
Abstract
Neoplastic adrenocortical lesions are common in humans and several species of domestic animals. Although there are unanswered questions about the origin and evolution of adrenocortical neoplasms, analysis of human tumor specimens and animal models indicates that adrenocortical tumorigenesis involves both genetic and epigenetic alterations. Chromosomal changes accumulate during tumor progression, and aberrant telomere function is one of the key mechanisms underlying chromosome instability during this process. Epigenetic changes serve to expand the size of the uncommitted adrenal progenitor population, modulate their phenotypic plasticity (i.e., responsiveness to extracellular signals), and increase the likelihood of subsequent genetic alterations. Analyses of heritable and spontaneous types of human adrenocortical tumors documented alterations in either cell surface receptors or their downstream effectors that impact neoplastic transformation. Many of the mutations associated with benign human adrenocortical tumors result in dysregulated cyclic adenosine monophosphate signaling, whereas key factors and/or signaling pathways associated with adrenocortical carcinomas include dysregulated expression of the IGF2 gene cluster, activation of the Wnt/β-catenin pathway, and inactivation of the p53 tumor suppressor. A better understanding of the factors and signaling pathways involved in adrenal tumorigenesis is necessary to develop targeted pharmacologic and genetic therapies.
Introduction
Adrenocortical neoplasms are common in humans and domestic animals. Postmortem studies showed that approximately 5% of people over the age of 50 years have at least one grossly visible adrenocortical nodule. 25 The vast majority of these “incidentalomas” are nonfunctioning adenomas, but others secrete steroid hormones that cause Cushing syndrome or other complications. 11 Adrenocortical carcinoma (ACC) is rare (approximately 1 case per million people per year), but it carries a grim prognosis because of its propensity to metastasize before detection. 7, 150 The overall frequency of adrenocortical neoplasia in dogs is similar to that in humans, although dogs are more prone to cortisol-secreting adrenal lesions. 49, 116 The frequency of functional adrenocortical neoplasms is even higher in certain gonadectomized animals, such as goats, ferrets, hamsters, and mice (Table 1). 132 These gonadectomy-induced adrenocortical tumors, which are more often benign than malignant, may produce ectopic sex steroids that cause significant morbidity.
Frequency of adrenocortical neoplasia in different species.
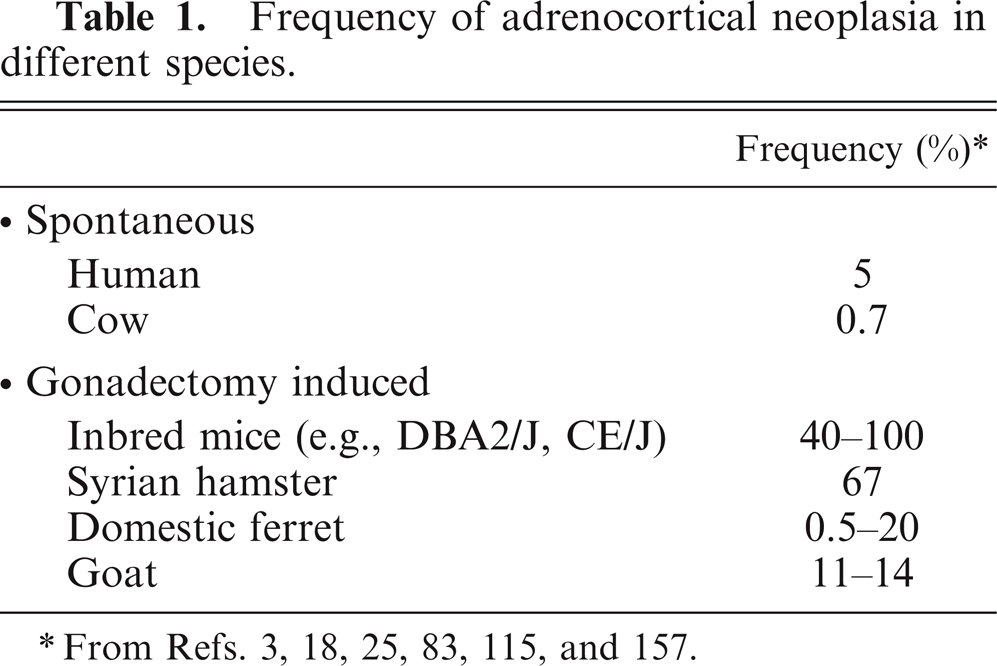
From Refs. 3, 18, 25, 83, 115, and 157.
The factors that account for the frequent occurrence of benign adrenocortical neoplasms and the low rate of ACC have been the subject of intense investigation over the past decade. 15, 54, 150 Recent advances in our understanding of the molecular pathogenesis of benign and malignant adrenocortical tumors have come from genetic analysis of sporadic and familial human adrenocortical tumors and from studies of naturally occurring and genetically engineered animal models. 7, 18, 72, 118, 145 In this article, we review the genetic and epigenetic events involved in adrenocortical tumorigenesis in humans and domestic animals, and we discuss the relevant animal models.
