assay on CHO Cells
CHO cells stably expressing mouse MAdCAM-1 were established as described previously (Kobayashi et al. 2009). Briefly, CHO cells were transfected with pcDNA1.1-mMAdCAM-1 and pcDNA3.1/Hygro at a ratio of 10:1 using Lipofectamine Plus (Invitrogen) and selected in 400 µg/mL of Hygromycin B (Invitrogen). Cells positive for MECA-367 antibody specific to mouse MAdCAM-1 (BD Pharmingen, Franklin Lakes, NJ) were cloned by limited dilution, resulting in CHO/mMAdCAM-1.
5 × 104 CHO/mMAdCAM-1 cells and control CHO cells were seeded onto each well of Lab-Tech 4-well chamber slides (Nalge Nunc International, Rochester, NY) 24 hours prior to assay. Lymphocytes were isolated from the spleen, and expression of integrin α4β7 and L-selectin was confirmed by FACS analysis using antibodies R1-2, FIB506.64, DATK32, and MEL-14 (BD Pharmingen), specific to mouse integrin α4 subunit, β7 subunit, α4β7 complex, and L-selectin, respectively (Andrew et al. 1994). 1 × 106 lymphocytes suspended in 500 µL of DMEM supplemented with 20 mM HEPES (pH 7.0) and 1 mM MnCl2 with or without serially diluted α4β7•IgG were overlaid onto CHO cells in each well and incubated at RT for 30 minutes with rotation at 60 rpm using a rotating shaker SRR-2 (AS ONE, Osaka, Japan). Nonadherent cells were removed by rinsing three times with 1 mL/well of DMEM. After fixation with phosphate-buffered saline (PBS) containing 1% glutaraldehyde, slides were observed with Nomarski differential interference optics using an AX-80 fluorescence microscope (Olympus). The number of CHO cells and adherent lymphocytes in five fields of view with ×200 magnification was determined, and the number of lymphocytes attached per 100 CHO cells was calculated.
Lymphocyte Adhesion Inhibition Assay on HEVs by Modified Stamper-Woodruff Assay
Lymphocyte adhesion inhibition on HEVs was evaluated by the Stamper-Woodruff assay (Stamper and Woodruff 1976) with modifications as described (Hirakawa et al. 2010). Frozen tissue sections of mouse PPs, MLNs, and PLNs were fixed with 20% neutral buffered formalin for 15 minutes. After three washes with distilled water, sections were blocked with buffer A (20 mM HEPES [pH 7.4], 150 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, 1 mM MnCl2) containing 3% BSA for 45 minutes. Freshly prepared splenocytes were labeled with CellTracker Green CMFDA (5-chloromethylfluorescein diacetate) (Invitrogen), as per the manufacturer’s instructions. 1 × 106 lymphocytes suspended in 200 µL of DMEM supplemented with 20 mM HEPES (pH 7.0) and 1 mM MnCl2 with or without α4β7•IgG were overlaid on a section and incubated at RT for 30 minutes with rotation at 60 rpm. After removing nonadherent cells by gentle tapping onto paper towels and dipping into buffer A in a 50-mL centrifuge tube for 10 seconds, sections were fixed in buffer A containing 0.5% glutaraldehyde at 4C for 15 minutes. After three washes with distilled water, sections were mounted with Vectashield mounting medium (Vector Laboratories), and lymphocyte adhesion was quantified under an AX-80 fluorescence microscope (Olympus) by counting the number of lymphocytes attached to 20 randomly chosen HEVs in each group.
Statistical Analysis
Differences between groups were statistically analyzed by the one-way analysis of variance (ANOVA) with Bonferroni posttest using InStat 3 software (GraphPad Software, San Diego, CA). p values less than 0.05 were considered significant.
Results
Formation of α4β7•IgG by Co-Transfection with α4•IgG and β7•IgG cDNAs
To determine whether co-transfection of HEK 293T cells with expression vectors harboring cDNAs encoding α4•IgG-FLAG and β7•IgG-HA results in formation of α4β7•IgG heterodimers, Western blot analysis was carried out under reducing and nonreducing conditions. Under reducing conditions, immunoblotting with anti-FLAG showed a single band migrating at 170 kDa and corresponding to α4•IgG-FLAG (Figs. 1B and 2, left panel). Immunoblotting with anti-HA showed a single band migrating at 160 kDa, corresponding to β7•IgG-HA. Immunoblot with anti-human IgG revealed two bands of similar intensity migrating at 170 kDa and 160 kDa, corresponding to α4•IgG-FLAG and β7•IgG-HA, respectively.
