
Abstract
Two opposing hypotheses of the origin of cancer have existed for many decades. One hypothesis postulates that the adult stem cell is needed to initiate the carcinogenic process, whereas the other claims a somatic differentiated cell can dedifferentiate or be reprogrammed to regain properties associated with cancer cells. Recent reemergence of the cancer stem cell hypothesis and the isolation of presumptive cancer stem cells from many types of cancer have forced a reexamination of these 2 hypotheses of the origin of cancer. In addition, normal embryonic and adult stem cells have now been isolated and partially characterized. Furthermore, the demonstration of embryonic-like stem cells, being isolated from adult-differentiated skin fibroblast cells of mice, monkey, and human beings, provides a newer opportunity to determine which of these 2 hypotheses might explain the cell type for initiating the carcinogenic process. The goal of this review is to integrate these recent findings, concerning the isolation of normal and cancer stem cells, with several of the classical concepts of carcinogenesis (initiation/promotion/progression; mutation/epigenetic; stem cell theory/dedifferentiation hypotheses; oncogenetumor suppressor theory). Although the weight of the evidence in this review seems to support the stem cell hypothesis, only future studies, probably using comparative animal and human oncologic studies, will determine if targeting the cancer stem cell, with individualized medical approaches, will improve cancer prevention and therapy.
Introduction: Potential Insights From Human Carcinogenesis to New Strategies for Treatment of Animal Cancers
The understanding of human and animal carcinogenesis will, no doubt, ultimately lead to more efficacious cancer prevention and treatment strategies. Today's understanding of human carcinogenesis is by no means complete. However, the use of animal experimental approaches (e.g., bioassay of carcinogens, genetically engineered animals, rodent in vitro studies), together with human genetic syndromes (e.g., xeroderma pigmentosum, retinoblastoma); dietary, occupational, and environmental epidemiology; and the use of modern molecular biologic tools, have provided new insights on the interactive roles of shared genetic and environmental factors in the carcinogenic process in animals and humans. Because there are a number of significant differences in evolutionary forces related to life span determination and the development of culture (e.g., altered physical environments, diet), 8, 12, 18, 64, 70, 80, 102, 108 there is not a one-to-one correspondence of the incidence of cancer in human populations and populations of specific breeds of domesticated animals, nor possibly in some molecular contributors to specific cancers. Today's view of the complexity of carcinogenesis might even seem to rule against the understanding of a single type of cancer in either the animal or the human. This is seen in both animal and human tumors, in that no 2 tumors of a given organ in either the animal or the human being would contain tumor cells that are uniformly genetically or phenotypically identical.
Yet, despite potential real differences between animal and human carcinogenesis, there do seem to be fundamental concepts in the understanding of carcinogenesis, shared by both animals and human beings. 109 Genes or environmental factors alone cannot cause cancer. Although there are genes (proto-oncogenes and tumor suppressor genes) in both animals and human beings, these genes must interact with specific classes of environmental factors in order for a normal cell to be converted to a cancer cell. This is beautifully illustrated with the interaction of the lack of DNA repair (nature/genetic) to ultraviolet light damage (nurture/environment) and the production of mutations in specific tumor suppressor gene in exposed skin cells of the xeroderma pigmentosum patient. 10
However, the production of DNA damage by various environmental agents and the ultimate production of mutations in either proto-oncogenes or tumor suppressor genes, while necessary, are insufficient for most cancers (exception being the production of teratomas). That single irreversibly altered cell must be clonally expanded so that additional rare events, some of which are not mutagenic but are epigenetic in nature, can occur. These epigenetic alterations of individual gene expression can occur at the transcriptional, translational, or posttranslational levels. Therefore, the concept of both mutation and epigenetic mechanisms must be involved in the carcinogenic process.
The multistage, multimechanism concept of the initiation/promotion/progression model of carcinogenesis can integrate the nature and nurture and mutation and epigenetic mechanism with the oncogene–tumor suppressor gene theory of carcinogenesis. 112 Here, the classic animal experiments, demonstrating that there are distinct phases of this carcinogenic process, showed that a single cell, when exposed to some agent that could induce an irreversible event in that cell (e.g., a DNA-damaging agent to cause a mutation), would allow that cell to be selectively amplified by agents that were not necessarily DNA damaging or mutagenic (i.e., tumor promoters, such as phorbol esters in croton oil). 6, 122
The ultimate question arose decades ago in the search for the cell that ultimately starts the multistage, multimechanism process of carcinogenesis: Is the “target cell” a stem cell or any somatic differentiated cell? (The stem cell theory of cancer.) 29, 33, 45, 65, 76, 82, 85, 99
Although the concept of stem cells has been a part of both plant and animal embryology, developmental biology, and even in cancer research for decades, its recent emergence in multiple fields of “cutting-edge” research has been extraordinary. The convergence of the reports of the cloning of Dolly 120 and the isolation of human embryonic stem cells 93, 103 has stimulated the field of “regenerative therapy” with stem cells. 69, 86 The use of embryonic stem cells for drug development and safety assessment, 21, 107 as well as for basic biology of gene regulation during development and differentiation, has also contributed to the understanding of the role these stem cells might play in carcinogenesis. In the cancer field, although the old concept of the stem cell theory of cancer∗ existed in cancer research, the recent discovery of cancer stem cells has restimulated the field. 1, 19, 47, 53, 95 This discovery has triggered multiple reports of cancer stem cells in multiple tumor types and cancer cell lines.† The convergence of stem cells in embryology and in cancer research should be no surprise; it was predicted by the embryologist, C. Markert, 66 when he stated the following:
Cells interact and communicate during embryonic development and through inductive stimuli mutually direct the divergent courses of their differentiation. Very little cell differentiation is truly autonomous in vertebrate organisms. The myriad cell phenotypes present in mammals, for example, must reflect a corresponding complexity in the timing, nature, and amount of inductive interactions. Whatever the nature of inductive stimuli may be, they emerge as a consequence of specific sequential interactions of cells during embryonic development. The first embryonic cells, blastomeres, of mice and other mammals are all totipotent. During cleavage and early morphogenesis these cells come to occupy different positions in the three-dimensional embryo. Some cells are on the outside, some inside. The different environments of these cells cause the cells to express different patterns of metabolism in accordance with their own developing programs of gene function. These patterns of metabolism create new chemical environments for nearby cells and these changed environments induce yet new programs of gene function in responding cells. Thus a progressive series of reciprocal interactions is established between the cellular environment and the genome of each cell. These interactions drive the cell along a specific path of differentiation until a stable equilibrium is reached in the adult. Thereafter little change occurs in the specialized cells and they become remarkably refractory to changes in the environment. They seem stably locked into the terminal patterns of gene function characteristic of adult cells. The genome seems no longer responsible to the signals that were effective earlier in development. Of course, changes can occur in adult cells that lead to renewed cell proliferation and altered differentiation as seen in neoplasms, both benign and malignant, but such changes are very rare indeed when one considers the number of cells potentially available for neoplastic transformation. Possibly, mutations in regulatory DNA of dividing adult cells can occasionally lead to new and highly effective programs gene function that we recognize as neoplastic or malignant. However, most genetic changes in adult cells can probably lead to cell death since random changes in patterns of gene activity are not likely to be beneficial.