Development of the Adrenal Cortex
The adrenal cortex and gonads are major sites of steroid production. Steroidogenic cells in the adrenal cortex and the gonads appear to arise from a common pool of mesodermal progenitors in the urogenital ridge. 79 During mammalian embryogenesis, progenitors fated to become adrenocortical cells associate with neural crest derivatives that will give rise to the adrenal medulla; cells destined to become gonadal stroma associate with primordial germ cells. Mutations in steroidogenic factor-1 (Sf1) and Wilms tumor-1 (Wt1), 2 transcription factor genes expressed in the urogenital ridge, disrupt development of both adrenal and gonadal steroidogenic cells, underscoring the close relationship between these lineages. 79, 144
In early human gestation, the fetal adrenal cortex consists of a large inner layer, termed the fetal zone and a thin outer rim of more immature cells, known as the definitive zone. 42 The principal function of the fetal zone is to produce the adrenal androgens, dehydroepiandrosterone (DHEA) and DHEA sulfate (DHEA-S), which are metabolized by the placenta into estrogens that serve to maintain pregnancy. 124 Accordingly, the fetal zone expresses enzymes and allosteric regulators required for androgen production, including cytochrome P450 side-chain cleavage (P450scc), cytochrome P450 17α-hydroxylase 17,20-lyase (P450c17), cytochrome b 5 (cyt b 5), and the steroid sulfotransferase SULT2A1 (Fig. 1). 124 3β-Hydroxysteroid dehydrogenase type 2 (HSD3β2), an enzyme required for synthesis of glucocorticoids and mineralocorticoids, is transiently expressed in the fetal zone from weeks 7 to 12 of gestation and serves to safeguard against virilization of the female genital anlage. 58 After birth, the fetal zone regresses, and the production of DHEA and DHEA-S ceases. 8 The definitive zone of the human adrenal cortex begins to partition into anatomically and functionally distinct compartments: the zona glomerulosa (zG), zona fasciculata (zF), and zona reticularis (zR). 42 Cells in the zG express HSD3β2 but lack P450c17 activity and consequently produce mineralocorticoids. zF cells express HSD3β2 and possess the P450c17 17α-hydroxylase activity but not P450c17 17,20-lyase activity and, therefore, produce cortisol. This absence of 17,20-lyase activity in the zF has been attributed, in part, to a lack of expression of its allosteric regulator, cyt b 5. 5, 153 Adrenal production of DHEA and DHEA-S resumes at adrenarche (ages 6–8 years) and localizes to the zR. 5
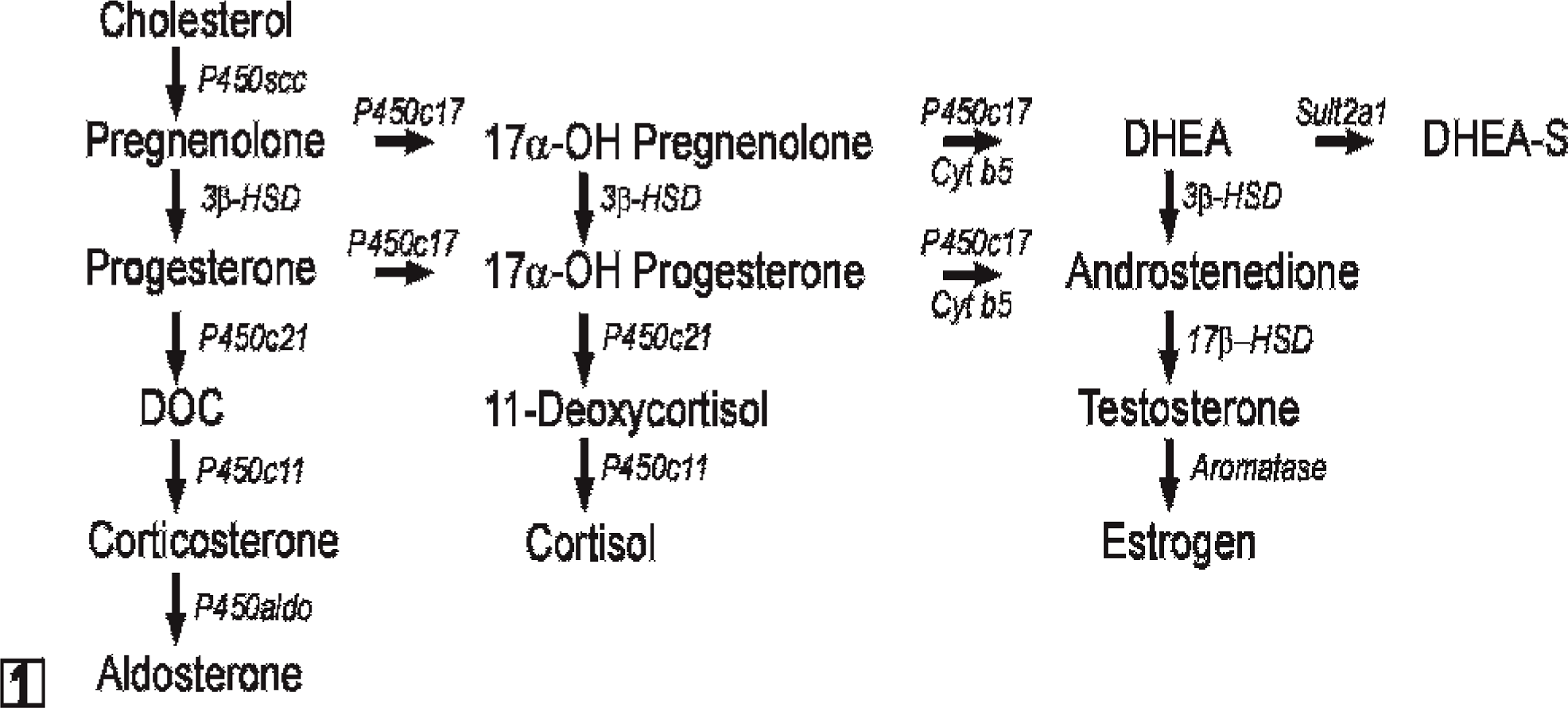
Steroid hormone biosynthetic pathways. All steroidogenic cells share the capacity to mobilize and cleave cholesterol. The repertoire of enzymes distal to P450scc determines the steroidogenic capacity of a given cell. Note that P450c17 has both 17α-hydroxylase and 17,20-lyase activities. Cyt b 5 selectively enhances the 17,20-lyase activity of P450c17 through allosteric effects. Abbreviations: 3β-HSD, 3β-hydroxysteroid dehydrogenase; 17β-HSD, 17β-hydroxysteroid dehydrogenase; cyt b 5, cytochrome b 5; DOC, deoxycorticosterone; DHEA, dehydroepiandrosterone; P450aldo, aldosterone synthase; P450c11, cytochrome P450 11β-hydroxylase; P450c17, cytochrome P450 17α-hydroxylase/17,20-lyase; P450c21, cytochrome P450 21-hydroxylase.