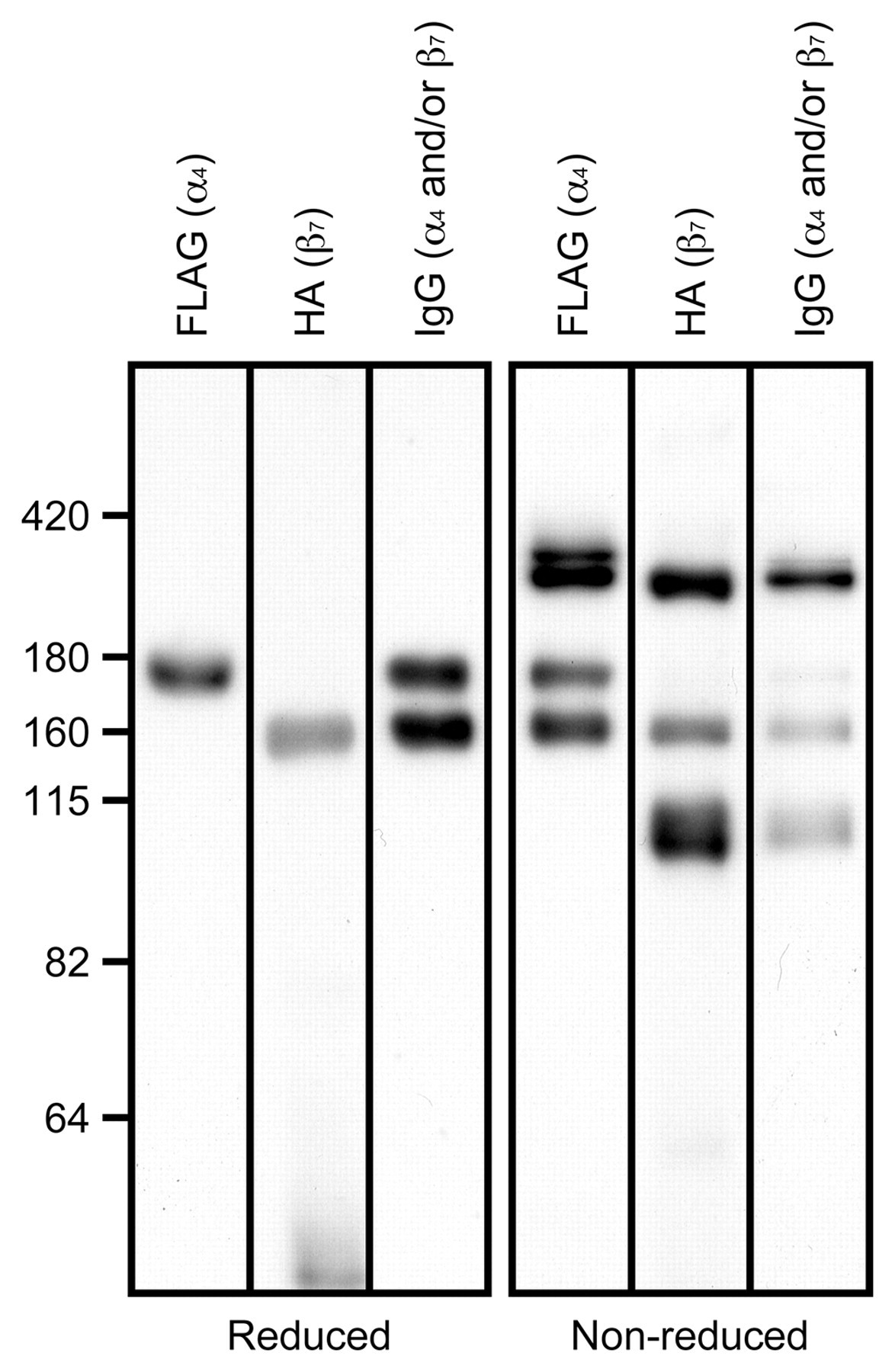
Expression and heterodimerization of α4•IgG-FLAG and β7•IgG-HA. Shown are immunoblots with respective anti-FLAG and anti-HA antibodies under reducing (left) and nonreducing (right) conditions. Under reducing conditions, blots show single bands migrating at 170 kDa and 160 kDa, corresponding to α4•IgG-FLAG and β7•IgG-HA, respectively, while the control anti-human IgG antibody detects equivalent levels of both bands. Under nonreducing conditions, in addition to bands seen under reducing conditions, a band migrating at 330 kDa is detected as an intense band following staining with anti-human IgG, anti-FLAG, or anti-HA. The number on the left vertical axis indicates molecular weight (kDa).
Under nonreducing conditions, chimeric proteins should remain dimerized due to disulfide bond formation at the IgG hinge (Stephens et al. 2000). As expected, in addition to bands seen under reducing conditions, a band migrating at 330 kDa was detected as the predominant band following immunoblotting with anti-human IgG, and this band was also detected with anti-FLAG and anti-HA antibodies (Fig. 2, right panel). The 330-kDa band detected by anti-HA antibody was slightly broader and smaller than those detected by anti-FLAG and anti-IgG antibodies. This band is most likely composed of two proteins: a 330-kDa α4β7•IgG heterodimer and a 320-kDa β7•IgG homodimer. As judged by immunoblotting with anti-human IgG, β7•IgG homodimers accounted for only a small fraction of this band; however, the number of HA epitope in a β7•IgG homodimer is twice as much as an α4β7•IgG heterodimer has; therefore, β7•IgG homodimers could be more strongly detected by anti-HA antibody compared to α4β7•IgG heterodimers, thus making these two bands seen as a single band with smaller molecular weight. In addition, a band migrating at 340 kDa detected by anti-FLAG and anti-human IgG but not by anti-HA and corresponding to α4•IgG homodimers was also detected. This homodimeric chimera also accounted for only a small fraction of IgG-positive protein as judged by immunoblotting with anti-human IgG. Additionally, a band migrating at 160 kDa detected by anti-FLAG and a band migrating at 90~100 kDa detected by anti-HA were also observed. Because these latter bands were not observed under reducing conditions, they likely represent anomalous forms of α4•IgG and β7•IgG. Overall, these findings indicate that transfection of HEK 293T cells with cDNAs encoding α4•IgG-FLAG and β7•IgG-HA results in formation of α4β7•IgG heterodimeric chimeras.
α4β7•IgG Functions as a Soluble Form of Integrin α4β7
To determine whether α4β7•IgG functions as a soluble form of integrin α4β7, CHO cells were transiently transfected with cDNAs encoding mouse MAdCAM-1, VCAM-1, and ICAM-1 and subjected to an α4β7•IgG binding assay. As shown in Figure 3, α4β7•IgG robustly bound to the membrane of CHO cells expressing MAdCAM-1 and, to a lesser extent, to cells expressing VCAM-1 but did not bind to cells expressing ICAM-1 or to mock-transfected control CHO cells. As shown in Figure 4, α4β7•IgG binding to CHO cells expressing MAdCAM-1 or VCAM-1, but not ICAM-1, as assessed by absorbance at 405 nm in a cell ELISA, was significantly higher than binding to mock-transfected control cells. Also, α4β7•IgG binding to CHO cells expressing MAdCAM-1 was higher than to any other CHO transfectants, with high statistical significance (p < 0.01 vs VCAM-1, p < 0.001 vs ICAM-1). Such binding was not observed in the presence of homodimeric chimeras α4•IgG or β7•IgG, expressed following single transfection of either pcDNA3.1/Hygro-α4•IgG or pcDNA3.1/Hygro-β7•IgG, respectively (data not shown). Moreover, α4β7•IgG binding to CHO cells expressing MAdCAM-1 and VCAM-1 was completely abolished in the presence of EDTA (Fig. 3, lower panels). These findings indicate that α4β7•IgG preferentially binds to MAdCAM-1 in a divalent cation–dependent manner and thus functions as a soluble form of integrin α4β7.