In addition, V. P. Potter, who coined the phrase “oncogeny as partially blocked ontogeny,” also viewed cancer as having its origin in an adult stem cell. 85 Yet the concept of cancer having its origin in an adult stem cell went almost nowhere, with the exception of leukemia research. 104 An equally valid opposing hypothesis stated that these cancers originated from the dedifferentiation of a mortal progenitor or even from a terminally differentiated somatic cell. 90 In addition, the recent successes of molecular oncology, with the discovery of oncogenes and tumor supressor genes, put the current research focus on DNA, rather than on any cell type. Advances in molecular biologic technology and completion of the delineation of the human genome and of the in vitro toxicology of carcinogens further pointed to mutations of oncogenes and tumor suppressor genes as leading the way to discover the causes of cancer.
However, in the wake of these exciting new discoveries was the fading of the older concepts of carcinogenesis, such as the multistage, multimechanisms hypothesis of carcinogenesis from the intellectual critical analyses of the newly generated data. The conflicting data, generated to test the mutation theory of cancer and the development of methodologies to measure epigenetic changes (e.g., methylation of DNA/ histone acetylation changes, DNA microarray analyses), seemed to show that epigenetic events were affected by nonmutagenic, tumor-promoting chemical toxicants. Redox changes or oxidative stress-induced intracellular signaling and regular, programmed-like gene expression changes occurred during tumor promotion. 110 These observations could not be explained by rare, random mutagenic changes. These detailed observations only created new disciplines, such as bioinformatics, and the further reductionalistic treatment of this complex disease process.
Teratogenesis and Carcinogenesis: Twin Diseases That Share Many Common Risk Factors
There was a time when genetic and epidemiologic analyses seemed to link these 2 disease processes together, further giving credence to Markert, Pierce, and Potter's ideas. 65, 82, 85 Many cancer genetically predisposed individuals were at risk for both cancer and developmental congenital events (e.g., Fanconi's, Down's, xeroderma pigmentosum). In addition, experimentally, some chemicals and drugs, being positive for causing both disease processes, have been shown, in some cases, such as retinoids, as being able to prevent some cancers while inducing birth defects under different conditions. 30, 37, 94, 118 Even a well-known nongenotoxic toxicant and tumor promoter, TCDD, has been documented as an antitumor promoter. 17 There have been many such cancer- and teratogen-causing chemicals that can have beneficial health effects, depending on the circumstances of exposure. 106, 115 One might view this as the classic Good News/Bad News story.
Agents, such as X-rays, which are mutagenic, can be teratogens. Many of these multidisease-effecting agents, such as thalidomide, have been shown to be non–DNA-damaging agents (at least for nuclear DNA) and nonmutagenic. The recent demonstration of the alteration dramatic phenotypic changes and of the gene expression patterns associated with methylation and/or histone acetylation modifications has confirmed the importance of epigenetic events during both development and carcinogenesis. 25
However, although all these old and new observations coexist, there seems to be no resolution of the initial problems of how developmental abnormalities and cancers really occur and how the mechanisms of mutagenesis and epigenetic alterations affect either process. As indicated before, in some cases, genes play the predominant role. In other experimental cases, environmental toxic chemicals and physical agents play a role. However, in the case of human birth defects and human cancers, diet seems to be a significant factor, either increasing or decreasing the risk to both diseases processes. 108, 109 In the case of human breast, prostate, and colon cancers, collision of biologic and cultural evolutionary factors via the diet seems to have been responsible, in large part, in contributing to these cancers. 16
Stem Cells in Normal and Abnormal Development Bring Teratogenesis and Carcinogenesis Into the Same Focus
Before one can rigorously discuss the potential role stem cells play in development and diseases, a common agreement on the definition of stem cells must be made. The following definition might not be universally accepted, as none currently exists. Yet there does seem to be some consensus. A stem cell is one that can proliferate either symmetrically or asymmetrically. That is, under one set of circumstances, a stem cell can be stimulated to divide, symmetrically, to produce 2 stem cells. In this manner, the stem cell pool can increase. These stem cells are considered by many, if not most, to be immortal, that is, they can proliferate indefinitely until they are induced to differentiate or apoptose. In asymmetric cell division, one daughter cell maintains stemness like its mother cell, while its sib, a progenitor cell, can start its role as a potential origin of a differentiated cell of that lineage, as well as the transit cell to expand the population of that particular lineage. This progenitor cell, either a fibroblast- or an epithelial-type cell, has a finite life span, as in the case of human fibroblasts (i.e., the Hayflick limit). 36 A terminally differentiated cell is one that will no longer proliferate (e.g., a red blood cell, a lens cell); it either senesces or dies by apoptosis.
A totipotent stem cell is, in the case of the human being, one that can give rise to all cells of a human being. A pluripotent stem cell, the one usually referred to as an embryonic stem cell, can give rise to all cell types but is unable to be reimplanted to form a human being. A multipotent stem cell is a cell that is further committed to produce limited cell types. A bipolar stem cell, such as the oval cell of the liver, can give rise to 2 lineages, such as the hepatocyte and bile ductal cells. 116
From the single fertilized egg (the totipotent stem cell) to the adult human, consisting of more than 200 cell types and approximately 100 trillion cells, a very delicate and complex cybernetic cell communication system has to be in place. First, it is a part of a genetically determined blueprint in the individual's genome. Second, it dynamically takes place during the historically unique exposure of environmental, medical, and dietary agents during the maternal, neonatal, adolescent, sexually mature adult and aging geriatric stages. This homeostatically regulated communication process ensures a minimum of any chronic disease, usually associated with the aging process. 108, 114 It consists of extracellular communication (e.g., secreted hormones, cytokines, growth regulators, extracellular matrices, stromal-epithelial factors) that can trigger intracellular communication to alter both gene expression and gap junctional intercellular communication (GJIC) (Fig. 1). 110
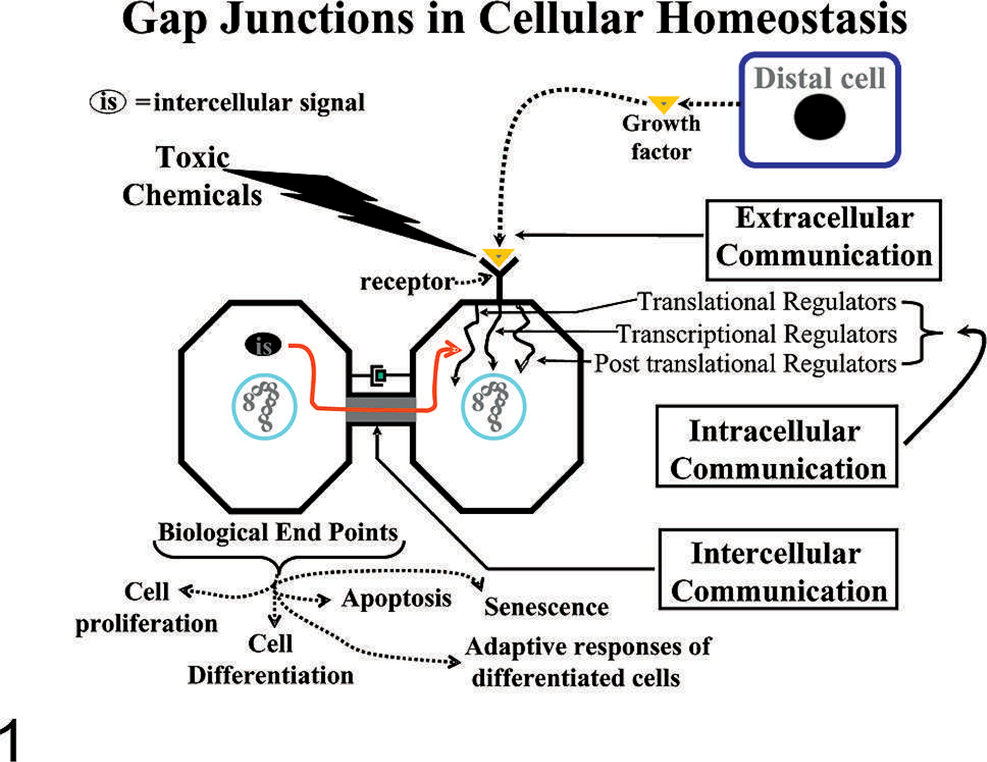
Gap junctions in cellular homeostasis. Extracellular signals, such as growth factors, toxicants, extracellular matrices, and cell adhesion molecules, interact with membrane receptors, which activate intracellular signal transduction pathways. These specific intracellular pathways operate under cascading systems that cross-communicate in controlling the expression of genes that direct cell behaviors. These multiple intracellular signaling checkpoints are further modulated by intercellular signals traversing gap junctions, thereby maintaining the homeostatic state of a tissue. Abnormal interruption of these integrated signalling pathways by food-related and environmental toxicants results in diseased states, such as the tumor promotion phase of carcinogenesis. 110 .