The adrenal cortex of most domestic animals resembles that of humans. Two less-prominent layers, the zona intermedia and zona juxtamedullaris, are also found in the adrenal cortex of the ferret and other carnivores; the functional significance of these layers is unclear. 70 Cortisol is the principal glucocorticoid secreted by the adrenal cortex of most domestic animals, although the adrenal glands of some species produce significant amounts of corticosterone. 55, 158
In contrast to humans, zonation of the adrenal cortex in the mouse is completed by birth. 79 The zG and zF are well defined in the mouse adrenal, but there is no discernible zR. The adrenal cortex of the young mouse contains an additional layer, termed the X-zone, which is adjacent to the medulla and regresses at puberty in males and during the first pregnancy in females. 79 The function and steroidogenic potential of the X-zone are unknown, although recent evidence indicates that it may be involved in progesterone catabolism. 67 Transgenic expression of LacZ driven by a specific Sf1 fetal enhancer element suggests that the X-zone is a remnant of the adrenal primordia that forms before the definitive adrenal cortex. 165 Unlike most other mammals, adrenocortical cells in the adult mouse lack P450c17, consequently, corticosterone is the principal glucocorticoid secreted by the mouse adrenal cortex, and, under physiologic conditions, androgenic steroids are not produced in this tissue. 81
Growth and Differentiation of Stem/Progenitor Cells in the Adrenal Cortex
In all mammals, the definitive adrenal cortex is a dynamic organ in which steroidogenic cells undergo constant turnover. Cells in each zone of the adult adrenal cortex are theorized to be derived from a common set of stem/progenitor cells in the subcapsular region. 18, 84, 128 These progenitors divide and give rise to daughter cells that differentiate, migrate centripetally, gain zone-specific characteristics, and replenish senescing cells. 84 As a result, the adrenal cortex is arranged in radial cords of clonal cells that extend from the zG to the zR. 113 This cell migration model is supported by experimental evidence, although alternative theories, such as the existence of undifferentiated stem cells exist within each zone, have been proposed to account for the constant renewal and zonal specification of the adrenal cortex. 82
The differentiation, growth, and survival of adrenocortical stem cells and their progeny are influenced by a diverse array of hormones and autocrine factors, including adrenocorticotropic hormone (ACTH), angiotensin-II, and members of the insulin-related growth factor (IGF) family. 82, 127 Endocrine hormones and paracrine factors traditionally associated with the function of gonadal steroidogenic cells, such as luteinizing hormone (LH) and members of the transforming growth factor-β (TGFβ) superfamily (e.g., TGFβ, activin, and inhibin), also affect the differentiation, proliferation, and function of cells in the adrenal cortex, both in physiologic 12 and pathophysiologic 13 states.
The molecular characteristics of quiescent subcapsular stem cells are unknown. However, differentiation markers characteristic of their descendants have been identified (Fig. 2). 18, 84 Proliferating early progenitors express SF1, a transcription factor that promotes cell growth, limits apoptosis, and activates a wide array of genes involved in both adrenal and gonadal steroidogenesis. 39, 50, 120 Sf1−/− mouse embryos exhibit aberrant adrenal and gonadal development, 164 and SF1 haploinsufficiency in mice impairs the compensatory adrenocortical cell growth that follows unilateral adrenalectomy. 14 In humans, heterozygous SF1 mutations cause adrenal insufficiency and male-to-female sex reversal. 1 Subcapsular progenitors that express Sf1 have limited steroidogenic capacity because of the SF1-dependent expression of Dax1, an X-linked gene that encodes a repressor of steroidogenic gene expression. 78, 95, 162 In response to ACTH, subcapsular progenitors downregulate Dax1 and cells differentiate into corticoid-producing cells that express GATA6, a transcription factor that acts in synergy with SF1 and other factors to enhance the expression of genes involved in corticoid biosynthesis. 149 DAX1 deficiency in humans and mice leads to excessive differentiation of subcapsular progenitors and eventual depletion of the stem/progenitor cell compartment. 1 Consequently, DAX1 deficient males may exhibit transient hypercorticism when young, followed by progressive and irreversible hypocorticism. 1 On histologic examination, the adrenals of older DAX1 deficient individuals are characterized by a disorganized steroidogenic cortex that contains cytomegalic cells. 61
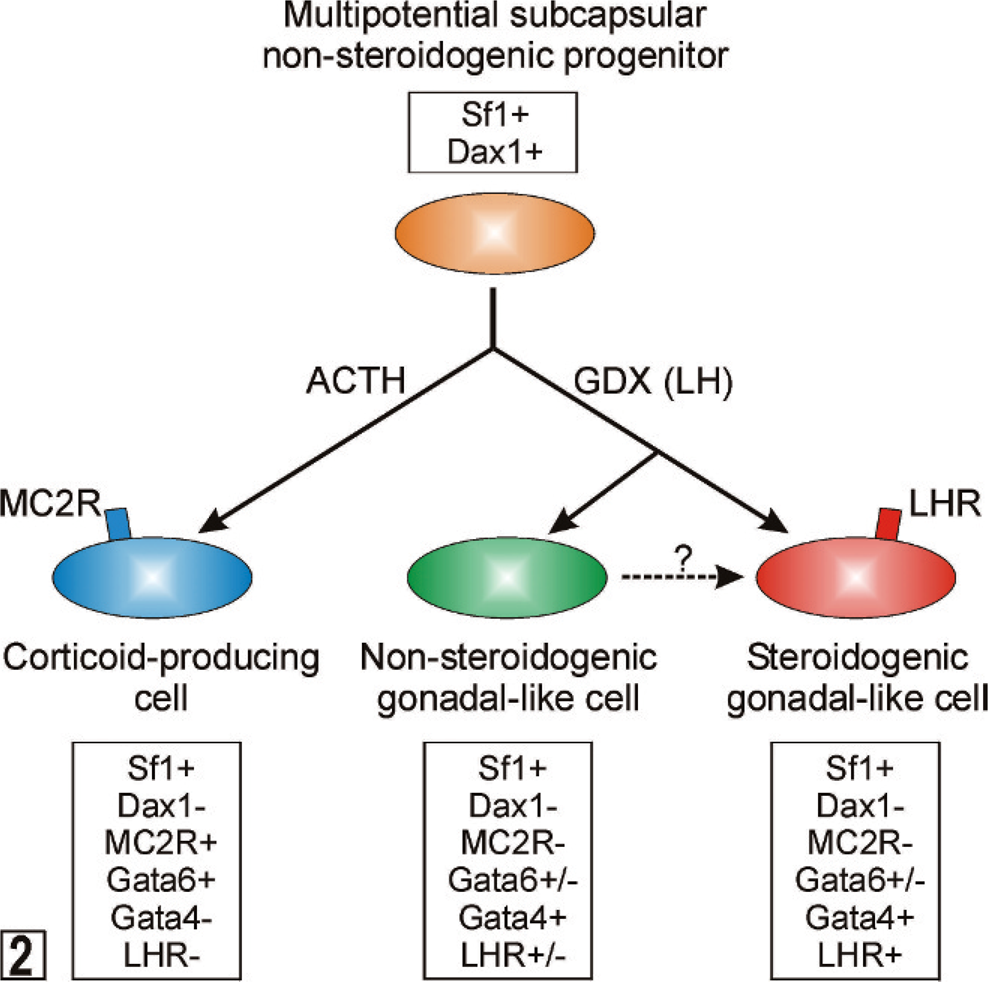
Differentiation of multipotential subcapsular adrenocortical progenitors into corticoid-producing or gonadal-like cells. See the accompanying text for details. ACTH = adrenocorticotropic hormone; GDX = gonadectomy; LH = luteinizing hormone; LHR = LH receptor; MC2R = melanocortin-2 receptor (ACTH receptor); Sf1 = steroidogenic factor 1.