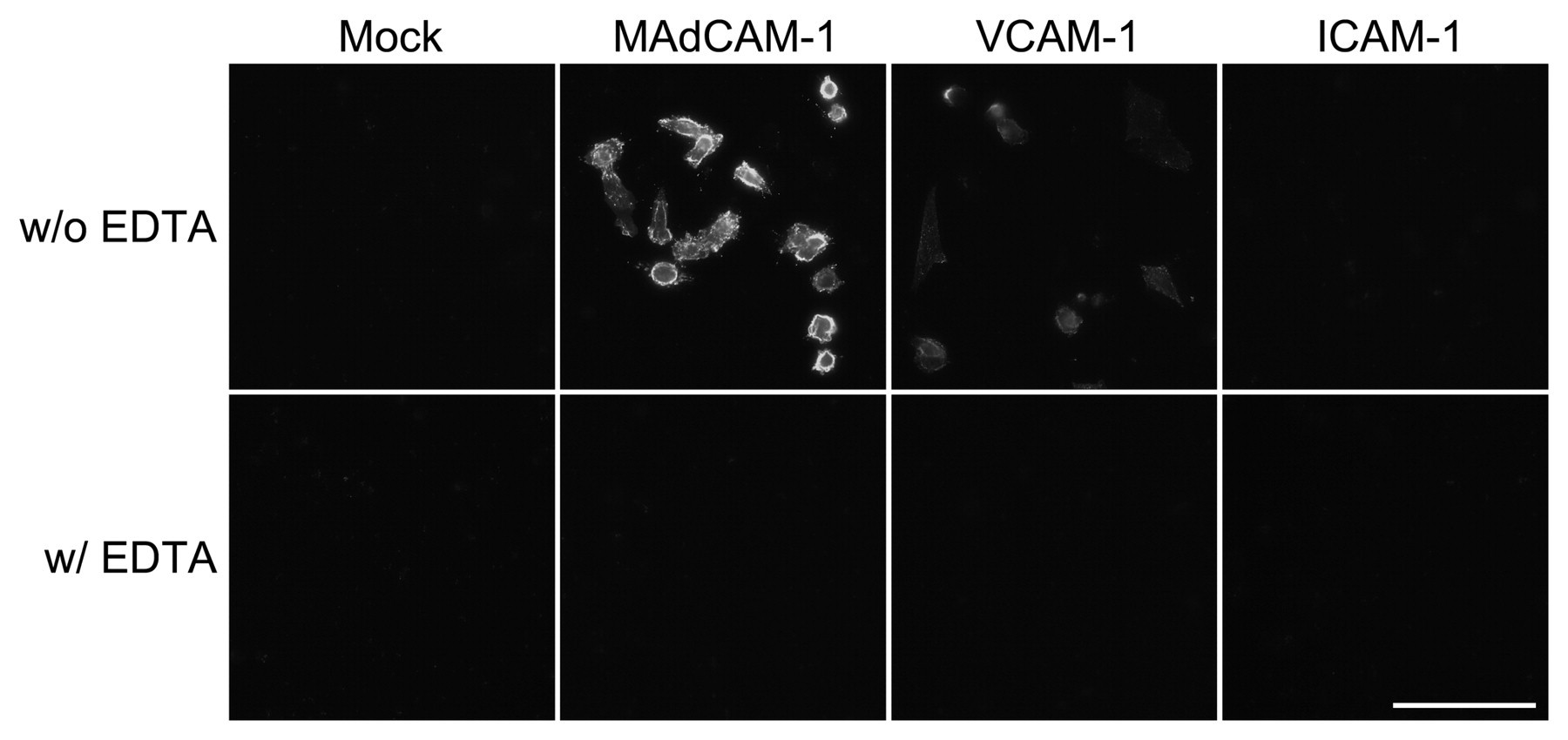
Binding characteristics of the recombinant α4β7•IgG heterodimeric chimera to MAdCAM-1, VCAM-1, and ICAM-1. CHO cells were transfected with pcDNA1.1-mMAdCAM-1, pcDNA1.1-mVCAM-1, pcDNA1.1-mICAM-1, or pcDNA1.1 (mock) and probed with α4β7•IgG, which was detected using Alexa Fluor 488–conjugated anti-human IgG. α4β7•IgG robustly binds to the membrane of CHO cells expressing MAdCAM-1 and, to a lesser extent, to those expressing VCAM-1 but does not bind to cells expressing ICAM-1 or to mock-transfected control cells. Such bindings are completely abolished in the presence of EDTA (w/ EDTA). Bar = 50 µm.
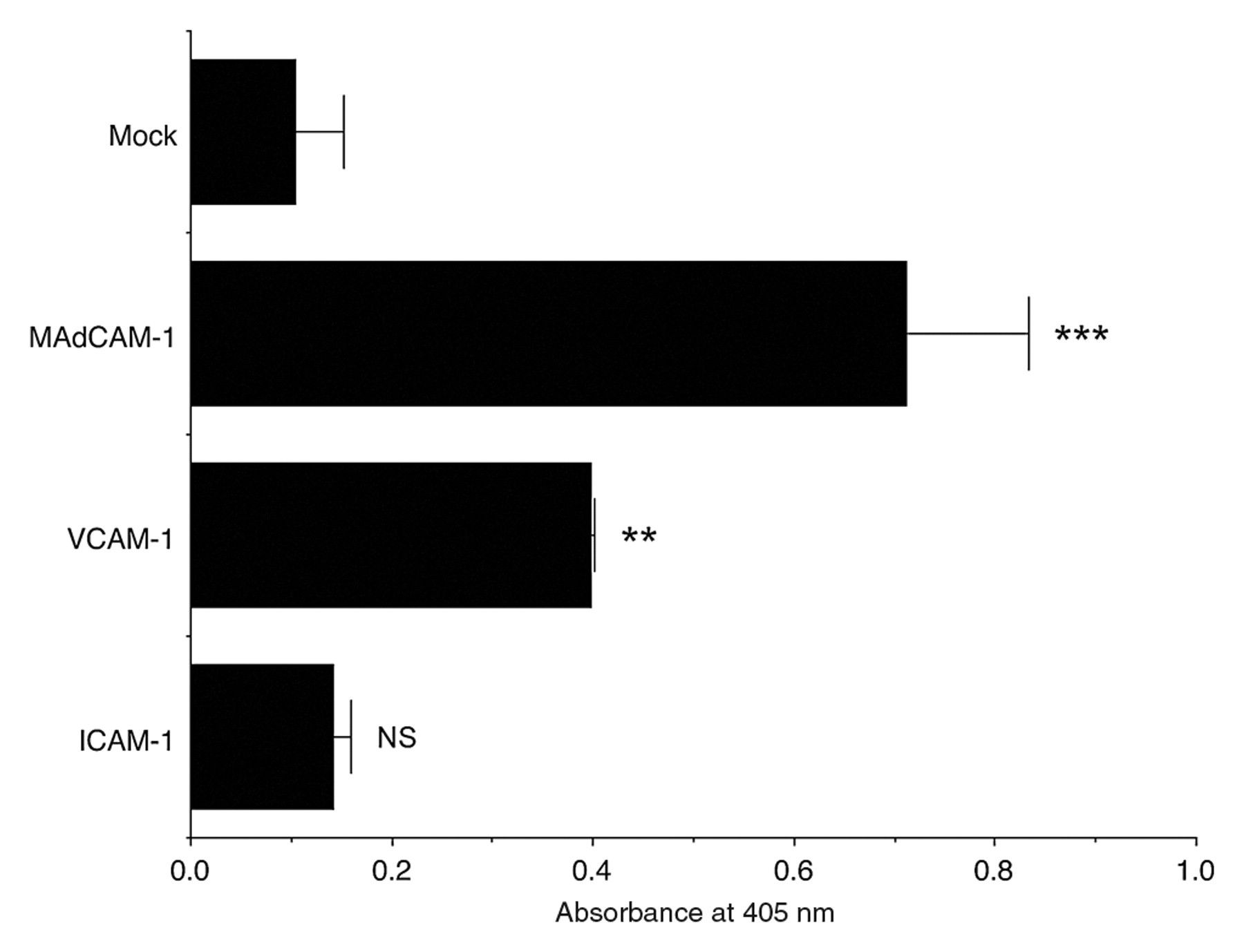
Binding affinity of recombinant α4β7•IgG heterodimeric chimeras to MAdCAM-1, VCAM-1, and ICAM-1. CHO cell transfectants shown in Figure 3 were subjected to cell ELISA for α4β7•IgG binding using HRP-conjugated anti-human IgG and its enzyme substrate. Product absorbance at 405 nm for each transfectant is shown. α4β7•IgG binding to CHO cells expressing MAdCAM-1 and VCAM-1, but not ICAM-1, is significantly higher than to mock-transfected control cells, and α4β7•IgG binding to CHO cells expressing MAdCAM-1 is higher than to any other CHO transfectants, with high statistical significance (p < 0.01 vs VCAM-1, p < 0.001 vs ICAM-1). Data are expressed as mean ± SD (n = 3 for each group). ***p < 0.001; **p < 0.01; NS = not significant (p > 0.05) versus mock-transfected CHO cells.