Within a given tissue, the communication must exist between all cell types (e.g., from the adult stem cells to their committed multipotent progenitor and with the terminally differentiated daughters).
Disruption of any of these highly integrated communication processes can influence the other communication processes in the network. From an evolutionally point of view, this was designed to be adaptive to control the various cell behavior choices. A cell can 1) proliferate, 2) differentiate, 3) apoptose, 4) senesce, or 5) if already terminally differentiated, it can adaptively respond, such as a pancreatic beta cell to produce insulin.
Therefore, during embryogenesis and fetal development, an incredible regulatory control must be functional in order to have the one fertilized egg cell rapidly proliferate and differentiate to start new tissues. The new tissues must form new organs and structures, which in turn must effect further proliferation, differentiation, and even apoptosis (such as the loss of the webbing tissue between our digits before birth). Too many or too few stem cells, mutated stem cells, and stem cells with altered gene expression or faulty communication between the stem cells and their lineage derivative progenitor and differentiated offspring could lead to embryo/fetal death or some structural or behavioral teratogenic event.
This is demonstrated in the formation of teratomas, caused by abnormal epigenetic control of cell differentiation. Highly malignant teratocarcinoma cells can be placed back into a normal microenvironment of a normal blastocyte and lead to normal development. This suggests that the original cause of the abnormal differentiation of the stem cells was not an irreversible mutation but a potentially reversible epigenetic change. 71 In addition, over expression of the Oct-4 stemness gene in a transgenic mouse can cause dysplasia of tissues. 38
In the case of carcinogenesis, one must consider a well-documented experimental concept of carcinogenesis that probably is relevant to human carcinogenesis, namely, the multistage, multimechanism concept. 83 This model consists of 3 operational definitions of distinct mechanistic stages of the carcinogenic process. The first stage involves a single normal cell being irreversibly converted to a premalignant or an initiated cell (Fig. 2). The current paradigm holds that this first step is the immortalization of a normal differentiated progenitor or differentiated somatic cell. 49 That implies that there must be a reprogramming of the genes of this somatic differentiated cell to go back to the embryonic-like state, including the ability to regain the status of an immortal-like stem cell but to lose this capacity to differentiate terminally.
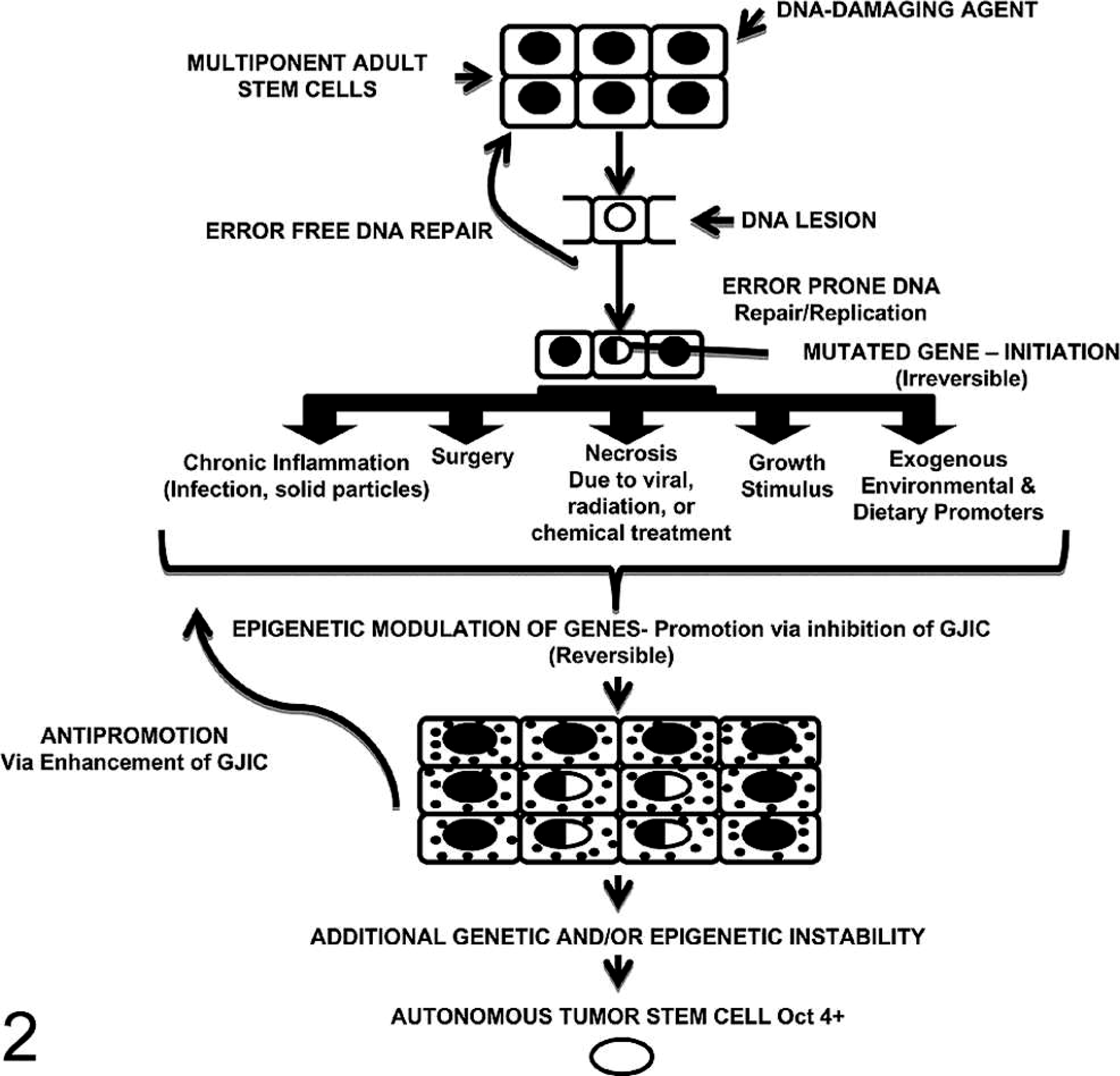
A diagrammatic scheme to depict the postulated mechanisms of the initiation and promotion phase of carcinogenesis. DNA lesions, induced by physical mutagens or by errors in DNA replication, are substrates in adult stem cells (Oct-4 positive) that can be fixed if they are not removed in an error-free manner prior to DNA replication. Promotion includes those conditions in which a pluripotent-initiated, but surviving, adult stem cell (Oct-4 positive) can escape the nonproliferative state. The build up of initiated cells allows them to “resist” the antimitotic influence of neighboring noninitiated cells. In addition, the changing microenvironment within the growing benign tumor will cause some of the initiated adult stem cells to partially differentiate into cancer nonstem cells. This, together with either additional mutations or stable epigenetic changes, might allow a given initiated adult stem cell to have autonomous, invasive properties of a malignant cell. 114 .