In a proper hormonal microenvironment, the multipotential stem/progenitor cells in the subcapsular region of the adrenal can differentiate into sex steroidogenic cells that resemble gonadal stroma. 18 In response to the hormone changes that accompany gonadectomy (increased LH, decreased inhibin, etc.), SF1-positive subcapsular progenitors downregulate Dax1 and upregulate GATA4, a transcription factor that drives expression of P450c17, and other genes involved in sex steroidogenesis (Fig. 2). 149
Subcapsular stem/progenitor cells are hypothesized to be the principal targets of neoplastic transformation in the adrenal cortex, 18, 39, 84 although it is possible that the adrenal cortex harbors other stem cell populations that undergo malignant transformation. Adrenocortical tumors that arise in young children usually express markers typical for the fetal zone and produce DHEA-S, which led investigators to hypothesize that these tumors arise from a stem cell in this zone. 156 Similarly, certain genetically engineered mouse strains develop adrenocortical tumors that appear to arise from progenitors in the X-zone. 18, 118 Efforts to purify stem-like cells from one or more of these anatomical sites within the adrenal cortex have met with limited success. 104
Conceptual Models for the Pathogenesis of Spontaneous Adrenocortical Neoplasia
Based on recent genetic and epigenetic data, two conceptual frameworks have been put forward to explain the origin and/or evolution of adrenocortical tumors: 1) the multistep, or clonal, genetic model and 2) the epigenetic progenitor model. These models are complementary rather than mutually exclusive. As discussed below and summarized in Table 2, most cases of adrenocortical tumorigenesis reflect a combination of clonal genetic changes and epigenetic alterations.
Evidence in support of 2 conceptual models of adrenocortical neoplasia.

The multistep (clonal) genetic model
X-chromosome inactivation studies showed that hyperplastic adrenal tissue usually consists of a polyclonal population of cells, whereas most adrenocortical adenomas (ACA) and all carcinomas consist of a monoclonal population of cells. 16, 20, 89 On the basis of these observations, it was proposed that adrenocortical tumorigenesis is a multistep process in which the initial event is the growth of a polyclonal neoplasm in response to activation of paracrine or endocrine signaling pathways. 101 Increased cell proliferation enhances the susceptibility of cells to oncogene or tumor suppressor gene mutations, which leads to the accumulation of specific genetic changes characteristic of these tumors. The clonal genetic model posits that these changes result in the sequential progression from normal cells to benign lesions and eventually to cancer. 34
Comparative genomic hybridization studies showed a positive correlation between adrenocortical tumor size and the number of chromosomal alterations, which supports the premise that chromosomal changes accumulate during tumor progression. 102, 131 Chromosomal amplifications and/or deletions are present in 28–61% of benign human adrenocortical neoplasias, including cortisol-producing nodular hyperplasias (Table 3). 68, 88, 131, 145 However, the average number of chromosomal aberrations per tumor cell is low in benign lesions. 145 The most common chromosomal gain is 9q (14%), and the most common loss is 1p (5%). 145 Similar chromosomal abnormalities have been detected in cases of adrenocortical hyperplasia and ACA (e.g., a gain of chromosome 17q), which suggests that hyperplastic lesions may evolve into adenomas. 145, 163
Genetic changes in benign human adrenocortical neoplasms.

In contrast to the limited chromosomal amplifications and/or deletions seen in benign adrenocortical tumors, widespread chromosomal alterations are evident in nearly all ACCs (Table 4). 88, 131, 145 The most common changes in human ACC are gains that affect chromosomes 4, 5, 12, and 19, and losses on chromosomes 1, 2, 11, 17, and 22. 74 Aneuploidy often accompanies these widespread chromosomal imbalances. 20 In addition to deletions or amplifications, a high percentage of ACC cells exhibit loss of heterozygosity (LOH) at key loci. 56, 90 Collectively, these findings support a multistep model of adrenocortical tumorigenesis in which genomic instability begins in benign lesions and progresses during malignant transformation. At present, however, there is no definitive evidence to either prove or refute the premise that ACAs evolve into ACC. 145
Genetic changes in malignant human adrenocortical neoplasms.

The epigenetic progenitor model
Epigenetic alterations occur in cancer cells as commonly as genetic mutations and can mimic the effects of the latter. 46, 47 The term epigenetics refers to non–sequence-based modifications of DNA or DNA-associated factors (e.g., histones) that are maintained during cell division. One such epigenetic alteration is global DNA hypomethylation, a hallmark of both benign and malignant tumors. 151 The consequences of widespread hypomethylation include increased expression of oncogenes, chromosome instability, and LOH. 151 Other epigenetic alterations associated with tumorigenesis include hyper- or hypomethylation of specific genes, chromatin modification, and loss of imprinting (LOI). 46, 47 Epigenetic alterations are thought to be central to tumorigenesis in many tissues. Indeed, it has been suggested that tumors would neither arise nor progress without a disruption of normal epigenetic programs. 45
Epigenetic alterations could be the consequence of tumor progression, but recent data suggest an alternative explanation, namely, that epigenetic alterations precede tumor formation and increase the probability of cancer when genetic changes arise. 46, 47, 151 According to the epigenetic progenitor model, the first step in tumorigenesis is epigenetic disruption of stem/progenitor cells in a tissue, which alters the normal balance between undifferentiated stem/progenitor cells and their progeny, which leads to polyclonal proliferation of “neoplasia-ready” progenitor cells. 46, 47
A set of “tumor-progenitor” genes is hypothesized to promote epigenetic disruption of stem/progenitor cells. 46, 47 The normal function of these genes might be to promote “stemness,” the tendency toward pluripotency and replication over the tendency toward differentiation and limited replication. 46, 47 An example of a tumor-progenitor gene is IGF2. In intestinal epithelium, LOI at the IGF2 locus leads to increased IGF-II expression, which, in turn, promotes expansion the stem/progenitor cell compartment and increases the probability of neoplasia. 77 Of note, IGF2 is the most commonly overexpressed gene in human ACC. 57, 102 This suggests that, in adrenocortical progenitors, as in intestinal epithelial progenitors, dysregulated expression of IGF2 may increase tumor risk by expanding the target-cell population and/or modulating the effect of subsequent genetic alterations on these cells.