α4β7•IgG Binds to MAdCAM-1–Expressing HEVs in GALT In Situ
To determine whether α4β7•IgG binds to MAdCAM-1 expressed on HEVs in GALT in situ, α4β7•IgG binding to HEVs was assayed on mouse frozen tissue sections. As shown in Figure 5, α4β7•IgG bound to HEVs in PPs and MLNs, where MAdCAM-1 is expressed, but not to HEVs in PLNs, which lack MAdCAM-1 expression. α4β7•IgG binding to MAdCAM-1–expressing HEVs in GALT was completely abrogated in the presence of EDTA. These findings indicate that α4β7•IgG preferentially binds to MAdCAM-1–expressing HEVs in GALT in situ and can be used to stain MAdCAM-1–expressing HEVs in GALT.
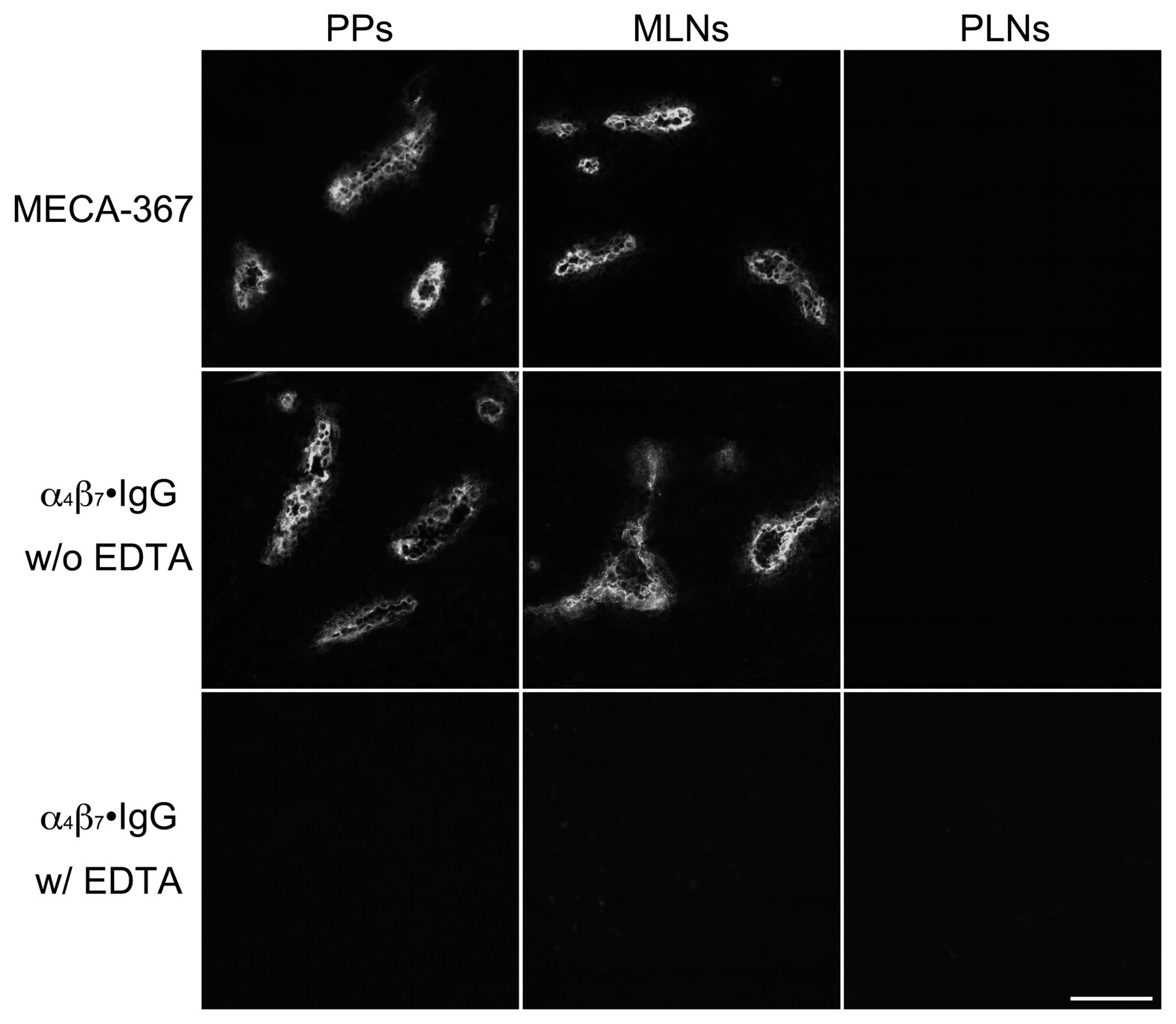
α4β7•IgG binding to mouse HEVs in situ. Mouse tissue sections of PPs, MLNs, and PLNs were probed with MECA-367 antibody, recognizing mouse MAdCAM-1 (upper panels) or α4β7•IgG in the presence (lower panels) or absence (middle panels) of EDTA. HEVs in PPs and MLNs express MAdCAM-1 as detected by MECA-367 antibody, while HEVs in PLNs do not. α4β7•IgG robustly binds to MAdCAM-1–expressing HEVs in PPs and MLNs in the presence of divalent cations (w/o EDTA). Such binding is completely abolished in the presence of EDTA (w/ EDTA). Bar = 100 µm.