The second step, which has to be mechanistically distinct from the initiation step, is referred to as the promotion stage. That is, the single initiated cell is clonally expanded by a mitogenic process of the initiated cell, the inhibition of apoptosis of the initiated cell, or both. 105 The combination of both of these mechanisms, mitogenesis and apoptosis, would lead to a preferential or differential expansion, in any tissue, of the initiated, immortal cell. The initiated cell would not terminally differentiate by asymmetric cell division or die by apoptosis, while any normal stem cell would divide asymmetrically and could die by apoptosis or eliminated by senescence. The third step, the progression step, is where a single initiated cell, within the clone of promoted initiated cells, accrues other changes needed to fulfil the requirements of having all the hallmarks of cancer. 35 Although the underlying mechanisms for these 3 stages are not unequivocally delineated, it is believed the initiation step is caused by an irreversible mutational event in that single normal cell, which inhibits its ability to asymmetrically divide under normal conditions, although it can symmetrically divide.
The promotion step, involving the mitogenic expansion of the initiated cell, is now thought to be an epigenetic mechanism, which allows the single initiated cell to escape the proliferation-suppressing effect of either secreted negative growth regulators or gap junction-mediated intercellular factors. 106 Most, if not all, promoting conditions, such as wound healing, surgery, cell death, inflammatory agents, growth factor hormones, and nonmutagenic chemicals, are inhibitors of gap junctions. 105 From experimental studies, promoters can be species specific, gender specific, and organ specific; can have threshold levels of action; and must be applied in a regular fashion over a long period. Promoters must be given to the initiated cell in the absence of agents that can prevent the downregulation of gap junctions in initiated progenitor cells or receptor signaling in stem cells. 106, 109, 112
On the other hand, nonadaptive alterations, in perturbing the integrated communication mechanisms, could lead to disruption of those signals controlling any of the 5 cellular choices. This could happen on several levels. The first is the disruption of a normal progenitor cell to proliferate abnormally, leading to hyperplasia, such as that described by Hochedlinger et al. 38 The second is to abnormally induce differentiation of stem cells, as might be the case when some teratogen, such as retinoid 62 and thalidomide, 51 is given to developing embryos or fetuses. It could decrease the stem cell pool. The third is abnormal induction of apoptosis, as was shown when the small intestinal stem cells died after ionizing radiation. 4 This observation also might explain reversible hair loss after chemical chemotherapy.
Now the real questions are: “What is that normal cell that is initiated?” and “Is it a normal somatic differentiated, mortal cell that must be dedifferentiated or reprogrammed to be an immortal stem cell?”; or “Is that normal cell a normal adult stem cell, which is already immortal, unable to mortalize or terminally differentiate because the initiation event blocks asymmetric cell division?”
Is Initiation an Immortalizing Event or a Blockage of a Mortalization or Differentiation Event?, or “Reprogramming or Blockage of Differentiation Programming”
Operationally, from classic experimental animal studies, after a single exposure to an agent that can cause an irreversible change in the genome of a single normal cell, the process has been called “initiation.” The assumption was previously made that the normal cell, which ultimately gives rise to a cancer, was a mortal somatic or differentiated cell, such as a keratinocyte in the mouse skin or hepatocyte in a rat liver (2 model initiation/promotion/progression systems for skin and liver tumors). Therefore, in that context, an initiated somatic differentiated cell, which has a finite lifespan, 36 was assumed to have been immortalized so that it could proliferate in an unlimited fashion to accrue all those hallmarks of cancer phenotypic changes. This assumption was indirectly supported by the fact that cell lines from tumors were classified as immortal. Initiation, then, was interpreted as an immortalizing step.
Conceptually, that means a somatic differentiated cell, with its set limited life span and proliferative capacity, had to be “reprogrammed” (at least partially) in terms of its differentiated gene expression pattern to one that resembles a stem cell. This initiated cell is both “primitive” in its differentiation phenotype and its ability to have unlimited proliferative capacity. However, unlike a normal stem cell that can divide both symmetrically and asymmetrically, this reprogrammed differentiated cell could only divide symmetrically to produce 2 initiated cells after mitogenic stimulation. That is why tumor promoters can increase the number of initiated cells. In addition, promoters also seem to prevent the apoptosis of these initiated cells. At least in studies of rat liver–promoted foci of initiated cells, there seems, initially, to be uniform phenotypes (enzyme altered foci) of these cells. 83 Later, upon further promotion, foci within the original foci appear. 83 Therefore, in this interpretation, initiation is the process that induces an irreversible change that prevents a normal somatic differentiated cell to divide symmetrically and to regain its ability to have unlimited proliferation capacity and to partially regain a stem-like phenotype.
A completely different interpretation comes from the stem cell hypothesis of carcinogenesis. That hypothesis assumes that in all organs, there exist adult stem cells, that is, multipotent stem cells that have been derived from pluripotent stem cells during development that are, at least, committed to differentiation into an organ-specific lineage of cells. These adult stem cells are assumed to be naturally immortal until they are induced to terminally differentiate or apoptose. During growth or wound healing, these adult stem cells serve to replenish or replace lost, diseased, or dead differentiated cells. The best example, of course, is the hematopoietic stem cell. However, skin and intestine are obvious organ sites of adult stem cells that are needed to replace the billions of differentiated cells lost every day in those organs.
In the stem cell hypothesis of carcinogenesis, initiation would, operationally, prevent the mortalization of a normal adult stem cell, rather than induce immortalization of a normal, mortal somatic cell. In operational terms, initiation of an adult stem cell means that it is now restricted to divide symmetrically (Fig. 3).