In addition to expanding the progenitor cell compartment, preexisting epigenetic alterations are thought to impact phenotypic plasticity, the ability of stem/progenitor cells to change their behavior in response to hormones and other environmental cues. 46 Epigenetic alteration of adrenal progenitor cells is followed by mutation of tumor suppressor genes (e.g., p53), which enhances genetic instability and increases the likelihood of additional mutations. If progressive, these changes can culminate in a malignant phenotype. In summary, this model proposes that epigenetic changes may contribute to adrenocortical tumorigenesis by modulating the size of the stem/progenitor population, altering phenotypic plasticity, and enhancing sensitivity to subsequent mutations.
Gonadectomy-induced Adrenocortical Neoplasia as a Model of Epigenetically Influenced Tumorigenesis
Approximately 60 years ago, researchers first noted that gonadectomy causes cells in the subcapsular region of the mouse adrenal cortex to transform into sex-steroid–producing cells that are histologically and functionally similar to gonadal tissue. 48 Subsequent experiments established that gonadectomy-induced adrenocortical neoplasia is highly strain dependent. 18 The most susceptible strain, CE/J, develops adrenocortical carcinomas, whereas DBA/2J, C3H, and NU/J mice develop adenomas. Other strains, including C57BL/6J and FVB/N, are resistant to gonadectomy-induced adrenocortical neoplasia. This process is believed to represent metaplasia of cells in the adrenal cortex, which, under the influence of continuous gonadotropin stimulation, transform into tissue that resembles gonadal stroma. This transformation is accompanied by ectopic expression of GATA4, a sex steroidogenic transcription factor that is normally absent from the adrenal cortex of adult mice. 18 Like other classic examples of metaplasia (e.g., Barrett's esophagus), gonadectomy-induced adrenocortical lesions arise in a self-renewing epithelium, are driven by chronic hormonal stimulation or tissue injury, and have the potential to evolve into frank neoplasia. 143 Gonadectomy-induced adrenocortical neoplasia is not limited to mice; sex-steroid–producing adrenocortical tumors are common in gonadectomized ferrets, hamsters, and goats (Table 1). 18
Gonadectomy-induced adrenocortical neoplasia may be viewed as an example of epigenetically triggered tumorigenesis. Stem/progenitor cells in the adrenal cortex are subject to epigenetic alteration, even in the absence of tumor formation, as illustrated by studies of mice that harbor a β-galactosidase transgene driven either by a P450c21 promoter 113 or a P450scc promoter. 73 X-gal staining of the adrenal glands from these mice demonstrates radial columns of cells in the cortex that either express or do not express β-galactosidase, which reflect random epigenetic activation (or silencing) of the transgene in subcapsular stem/progenitor cells. Each of these columns of cells is presumed to be derived from a distinct, epigenetically defined stem/progenitor cell. In cases of gonadectomy-induced adrenocortical neoplasia, preexisting epigenetic alterations might impact the phenotypic plasticity of stem/progenitor cells, which allows them to respond to the hormonal changes associated with gonadectomy. This may explain why gonadectomy leads to discrete cords of proliferating cells in the adrenal cortex.
The rapidly expanding database of genomic DNA sequences and single nucleotide polymorphisms in various strains of mice afford a means to characterize the alleles and genetic modifiers that influence gonadectomy-induced adrenocortical neoplasia (i.e., the “tumor-progenitor” genes is that promote epigenetic disruption of stem/progenitor cells). Linkage analysis of crosses between a susceptible (DBA/2J) and a nonsusceptible (C57BL/6J) strain of mice has shown that postgonadectomy tumorigenesis in DBA/2J mice is a dominant trait and that a major locus for tumorigenesis resides on chromosome 8. 9 One of the candidate genes in the linkage region is secreted Frizzled-related Protein-1 (sFRP1), a tumor suppressor that inhibits the Wnt/β-catenin signaling pathway (see below). 9 Although Sfrp1 is an attractive candidate gene for tumorigenesis, no causal mutations have been identified in the coding or noncoding regions of the gene in DBA/2J mice. Remarkably, a significant proportion of nonaffected F2 animals carry the susceptible DBA/2J allele on chromosome 8, and epistasis analysis suggests that multiple interacting genetic loci contribute to the phenotype. More than 30% of tumorigenic F2 animals were homozygous for the nonsusceptible C57BL/6J allele on chromosome 8, which indicates that other genes were involved in tumorigenesis. Unbiased genetic approaches such as this should yield valuable insights into the pathogenesis of adrenocortical neoplasia.
Role of Telomeres in Adrenocortical Tumorigenesis
Telomeres, the ends of chromosomes, are specialized nucleoprotein structures that consist of tandem arrays of G-rich repetitive DNA sequences bound to accessory proteins. 59 Telomere-associated proteins protect the ends of chromosomes and also function to regulate telomerase, the enzyme responsible for the addition of the G-rich DNA repeats. Telomerase is a ribonucleoprotein complex that comprises a reverse transcriptase (TERT), an RNA template (TERC), and other proteins. 32, 147 Most somatic cells, including adrenocortical cells, lack telomerase activity, so telomeres shorten by 50 to 200 base pairs with each cell division because of the end replication problem, the inability of DNA polymerase to fully replicate the lagging DNA strand. Telomere dysfunction, owing to progressive shortening and/or loss of telomere-associated proteins, triggers a tumor suppressor p53-dependent DNA damage response that results in cell-cycle arrest, replicative senescence, or apoptosis, eventually leading to impaired tissue function. 148 Telomere extension by telomerase is essential for normal stem/progenitor cell function; proper telomere maintenance ensures that stem cells can divide an unlimited number of times and avoid replicative senescence.
Besides its well-documented role in telomere maintenance, one of the components of the telomerase complex, TERT, appears to directly enhance the entry of quiescent stem cells into the cell cycle. 31, 53 This noncanonical function of TERT does not require TERC (i.e., it is independent of reverse transcriptase activity). 31 TERT binds Brg-1, a chromatin remodeling protein that interacts with and activates T-cell factor/lymphoid enhancer factor (TCF/LEF), a key downstream mediator of Wnt/β-catenin signaling (see below). 31 Thus, TERT can directly activate tissue stem cells by controlling gene expression of proliferation genes, e.g., MYC, through the Wnt/β-catenin pathway.
In addition to triggering senescence, telomere dysfunction leads to end-to-end chromosome fusions followed by DNA breakage-fusion-bridge cycles, which result in chromosomal duplications and deletions that are characteristic of cancers. 6, 43, 69 In the setting of an impaired p53 checkpoint, the genomic instability caused by dysfunctional telomeres can initiate malignant transformation, particularly in self-renewing tissues, e.g., the adrenal cortex. 4, 117 Telomere-based crisis provides a mechanism of chromosomal instability, which leads to regional amplifications and deletions that drive tumorigenesis.