α4β7•IgG Inhibits Lymphocyte Binding to MAdCAM-1
The above findings prompted us to determine whether α4β7•IgG inhibits binding of integrin α4β7–expressing lymphocytes to MAdCAM-1. To do so, we carried out a lymphocyte adhesion inhibition assay on CHO/mMAdCAM-1 cells. Prior to the assay, expression of integrin α4β7 as well as that of L-selectin on lymphocytes isolated from the spleen was confirmed by FACS analysis (Fig. 6). As shown in Figures 7 and 8, in the absence of the chimera, lymphocytes adhered robustly to CHO/mMAdCAM-1 cells, and such binding was completely abrogated with EDTA. By contrast, in the presence of the chimera, the number of lymphocytes attached per 100 CHO/mMAdCAM-1 cells was decreased, with high statistical significance compared to that seen in the absence of the chimera (p < 0.001). The number of lymphocytes attached was increased in a stepwise fashion when serially diluted α4β7•IgG was administered. These findings indicate that α4β7•IgG inhibits α4β7 integrin–expressing lymphocyte binding to MAdCAM-1 in a dose-dependent manner, most likely by competing with native α4β7 integrin expressed on lymphocytes.
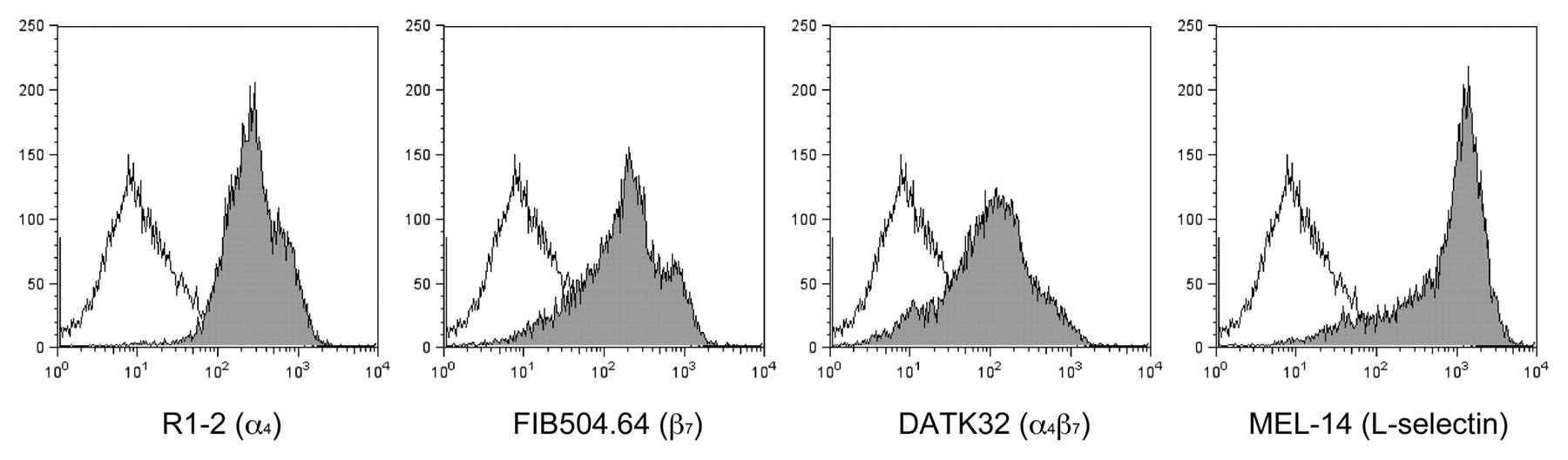
Expression of integrin α4β7 and L-selectin on lymphocytes. Lymphocytes isolated from the spleen were subjected to FACS analysis for expression of integrin α4 subunit (R1-2), β7 subunit (FIB504.64), α4β7 complex (DATK32), or L-selectin (MEL-14) (closed gray histograms). Open histograms show control experiments omitting the primary antibody. The x- and y-axes indicate fluorescence intensity and numbers of the events (cells), respectively.