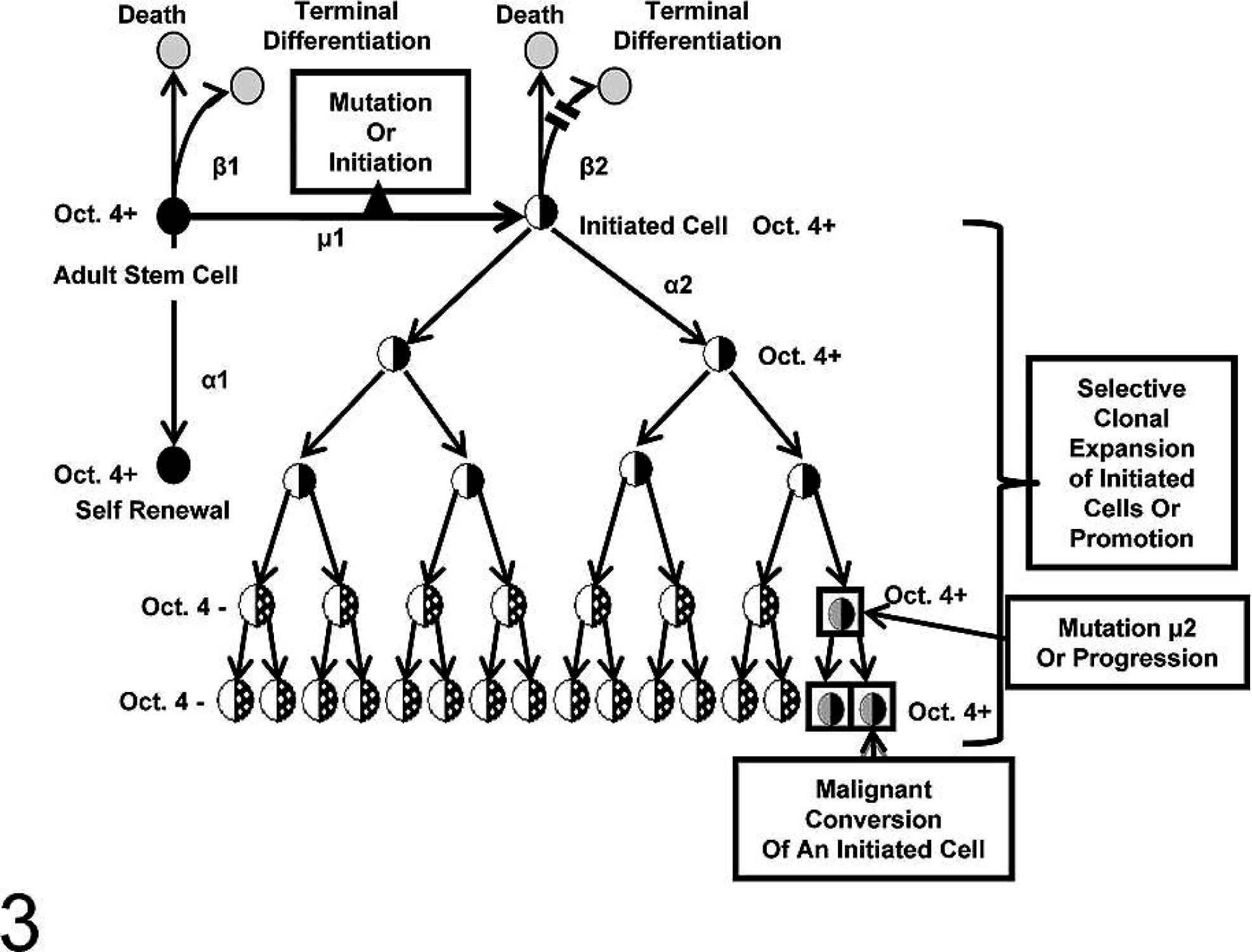
In this diagram, a normal adult stem cell is shown dividing asymmetrically to form one daughter that is committed to ultimately terminally differentiate. The other daughter is designated to be identical to its mother adult stem cell (Oct-4 positive). If that adult stem cell is exposed to some condition that prevents asymmetrical cell division but does not suppress the Oct-4 expression, it is operationally an initiated cell. These adult-initiated stem cells are still Oct-4 positive or benign cancer stem cells. As these initiated Oct-4–positive cells are stimulated to proliferate and resist apoptosis, the growing benign tumor microenvironment changes and some of these initiated Oct-4–positive cells can partially differentiate into “cancer nonstem cells” (Oct-4 negative). Eventually, additional stable mutational or epigenetic events occur, allowing the benign Oct-4–positive cancer stem cells to become invasive, metastatic “cancer stem cells.” 114 .
Upon ordinary microenvironment conditions of mitogenic stimulation, it would only divide to produce 2 initiated adult differentiated cells. At this point, it seems the 2 hypotheses converge. However, it must be kept in mind that the adult stem cell is already in the immortal mode, whereas the somatic differentiated mortal cell must be reprogrammed, first during initiation to return to this stem-like state. Initiation, on the other hand, for the normal, immortal adult stem cell is not a reprogramming process but one that actually blocks programming to a differentiated state.
One area of cancer research that might have a bearing on determining if the normal immortal adult stem cell or the normal, mortal somatic cell is the “target” for carcinogenesis is that of research with immortalizing viruses. In vitro neoplastic transformation studies in rodent cells have been developed for many decades because they have been relatively easy to transform, neoplastically. The same cannot be said for in vitro human neoplastic transformation studies. 48, 88 As will be shown below, the rodent studies have not always been consistent, and they have not been particularly good at classifying the agent inducing the transformation. 115
The study by Nakano et al. 73 was designed to understand the basis for inconsistencies in the recovery of neoplastic Syrian hamster embryo fibroblast cells after exposures to carcinogens. From day to day or lab to lab, they found that, in order to find neoplastic transformants after exposures, the treated population had to have rare contact-insensitive cells. These contact-insensitive cells were normal, but they behaved like neoplastic-transformed cells that are contact insensitive. 9 Here, the field of GJIC had something to contribute. Loewenstein 60 and Lowenstein and Kanno 61 showed that a major difference between normal cells and neoplastically transformed or cancer cells was the absence of functional GJIC in the cancer cells. It was that observation, in large part influenced by the late Van R. Potter's concept that “oncogeny is partially-blocked ontogeny,” that stimulated our group 15 to try to isolate normal, contact-insensitive adult cells from human biopsied tissue. It was based on the assumption that if stem cells gave rise to cancer and cancers could be found in most organs, then there must have existed adult stem cells. In addition, these adult stem cells must not express the gap junction or connexin genes because it had been shown that gap junctions seemed to be required for development and differentiation. 58 Based on these assumptions and the Nakano observations, we were able to isolate normal human kidney adult stem cells. Later, we and others isolated human breast, pancreas, limbral, mesenchyme, intestine, skin, and liver adult stem cells. 42, 55, 56, 67, 68, 123 They all seemed to be characterized by the lack of expressed connexin genes or functional GJIC. 111
The converge of these studies and the studies on the use of “immortalizing viruses” happened when our group exposed normal human adult breast stem cells to SV40. What we found was that we could easily prevent the normal adult breast stem cell from differentiating (i.e., we blocked mortalization) and keep these cells in the immortal state. On the other hand, we were never able to obtain “immortal” or “rereprogrammed” cells when normal differentiated breast epithelial cells were treated with SV40. 98 In addition, with these nontumorigenic SV40-transformed adult stem cells, which were further treated with X-rays, weakly tumorigenic clones were obtained. 41 When these SV40, X-ray–treated, weakly tumorigenic cells were genetically modified with the ERB2/neu oncogene, they became highly tumorigenic.
In all the studies, using immortalizing viruses to obtain immortal human cells, one first treats an early passage of a primary explant of normal tissues. After the population is exposed to SV40 or human papilloma viruses, the population goes through “crisis.” A few clones of immortalized human cells can be obtained, usually at frequencies similar to mutation induction. It is thought that these immortalizing viruses either have caused an insertional mutation, turning on genes for immortalization, or have reprogrammed these normal mortal cells to the immortalized, stem-like state.