Most cancer cells avoid replicative senescence associated with telomere dysfunction and chromosomal imbalances by activating mechanisms of telomere maintenance, either through ectopic expression of TERT or a recombination-based mechanism known as alternative lengthening of telomeres. 63 More than 90% of ACC samples display signs of active telomerase maintenance mechanisms, whereas benign adrenocortical tumors do not. 41 The importance of telomere maintenance of the malignant phenotype is underscored by a series of xenotransplantation studies with telomerase-negative bovine adrenocortical cells rendered tumorigenic by transfection with Ha-Ras (G12V) and SV40 large T antigen. These cells do not require exogenous TERT for initial tumor formation 27 ; however, the cells exhibit a progressive loss of malignant behavior, which can be restored by transfection with TERT. 137 Transfection of tumorigenic bovine adrenocortical cells with TERT triggers a progenitor-like state, characterized by increased cell proliferation regulated by the IGF2 pathway. 119
Tumor Suppressor Genes Involved in Adrenocortical Tumorigenesis
p53
The p53 tumor suppressor pathway functions as a feedback loop that integrates with other signal transduction networks, including the IGF-AKT pathway (see below). 99 p53 itself acts as a transcriptional regulator of the cell cycle, inducing cell cycle arrest (G1 or G2), senescence, or apoptosis in response to DNA damage and other stressors. 135, 161 DNA damage triggers a series of post-translational modifications of p53, including its phosphorylation by the kinases ATM (ataxia telangiectasia mutated) and CHK2 (checkpoint kinase 2). These modifications render p53 resistant to inactivation by MDM2 (mouse double minute 2), an E3-ubiquitin ligase that binds p53 and targets it for degradation in the proteosome. For efficient degradation of p53, MDM2 needs to be phosphorylated, which can be accomplished by the AKT protein kinase.
There is strong selective pressure for emerging tumors to disrupt the p53 signaling pathway, either through mutation of the p53 gene or overexpression of MDM2 or other negative regulators of p53 function. 135, 161 Somatic inactivating mutations of TP53, the gene encoding p53, are observed in one third of human ACCs. LOH at the TP53 locus on 17p13 is found in only 14% of cases of human ACA 134 but is associated with a worse prognosis in these tumors. 56, 103 LOH at 17p13 is evident in approximately 80% in ACC. 103, 134 Fine mapping of 17q13 LOH in ACC suggests that additional tumor suppressor genes besides TP53 may reside in this locus and contribute to tumorigenesis. 134
Individuals with Li-Fraumeni syndrome (LFS), an autosomal dominant tumor predisposition syndrome generally caused by germline mutations in TP53, develop ACC, albeit rarely. An analysis of 91 families with LFS revealed that breast cancer, brain cancer, and sarcomas were common, but ACC was evident in only 1% of the patients. 91 Of interest, a specific germline missense mutation in TP53 (R337H) has been observed in a group of children with adrenocortical tumors in southern Brazil. 51, 125 In contrast to most somatic mutations that affect the DNA binding region of p53, the TP53 R337H mutation affects the oligmerization motif in p53, which leads to pH-dependent instability. Carriers of the TP53 R337H mutation have a 20,000-fold increased risk of ACC, but they are not predisposed to other tumor types typical of LFS. Penetrance is incomplete; only 10% of carriers develop ACC. The loss of chromosome 17 that harbors the normal TP53 allele is crucial for ACC development in patients with the R337H mutation. 121 Other low penetrance constitutional TP53 mutations associated with childhood ACC have also been identified. 51, 125
MEN1
A heterozygous inactivating germline mutation in MEN1, a tumor suppressor gene located at 11q13, is found in about 90% of patients with multiple endocrine neoplasia type 1, an autosomal dominant syndrome characterized by predisposition to parathyroid, endocrine pancreas, pituitary, and adrenocortical tumors. 98 These are typically nonfunctional ACAs; ACC is uncommon. Somatic mutation of the MEN1 gene is very rare in sporadic adrenocortical tumors, 66, 129 but LOH at 11q13 is present in more than 20% of ACA and 90% of ACC. 66, 90, 129 Mice with a germline mutation in the Men1 gene are predisposed to benign tumors in the adrenal cortex and other tissues. 106 Tumors in all sites showed LOH at the Men1 locus. 106 Menin, the protein encoded by the MEN1 gene, interacts with Smad3 and other binding partners, and menin deficiency elicits widespread effects on transcription, chromatin remodeling, and cell-cycle progression. 114
Fumarate hydratase
Hereditary leiomyomatosis and renal cell cancer (HLRCC) is an autosomal dominant disorder caused by mutations in the fumarate hydratase (FH) gene on chromosome 1q42.3–43. Patients with HLRCC are also predisposed to massive macronodular adrenocortical disease (MMAD). MMAD is a heterogeneous condition associated with Cushing syndrome and bilateral hyperplasia of the adrenal glands. 97, 109 Tumor tissue from these patients harbors both a germline FH mutation and allelic loss at the 1q42.3–43 FH locus. The mechanism through which FH, a mitochondrial protein, participates in adrenocortical tumorigenesis, is unknown.
Tpp1
Tpp1, one of the accessory proteins associated with the telomere cap, has been directly linked to adrenocortical stem-cell maintenance and tumorigenesis. Mice homozygous for a hypomorphic allele of Tpp1 exhibit a complex phenotype that includes adrenocortical dysplasia and hypofunction. 69, 80 This allele, termed Tpp1acd (for the adrenocortical dysplasia strain), results in telomeres that are dysfunctional, which leads to an increase in senescence-associated markers in adrenocortical cells. When telomere dysfunction-associated senescence is prevented by introducing mutant p53 alleles into Tpp1acd/adc mice, there is a partial normalization of adrenal weight and morphology. 30 However, preliminary studies suggest that p53 deficiency rescues the adverse effects of telomere dysfunction at the cost of accelerating carcinogenesis. 4 Collectively, these findings suggest that Tpp1 is essential for proper function of progenitor cells in the adrenal cortex and functions as a tumor suppressor.
Signaling Pathways Involved in Adrenocortical Tumorigenesis
In the adrenal cortex, as in other tissues, oncology recapitulates ontogeny. Signaling pathways involved in embryonic development and in stem cell maintenance and/or activation are often deranged in adrenocortical tumors.