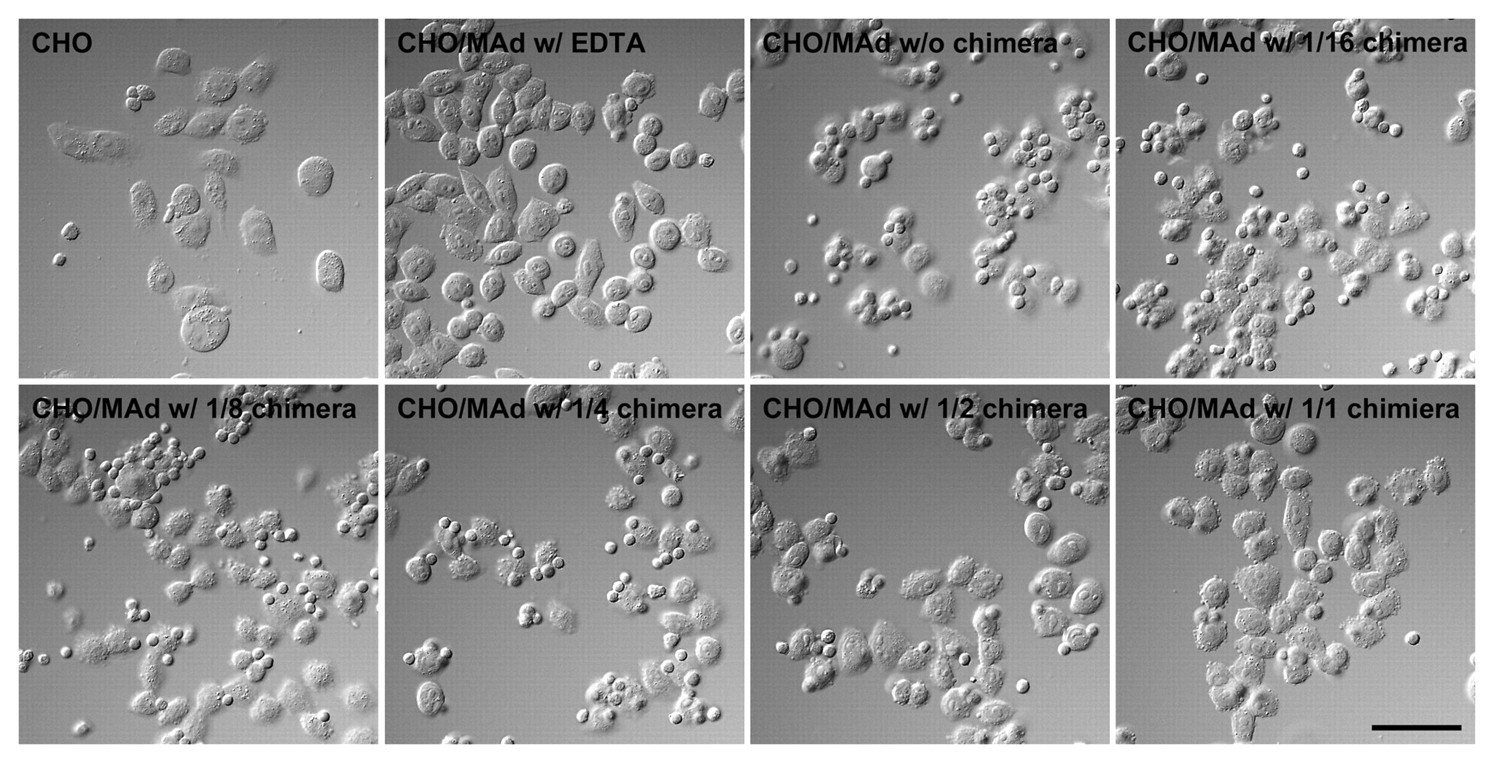
Lymphocyte adhesion to CHO/mMAdCAM-1 cells (CHO/MAd) in the presence (w/ chimera) or absence (w/o chimera) of the chimera, as observed by Nomarski differential interface optics. The chimera administered was serially diluted up to 1/16. Lymphocyte adhesion to CHO/MAd is inhibited by α4β7•IgG and is completely abolished in the presence of EDTA (w/ EDTA). Bar = 50 µm.
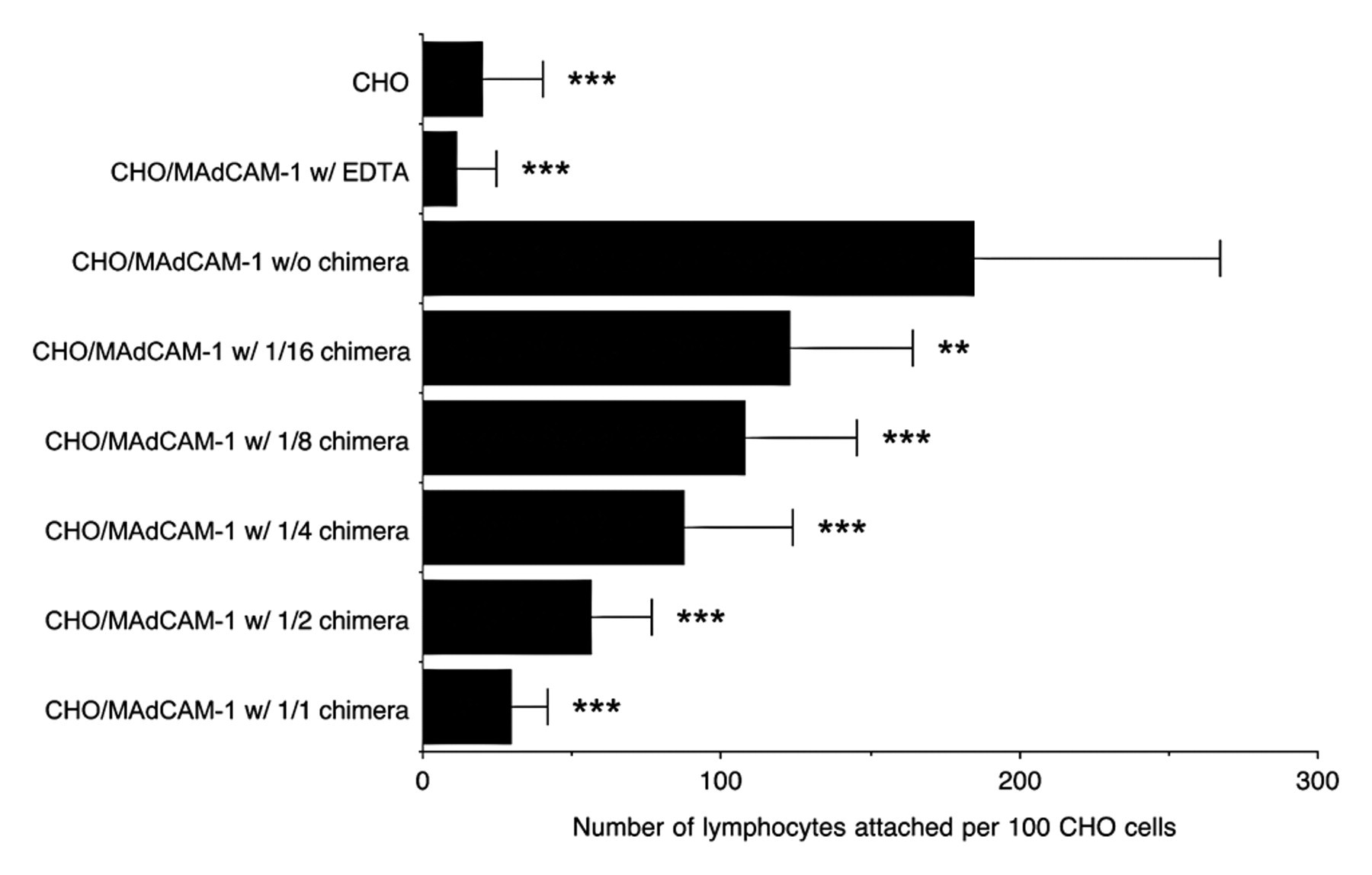
Quantification of data shown in Figure 7. The number of lymphocytes attached per 100 CHO cells was determined by examining five fields at ×200 magnification. Lymphocyte adhesion to CHO cells is inhibited by α4β7•IgG in a dose-dependent manner. Data are expressed as mean ± SD. ***p < 0.001; **p < 0.01 versus CHO/MAdCAM-1 in the absence of chimera.