Another interpretation, based on the Nakano et al. observation and our observations of obtaining normal adult human stem cells, is that in these heterogeneous primary human cell cultures, there exist a few adult stem cells. It is likely that the viruses infected or transduced all the cells in the population, but in only the few adult stem cells did the viral genes prevent the differentiation of those already immortal normal stem cells. In effect, the immortalizing event might simply be the result of viruses blocking the mortalization of the adult stem cells in any tissue or cell population. If this interpretation is correct, then this probably provides the explanation for the association of cancer-associated viruses, such as papilloma, hepatitis, and herpesviruses, and even SV40 with human cancers. 129 In the human body, adult stem cells do exist. If these cancer-associated viruses infect these few adult stem cells, then they might be viewed as being initiated (not yet cancerous). However, they can be expanded by mitogenic stimulation, and some ultimately could give rise to a cancer. This probably explains the rationale to reduce the risk to cervical cancers with vaccines against the human papilloma virus.
Cancer Stem Cells: From Adult Stem Cells or Reprogrammed Somatic Differentiated Cells?
The recent excitement about the discovery of breast cancer stem cells and demonstration in several types of cancer stem cells in tumors and tumor cell lines‡ has stimulated much speculation to explain both the difficulties in cancer therapies and the emergence of tumor-resistant cells. 24 Starting from above, it is a fact that adult stem cells do exist. However, that does not prove that these cells are the target cells for carcinogenesis, let alone the origin of cancer stem cells.
One solid line of evidence that adult organ-specific stem cells exist is that of the isolated normal human breast stem cell. This normal human epithelial cell was isolated and shown to lack any connexin expression or functional gap junctions. It was estrogen receptor alpha positive, cytokeratin18 positive, epithelial membrane antigen positive, Cx43 negative, and capable of independent growth. It could be induced to differentiate and to change this battery of gene expression to Cx43, cytokeratin14 positive, and epithelial membrane antigen negative and have functional gap junctions. 14 Most dramatically, when pure cultures of the presumptive breast stem cell were mixed one-to-one with a pure culture of the stem cell's differentiated breast epithelial daughter cells, within 24 hours on Matrigel, a 3-dimensional ductal structure was formed (Fig. 4). In a matter of a few weeks, this structure was highly organized into a structure that mimicked those found in human breast tissue.
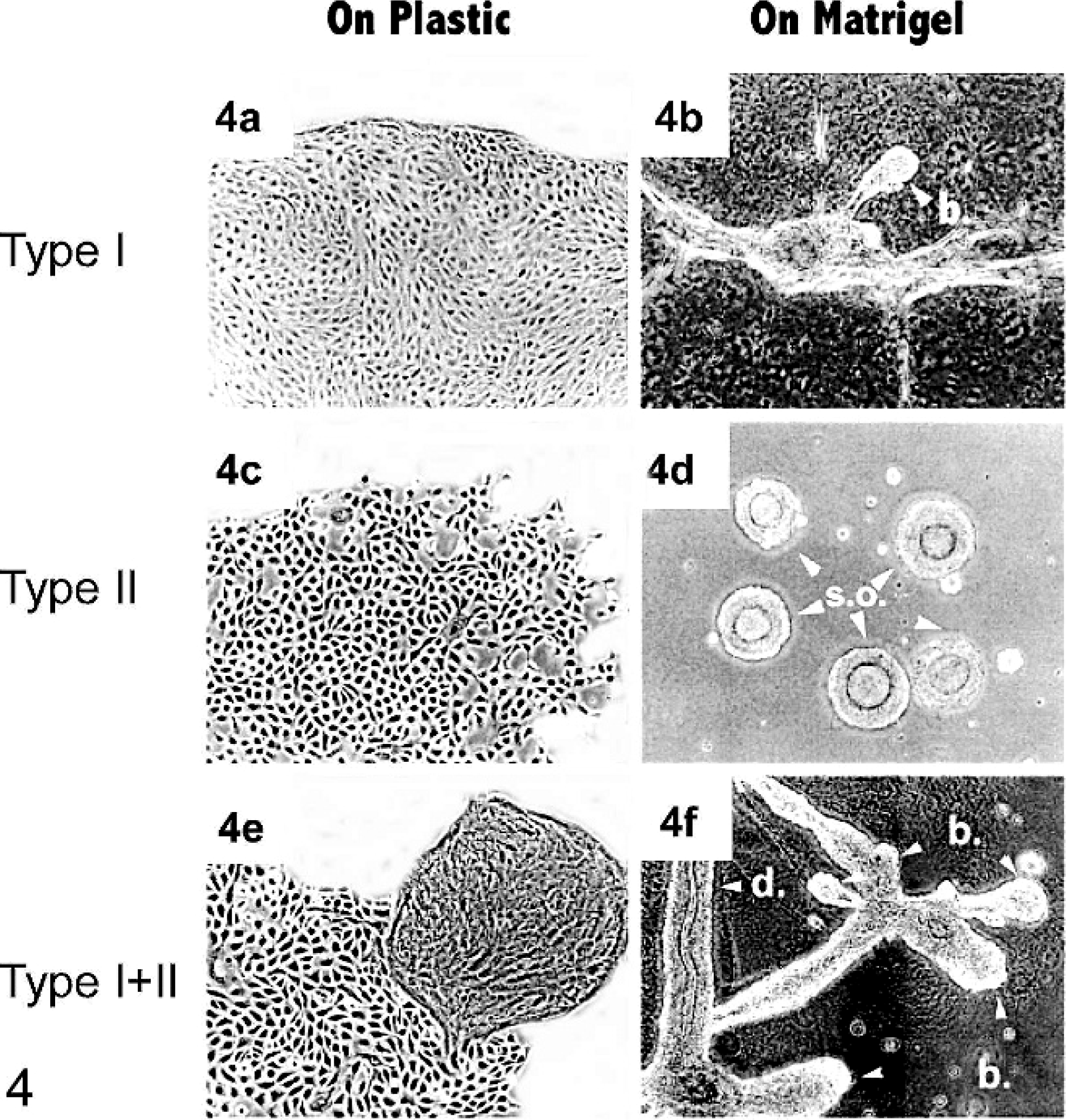
Human breast epithelial cell colonies on plastic and organoids on Matrigel formed by 2 types of normal human breast epithelial cells. Type I and type II colonies developed on plastic (a and c, respectively) are morphologically distinguishable. On Matrigel, type II cells typically formed spherical organoid (d), whereas type I cells formed a limited number of bud-like (b) structures and acini (not shown). The combination of type I and type II (E: 2 types of cells on plastic) in 1 : 2 or 1 : 3 ratios can generate many budding (b) ductal (d) structures in Matrigel (f) in 2–3 weeks. 98 .
At this point of trying to resolve either the stem cell hypothesis or the reprogramming of a differentiated somatic cell hypothesis, one must consider both the heterogeneous nature of the cells within the tumor and the concept of the cancer stem cells. How do either hypotheses deal with those issues? The question whether a differentiated somatic cell can be reprogrammed back to an embryonic state is of fundamental importance. Recently, 6 papers appeared, in which evidence was presented that suggests adult mouse, monkey, or human skin fibroblast cells could be reprogrammed to an embryonic-like state to form induced pluripotent stem cells. 11, 78, 81, 100, 101, 119, 125 The evidence includes, among other things, that the gene expression pattern resembled that of the normal mouse embryo rather than the adult mouse skin fibroblast. In addition, these cells were capable of giving rise to chimeric mice, some of which could pass on the original reprogrammed genome.