Wnt/β-catenin
Wnt signaling pathway regulates a variety of developmental processes, such as cell proliferation, adhesion, and cell-fate determination. 122 Members of the Wnt family of signaling molecules bind to Frizzled receptors on the cell surface and initiate a cytoplasmic signaling cascade. This results in disruption of a β-catenin degradation complex composed of glycogen synthase kinase 3β (GSK3β), the adenomatous polyposis coli (APC) tumor suppressor, and auxin. The net effect of disruption of this complex is stabilization of β-catenin. 122 Once translocated to the nucleus, β-catenin can enhance gene expression through interactions with various transcription factors or coactivators, including TCF/LEF family members, Smad factors, and SF1 in steroidogenic cells. 60 Among the targets of activation by the TCF/β-catenin complex are genes that drive cell proliferation, such as c-Myc. 64, 141
Wnt2b is expressed in subcapsular cells and might regulate progenitor fate, 105 whereas Wnt4 localizes to the zG and appears to regulate zonal identity. 65 Introduction of Wnt4 into primary cultures of adrenocortical cells was shown to alter steroidogenesis. 28 Wnt4 null mice have decreased expression of P450aldo in the adrenal and ectopic expression of the adrenal-specific marker P450c21 in gonadal tissue. 65, 75 Humans homozygous for Wnt4 loss-of-function mutations exhibit adrenal dysgenesis and female-to-male sex reversal. 108
Tissue-specific knockout of β-catenin in adrenocortical progenitors (by using an Sf1 promoter-Cre transgene) produces mice with adrenal glands that function normally at birth but fail by 30 weeks of age. 85 As with Dax mice, it is theorized that impairment of the Wnt/β-catenin signaling pathway leads to depletion of adrenocortical stem/progenitor cells. Thus, Wnt/β-catenin signaling pathway appears to function as a gatekeeper that serves to maintain the undifferentiated stem cell pool.
Overexpression of genes related to the Wnt/β-catenin signaling pathway was reported in adrenocortical hyperplasia. 23 Abnormal cytoplasmic or nuclear accumulation of β-catenin is seen in 38% of human ACAs and in 85% of adrenocortical carcinomas. 142 Somatic activating mutations of the β-catenin gene are present in approximately 30% of human ACAs and carcinomas 139, 142 and were linked to bilateral adrenal hyperplasia in humans. 140 β-Catenin immunoreactivity was also observed in the nuclei of neoplastic cells that accumulate in the adrenal cortex of gonadectomized inbred mice, which suggests that the Wnt signaling pathway may participate in gonadectomy-induced adrenocortical neoplasia. 17 The TCF/β-catenin antagonist PKF115-584 inhibits proliferation of adrenocortical carcinoma cells in vitro, which suggests that inhibitors of the Wnt signaling pathway might be useful in the treatment of adrenocortical tumors. 38
Individuals with familial adenomatous polyposis (FAP) have constitutive activation of the Wnt/β-catenin signaling pathway because of germline loss-of-function mutations of the APC gene. These individuals are predisposed to both colonic and extracolonic tumors, including adrenocortical tumors. 19 Most of the adrenocortical neoplasms in patients with FAP are benign. 136
IGF1R-AKT
Two members of the IGF family, IGF-I and IGF-II, act as adrenocortical mitogens by promoting progression through the G1/S phase of the cell cycle. IGF-II expression is normally limited to the embryonic and fetal stages of adrenal development. Both IGF-I and IGF-II bind the tyrosine kinase receptor IGF1R, which, in turn, phosphorylates the insulin receptor substrates (IRS) IRS-1 and IRS-2. Phosphorylated IRS recruits phosphatidylinositol 3-kinase (PI3K), which results in activation of the AKT protein kinase, which then phosphorylates a wide range of target proteins, including GSK3β and MDM2. GSK3β phosphorylation leads to activation of the Wnt/β-catenin pathway. Mice with null mutations in Igf1 or Igf2 appear to have normal adrenocortical function, but mice overexpressing IGF-II develop early adrenal hyperplasia, which suggests that this growth factor can drive adrenocortical cell growth in vivo. 26
The IGF2 locus (11p15) is parentally imprinted, and normally only the paternal allele is expressed. 37 Two other imprinted genes in this region, H19 and p57KIP2 , are expressed only from the maternal allele. The H19 mRNA is not translated and serves to inhibit IGF2 expression. The p57KIP2 gene encodes a cyclin-dependent kinase inhibitor involved in the G1/S phase of the cell cycle, and p57KIP2 is primarily found in differentiated cells. Mutation and/or aberrant expression of imprinted genes in the IGF2 locus cause Beckwith-Wiedemann syndrome (BWS), an overgrowth syndrome associated with adrenal hyperplasia and ACC. 36 BWS resembles late offspring syndrome (LOS), an overgrowth syndrome that affects ruminants. 133, 159, 160 Interestingly, both BWS and LOS are complications of assisted reproduction technology. In vitro culture of embryos during assisted reproduction is believed to cause abnormal genetic imprinting at the IGF2 gene cluster, which leads to overgrowth syndromes in children, ruminants, and probably other mammals. 100, 126
Overexpression of IGF2, caused by genetic or epigenetic alterations at chromosome 11p15, is evident in approximately 90% of cases of human ACC. 57, 102 Transcription analysis has shown that IGF2 is the most overexpressed gene in human ACC compared with ACAs or normal tissue, 2, 20, 156 and downstream AKT phosphorylation is more pronounced in ACC than in ACAs. 44 On the basis of these observations, it has been suggested that activation of IGF2 is essential for ACC growth. The clonal genetic model of tumorigenesis argues that genetic changes at the IGF2 locus are a late event in the evolution of ACAs into carcinomas, whereas the epigenetic progenitor model discussed below posits that LOI at the IGF2 locus is an early event in tumorigenesis.