α4β7•IgG Inhibits Lymphocyte Binding to HEVs in PPs and MLNs
We then employed a modified Stamper-Woodruff assay to further assess whether chimeric protein inhibits lymphocyte binding to HEVs in GALT. As shown in Figures 9 and 10, in the absence of the chimera, lymphocytes adhered on HEVs in PPs, MLNs, and, to a lesser extent, PLNs. On the other hand, in the presence of the chimera, the number of lymphocytes attached per HEV in PPs and MLNs was decreased, with high statistical significance compared to that seen in the absence of the chimera (p < 0.001 for both PPs and MLNs), while lymphocyte attachment to HEVs in PLNs was not altered significantly. These findings confirm that α4β7•IgG inhibits lymphocyte binding to HEVs in GALT, suggesting the potentiality of this chimeric protein as a new GALT-specific anti-inflammatory drug.
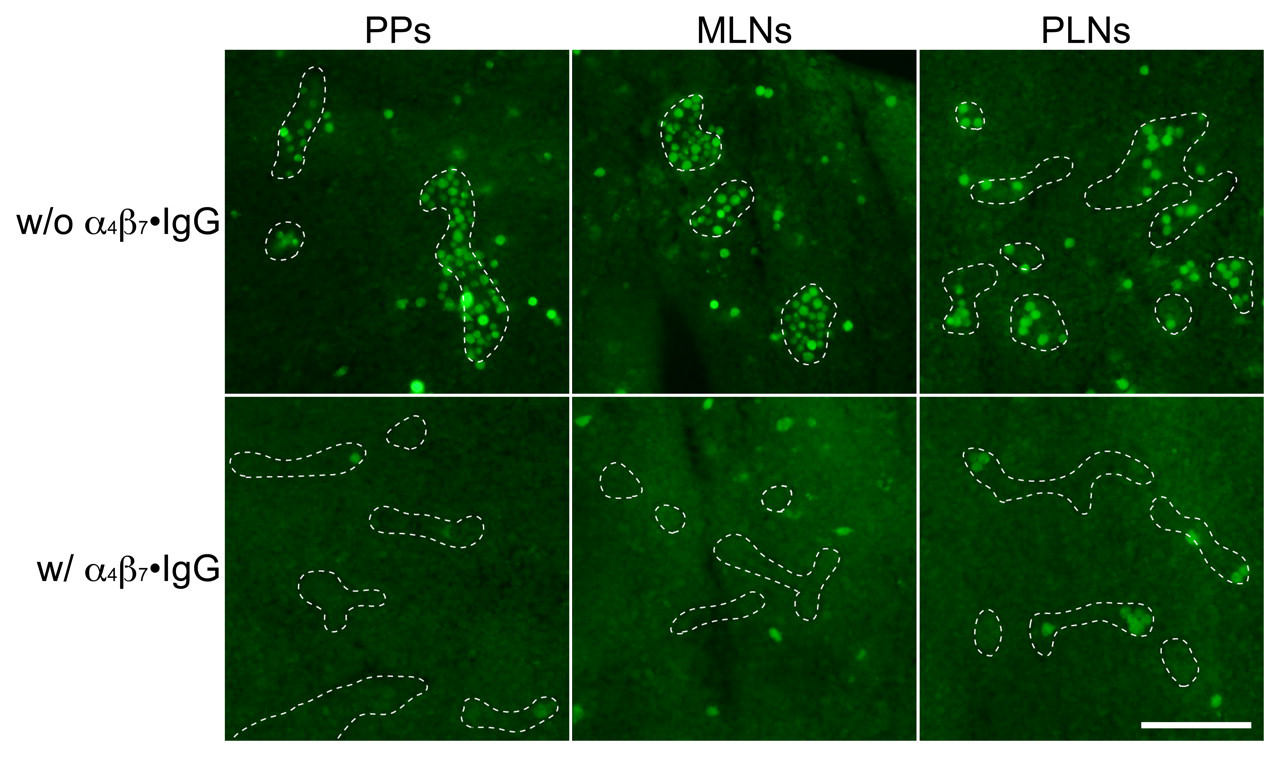
Lymphocyte adhesion to HEVs in PPs, MLNs, and PLNs in the presence (lower panels) or absence (upper panels) of the chimera, as detected by fluorescence originating from CMFDA-labeled lymphocytes. Lymphocyte adhesion to GALT HEVs is almost completely inhibited by α4β7•IgG. Dotted lines show outline of HEVs. Bar = 50 µm.
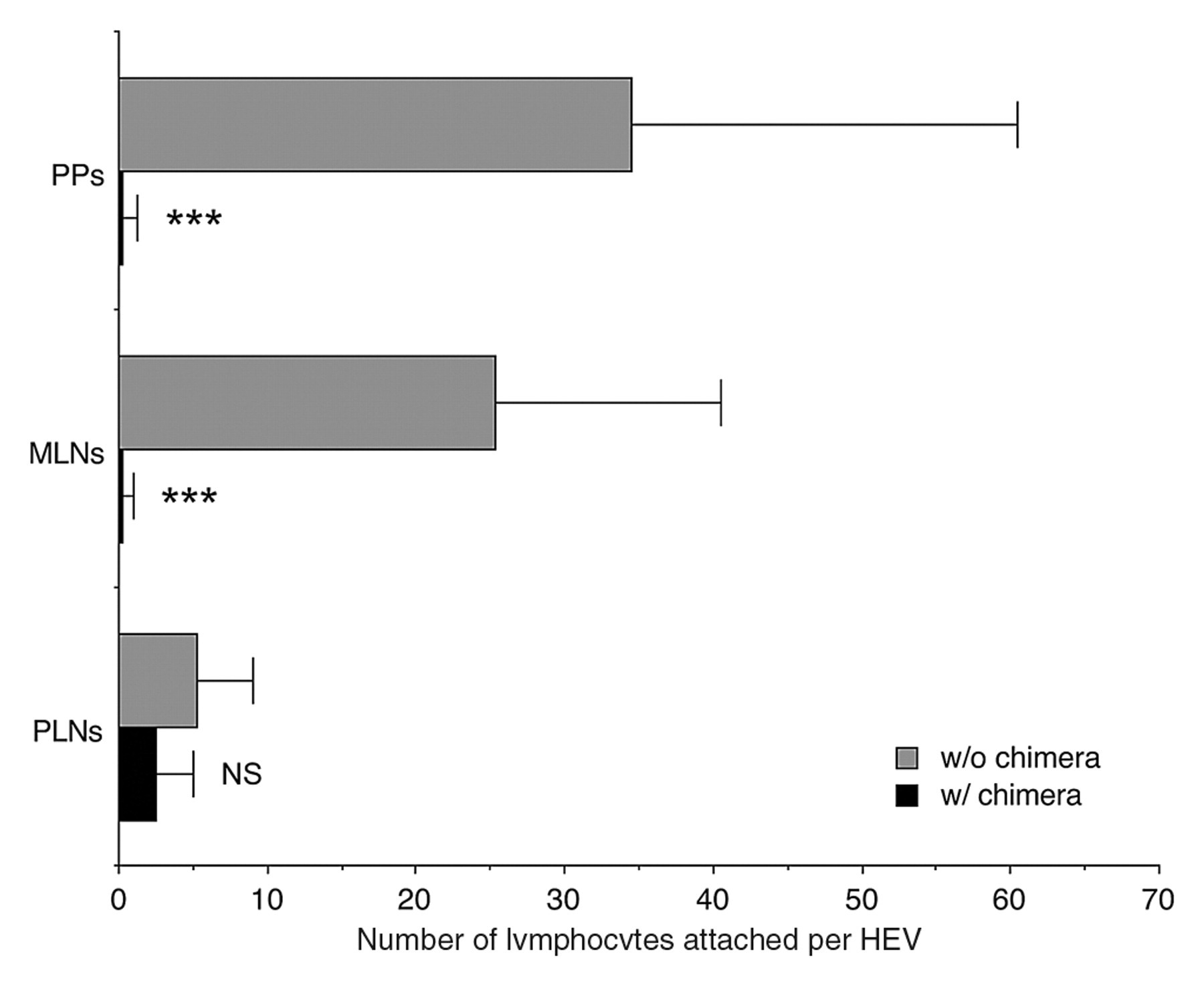
Quantification of data shown in Figure 9. The number of lymphocytes attached to HEV in the presence (w/) or absence (w/o) of the chimera was determined by counting the number of lymphocytes attached to 20 randomly chosen HEVs in each group. Data are expressed as mean ± SD (n = 20). ***p < 0.001; NS = not significant (p > 0.05) versus in the absence of chimera in each group.
Discussion
In the present study, we developed an antibody-like soluble integrin α4β7 heterodimeric IgG chimera by transfection of HEK 293T cells with a combination of cDNAs encoding the extracellular region of integrin α4 and β7 subunits, each connected to a human IgG constant region, utilizing the hinge region to drive heterodimer formation. This antibody-like heterodimeric molecule is functional because it preferentially binds to CHO cells expressing MAdCAM-1 in a divalent cation–dependent fashion. α4β7•IgG binds to CHO cells expressing VCAM-1 as well; however, binding affinity to VCAM-1 as assessed by cell ELISA is much less than affinity to MAdCAM-1, a finding consistent with previous reports (Ruegg et al. 1992). Furthermore, α4β7•IgG also bound to HEVs in frozen mouse tissue sections of PPs and MLNs, which express MAdCAM-1, in the presence of divalent cations. These findings indicate that α4β7•IgG is a useful probe for functional MAdCAM-1 expressed on HEVs in GALT.
Two decades ago, Watson et al. (1990) developed an L-selectin•IgG chimera as a probe for functional L-selectin ligand carbohydrates in lymph nodes. Since then, L-selectin•IgG and L-selectin•IgM chimeras have been used to analyze distribution of functional L-selectin ligands in N-acetylglucosamine-6-O-sulfotransferase 1 (GlcNAc6ST-1) and GlcNAc6ST-2 double-knockout mice (Kawashima et al. 2005; Uchimura et al. 2005) and core 1–extending β1,3-N-acetylglucosaminyl-transferase