On the other hand, an alternative hypothesis can be offered to explain those amazing results of the reprogramming of adult somatic, differentiated skin fibroblasts to embryonic-like stem cells. An alternative explanation is that somatic skin stem cells exist in all mice, monkeys, and humans. If, by rare chance, these investigators transferred the induced pluripotent stem nuclei of the adult skin stem cells to enucleated eggs, then there was, essentially, little reprogramming, especially of the genes needed for immortalization. These adult stem cells would, by definition, be assumed to be immortal until induced to terminally differentiate. This is a major challenge to the current assumption that a normal differentiated cell can be reprogrammed all the way back, perfectly, to the embryonic stem cell state.
Do Rare Cancer Stem Cells Really Exist?
The discovery, that only a few cells within human breast cancer could give rise to perpetuating the tumor, 1, 84 was further supported by reports of cancer stem cells in other organs and tumor cell lines.§ The concept of cancer stem cells has now been used to explain why current cancer therapies are relatively ineffective. This led to the idea suggesting this new discovery would pave the way for new cancer therapies directed to these rare but important cancer stem cells. 24
However, a recent challenge to this idea suggested that the current manner of testing cancer stem cells is unable to rigorously examine if these rare cancer stem cells really do exist. 43 This report, using 3 independent lymphoma cell lines in an isogenic, nonirradiated host animal (to minimize genetic and other microenvironmental factors that might select against other cancer cells that also could perpetuate a tumor), showed that many cancer cells in these tumors could perpetuate the growth of tumors. These limited results do seem to challenge the rare cancer stem cell hypothesis. In addition, their call for more rigorous tests to be made when trying to identify these cancer stem cells is reasonable. However, there was evidence, not cited in that challenge paper, 3, 99, 117 which suggests that cancer stem cells do exist and they are not uniformly rare in all tumors.
That evidence for the existence of cancer stem cells comes, at present, from many sources: 1) leukaemia, for which an excellent review has been made; 104 2) examination of classic cancer cell lines, MCF-7 and HeLa; 99 3) isolation from solid tumors; 2, 3, 23, 27, 72, 117 and 4) the stem cell theory. The first solid tumor, derived from a human stem cell example, came from the isolation and characterization of normal human adult breast stem cells that could be prevented from mortalization and later neoplastically transformed. 14 These cells were shown to express Oct-4 both as in the normal stem cell and in immortalized but not in tumorigenic cells and later in weakly and highly metastatic cell lines (Fig. 5).
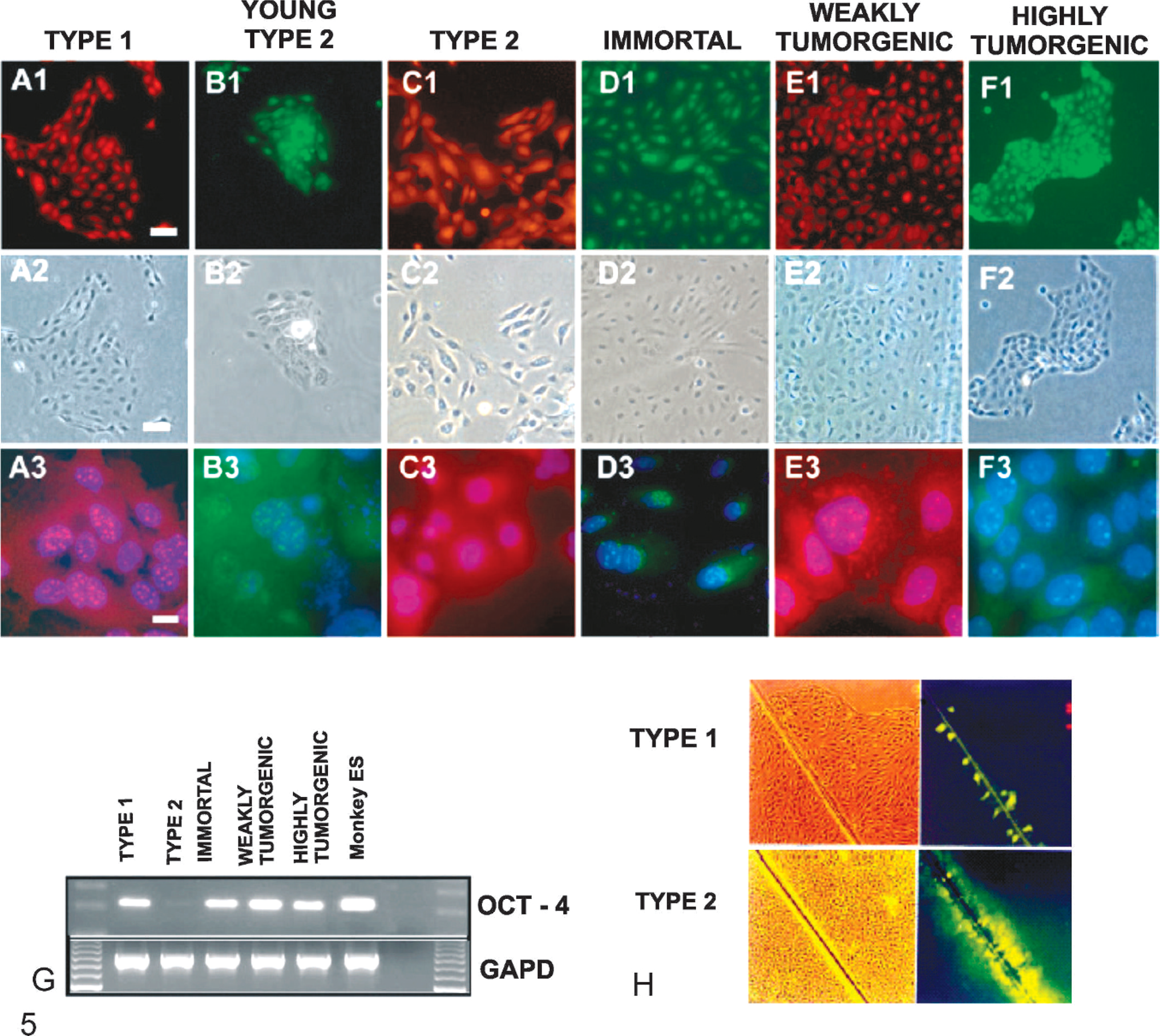
Expression of Oct-4 in human breast epithelial cells. Breast stem cells type I (A), differentiated daughter cells young and mature type II (B, C), SV40 “immortalized” breast stem cells type I (D), irradiated, weakly tumorigenic breast stem cells (E), neu/ErB-2 transduced highly tumorigenic cell line (F). Fluorescence for Oct-4 in green or red (A1–F1), phase contrast (A2–F2) and higher magnification images of A1–F1 superimposed on 4′,6′-diamidino-2-phenylindole dihydrochloride blue nuclear stain (A3–F3). Punctate staining of Oct4 was observed in the nucleus in most cell types, except for mature type II cells (C3). The reverse transcriptase polymerase chain reaction analysis of Oct-4 (G) correlated with the immunofluorescence data (A3–F3). Normal breast adult stem cells have no functional gap junctional intercellular communication (GJIC), whereas young and mature type II cells are efficient in GJIC (H), as measured by fluorescent scrape loading-dye transfer assay. 114 .