Supporting the notion of an IGF-II autostimulatory loop, overexpression of the IGF-1 receptor (IGF1R) was documented in cases of human ACC. 52, 154, 155 The biological effects of IGFs are modulated by a series of IGF binding proteins (IGFBP). Adrenocortical tumors with IGF-II overproduction were shown to express large amounts of IGFBP-2, which may regulate the effects of IGF-II in these tumors. 22 Interestingly, IGFBP-2 levels correlated with the stage of ACC and are inversely correlated with survival. 21 However, loss of function of a second receptor for IGF-II, IGF2R, is associated with malignant adrenocortical tumors. IGF2R is thought to be a tumor suppressor because of its ability to bind and degrade IGF-II and promote activation of TGFβ. 33, 96 Recent studies suggest that LOI at the IGF2 locus increases cancer risk in another way, by increasing the sensitivity of IGF-II signaling pathways. 77
Because Igf2 is the highest upregulated transcript in sporadic ACC, this growth factor and its receptors are logical candidates for molecularly targeted drug therapy. IGF1R antagonists cause growth inhibition in vitro in human ACC xenografts in nude mice, 62 and early clinical testing in humans is underway.
cAMP signaling
Activation of G-protein coupled receptors on the surface of adrenocortical cells elicits pleiotropic effects on cell growth, differentiation, and steroid production. These effects are mediated, in part, through the cAMP-protein kinase A signaling pathway, which culminates in the phosphorylation (i.e., activation) of transcription factors and enhanced expression of steroidogenic enzyme genes. The binding of ACTH to its G-protein coupled receptor, MC2R, drives differentiation of adrenocortical progenitors into corticoid-producing cells (Fig. 2). Disruption of MC2R leads to adrenocortical atrophy and reduced production of glucocorticoids and aldosterone. 29 A constitutively active, germline mutation of the MC2R gene has been observed in an individual with bilateral adrenal hyperplasia and Cushing's syndrome. 138 Genetic experiments with mice and observations on ferrets suggest that the binding of LH to its G-protein coupled receptor, LHR, can redirect differentiation of multipotential stem/progenitor cells in the subcapsular region of the adrenal from corticoid-producing cells fate to a sex steroidogenic cells (Fig. 2). 18 LHR is expressed in the fetal but not adult mouse adrenal cortex, a developmental expression pattern that resembles those of transcription factor GATA4 and P450c17. It is conceivable, albeit unproven, that a subset of adrenocortical stem/progenitor cells express functional LHR and proliferate in response to LH stimulation.
Many of the mutations associated with human adrenocortical hyperplasias and adenomas result in dysregulation of the cAMP-dependent protein kinase signaling pathway. Cases of benign multinodular hyperplasia and ACA in humans were linked to ectopic expression of certain transmembrane G-protein coupled receptors, including receptors for LH, gastric inhibitory polypeptide, vasopressin, β-adrenergic agonists, and interleukin-1 (Table 3). 35, 86, 92, 94, 136, 152 Enforced expression of LHR or the gastric inhibitory polypeptide receptor in bovine adrenocortical cells is a sufficient genetic event to induce benign adrenocortical tumors in a xenotransplantation model. 110– 112
Trimeric G-proteins transduce signals from LHR and other G-coupled transmembrane receptors. Certain benign multinodular adrenocortical hyperplasias have been linked to activating mutations in the gene for Gsα or inactivating mutations in gene for Gi2α, both of which cause excess cAMP signaling. Loss-of-function germline mutations in the genes encoding PDE8 and PDE11, 2 phosphodiesterases that degrade cAMP, were linked to bilateral adrenocortical hyperplasia and sporadic ACA. 71, 136 Inactivating mutations in the protein kinase A regulatory subunit (PRKAR1A) cause Carney complex, a multiple endocrine neoplasia syndrome associated with primary pigmented adrenocortical disease, abnormal pigmentation of the skin, myxomas, and other neoplasms. Somatic inactivating mutations or allelic losses of the PRKAR1A locus at 17q22–24 are also seen in sporadic cases of ACAs and ACCs. 10
TGFβ signaling
Members of this superfamily use a signaling pathway that includes their type I and type II receptor kinases and their downstream mediators, the Smad proteins. 130 Activation of the receptors leads to phosphorylation of Smad2 and Smad3. Phosphorylated Smad2 and Smad3, in conjunction with Smad4, are translocated to the nucleus, where they activate transcription of specific genes.
TGFβ1 and TGFβ2 are autocrine growth factors produced by normal adrenocortical cells. TGFβ1 and TGFβ2 have no effect on steroidogenic cell proliferation, but they limit steroidogenesis via downregulation of StAR, P450c17, and other genes. 24 Both TGFβ2 and its receptor kinase TGFβ-R1 are overexpressed in human ACC compared with ACA. 145
Two other members of the TGFβ superfamily, activin and inhibin, were shown to impact adrenocortical growth and tumorigenesis. Activin receptors are expressed in the murine adrenal gland, and activins were shown to inhibit adrenocortical cell growth and steroid production and to enhance apoptosis of X-zone cells. 12, 93 Inhibins have no direct effect on steroidogenesis or cell survival but can antagonize activin signaling by competing for activin receptors. 146 Differentiation of adrenocortical progenitor cells into corticoid-producing versus sex-steroid–producing cells is regulated by inhibin, 107 which serves to limit the expansion of LH-primed GATA4-positive progenitors destined for a gonadal fate. 84 Inhibins are produced primarily in gonadal somatic cells. Gonadectomy leads to a profound and rapid fall in circulating inhibin levels, which facilitates differentiation of gonadal-like stroma in cases of gonadectomy-induced adrenocortical neoplasia.
Other growth factors
Two members of the fibroblast growth factor (FGF) family, FGF-1 and FGF-2, are expressed in normal adrenal cortex and are potent mitogens in this tissue. FGF-1 and FGF-2 bind to a family of 4 FGF receptor tyrosine kinases. Two members of this kinase family, FGFR1 and FGFR4, are overexpressed in adrenocortical tumors. 156 In pediatric patients, expression of FGFR4 is much higher in ACC than in ACA. 145 Epidermal growth factor receptor (EGFR) overexpression was demonstrated in more than 90% of ACCs. 40, 76, 87, 123 Although the ligand EGF is not overexpressed in ACC, the receptor is known to bind TGFα, which is often found in adrenocortical tumors. 87 Inhibition of the EGFR signaling pathway inhibits proliferation in the ACC cell-line NCI-H295. 123 The general EGFR tyrosine kinase inhibitor erlotinib is being studied in combination with other chemotherapies for the treatment of advanced ACC, although preliminary results were disappointing. 123
Future Directions
Over the past decade, research on adrenocortical neoplasia has been dominated by gene expression profiling of tumor specimens and by analysis of human genetic disorders associated with a predisposition to these tumors. Although these studies have identified gene mutations and signaling pathways that are dysregulated in adrenocortical tumors, the molecular events accounting for the frequent occurrence of benign neoplasms and low rate of malignant transformation remain unknown. We are entering a new era in adrenocortical tumor research. The availability of inbred and engineered mouse models is shifting the research focus to genetic and epigenetic events that impact the growth and differentiation of adrenocortical stem/progenitor cells. Once these events are delineated, it may be possible to design rational therapeutic interventions that are specific for benign or malignant adrenocortical neoplasms.
Footnotes
Acknowledgements
This work was supported by NIH grants DK075618 and DK52574 and by the Sigrid Juselius Foundation and the Finnish Cancer Society.