and core 2–branching β1,6-N-acetylglucosaminyl-transferase 1 double-knockout mice (Mitoma et al. 2007). Moreover, we also applied the L-selectin•IgM chimera for functional detection of PNAd induced in human pathological states such as chronic Helicobacter pylori gastritis (Kobayashi et al. 2004) and the active phase of UC (Suzawa et al. 2007). Using the α4β7•IgG developed here, we can now analyze distribution of functional MAdCAM-1–expressing HEVs in GALT as well as in intestinal lamina propria in cases of chronic gastrointestinal inflammatory diseases including IBD.
We also found that α4β7•IgG substantially blocks lymphocyte binding to HEVs in GALT but not those in PLNs, indicating this chimeric molecule antagonizes native integrin α4β7 expressed on lymphocytes. Here, we employed a modified Stamper-Woodruff assay, which required incubation of lymphocytes with mouse tissue sections at RT. An RT incubation temperature is reportedly optimal to preserve protein-protein interactions such as integrin α4β7 and MAdCAM-1, while a 4C incubation temperature is optimal to maintain protein-carbohydrate interactions, such as L-selectin and PNAd (Lewinsohn et al. 1987; Lamagna et al. 2005). Because we focused on integrin α4β7–MAdCAM-1 interactions, the lymphocyte binding inhibition assay was carried out at RT. The small number of lymphocytes observed attached to HEVs in PLNs in the absence of α4β7•IgG is likely due to this temperature condition.
In addition to the modified Stamper-Woodruff assay, we performed an in vivo homing inhibition assay as described previously (Mitoma et al. 2007); however, the chimeric protein did not significantly inhibit lymphocyte homing to either PPs or MLNs (data not shown), most likely due to low concentration of α4β7•IgG administered. Establishing large-scale α4β7•IgG preparation conditions is indispensable for further in vivo studies.
In conclusion, we developed a soluble integrin α4β7•IgG heterodimeric chimera that preferentially binds to MAdCAM-1 and can be used as a probe for functional MAdCAM-1 expressed on HEVs in GALT. Moreover, because α4β7•IgG inhibits lymphocyte binding to HEVs in GALT, this molecule could serve as an effective anti-inflammatory drug, inhibiting GALT-specific lymphocyte migration.
Footnotes
Acknowledgements
The authors thank Drs. Hiroto Kawashima, Kenji Uchimura, and Tomoya Akama for useful discussion; Ms. Kayo Suzuki for technical assistance; Dr. Tsutomu Nakada for superb illustration; and Dr. Elise Lamar for critical reading of the article.
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research and/or authorship of this article: This work was supported by a Grant-in-Aid to Young Scientists for Exploratory Research from Shinshu University (to MK) and in part by a Grant-in-Aid for Young Scientists B-22790343 from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to MK), Grants-in-Aid for Scientific Research C-20570140 (to JM) and B-21390104 (to JN) from the Japan Society for the Promotion of Science, and a grant PO1 CA71932 from the National Institutes of Health (to MF).