After this demonstration, another example came from identifying Oct-4–positive cells in 83 canine tumors from 21 different organ sites (Fig. 6). 117 Independently, another example came from the examination of 36 human bladder tumors to find 35 had Oct-4–positive cells in the tumors. 3 Furthermore, a validation was made of the existence of cells that were previously claimed to be breast cancer stem cells 84 and then demonstrated to have tumor-perpetuating activity, as well as expression of Oct-4. In the cases of solid tumors, the ratio of Oct-4–positive to Oct-4–negative cells within the tumors varied widely from tumor to tumor or cancer cell line to cancer cell line. 3, 117
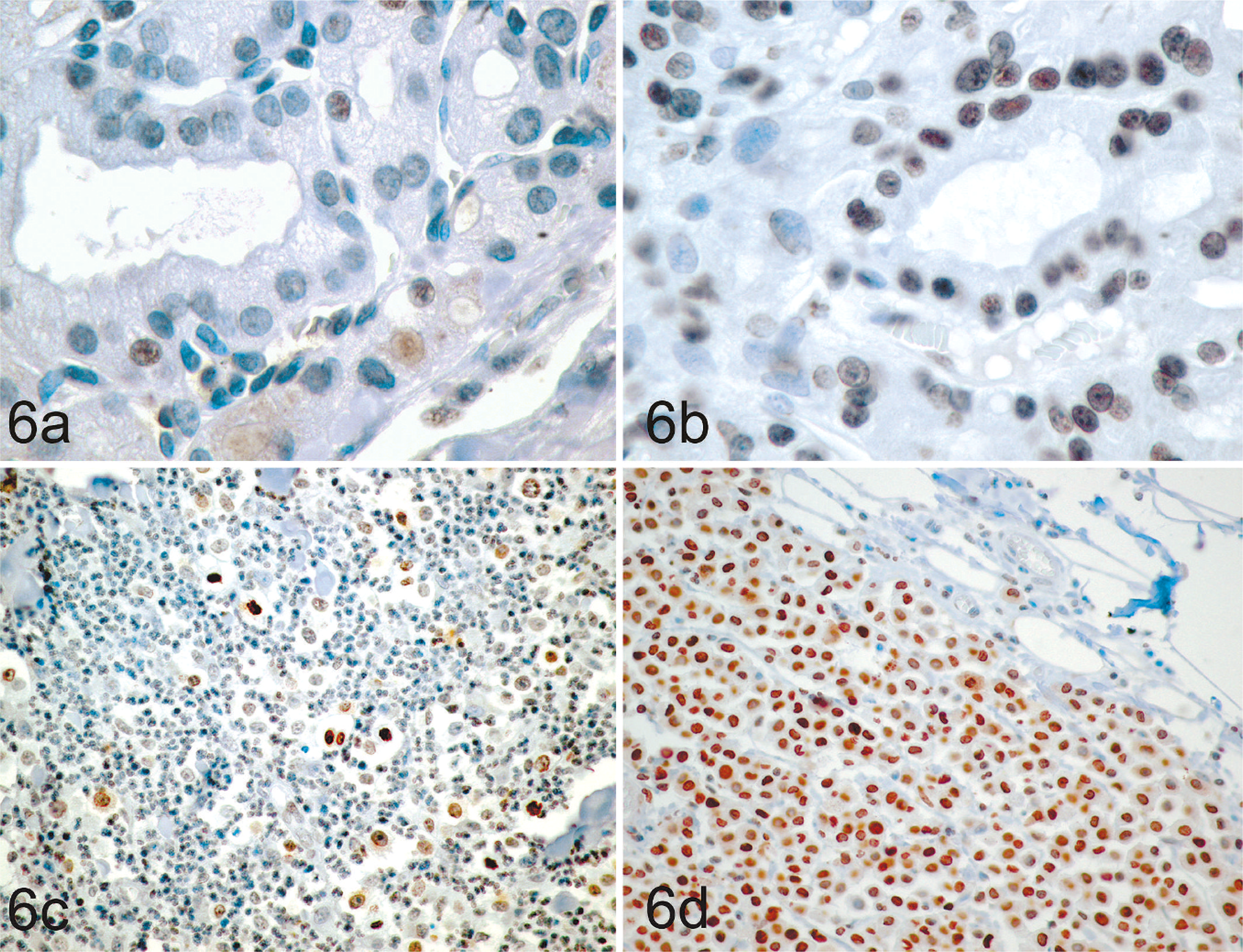
Oct4 expression in canine thyroid carcinoma (a, b) and mast cell tumors (c, d). Oct4 expression (brown nuclear staining) varied between individual tumors, with some tumors having relatively low proportions of Oct4-positive cells (a, c) and other tumors having relatively high proportions of Oct4-positive cells (b, d). Immunohistochemical staining with mouse anti-human Oct4 monoclonal antibody visualized with the substrate 3,3′-diaminobenzidine with hematoxylin counterstain. 117 .
The explanation for the wide variation of Oct-4–positive (cancer stem cells) and Oct-4–negative (cancer nonstem cells) is probably due to microenvironmental conditions. The Oct-4–positive cancer stem cells that normally divide asymmetrically can, if the microenvironment conditions change, divide symmetrically to 2 Oct-4–positive cells. This could be used as indirect evidence that the Oct-4–positive cell is a cancer stem cell because it can divide both symmetrically and asymmetrically.
As to the potential factors that cause the shift from the normal asymmetric division pattern of initiated and cancers stem cells, one must consider the holist picture of a tumor in a host (or tissue culture conditions of immortalized or cancer cell lines). As a tumor grows, the microenvironment within the tumor changes, which is caused by decreasing oxygen tension and blood nutrient availability. The tumor in the host can be influenced by the gender (hormone status), diet, environmental chemicals, and physiological state to produce factors that shift asymmetric cell division to symmetric cell division.
The implication of this kind of observation could lead to the use of changes in Oct-4 expression (positive or negative) in 3-dimensional organoids, produced by the use of cancer cell lines with cancer stem cells in their population to screen for either tumor promoters or chemoprevent/chemotherapeutic agents. 22, 57, 79, 96 The initiated cell (Oct-4 expressing) would normally divide symmetrically. However, as the population of cancer stem cells increases, the changed microenvironment induces asymmetric divisions of the cancer stem cells. This would lead to no increase of the initiated stem cell but to the production of partially differentiated cells. If, however, this population of a mixture of Oct-4–positive and Oct-4–negative cells is exposed to agents that stimulates symmetrical cell division and blocks asymmetrical cell division, the organoid or tumor would be expected to grow. An initiated population of Oct-4–positive cells should be evident in small promoted lesions. However, because the initiated population is expanded, the microenvironment changed. This could signal these symmetrically dividing cells to asymmetrically divide, thereby shutting off Oct-4–positive cells in some cells and allowing them to partially differentiate. The same could happen in the malignant cell, which is Oct-4 positive, such that, as this malignant population grows, some cells will symmetrically divide, whereas others would asymmetrically divide. The internal and external microenvironments to these cancer stem cells will determine the ratio of Oct-4–positive to Oct-4–negative cells in a tumor or cancer cell line in culture.
That leads, finally, to the understanding of primary cell strains and immortal cell lines in culture. The primary cell cultures, most likely, contain a few normal adult stem cells. However, under traditional culture conditions, the culture media and atmospheric conditions (high oxygen content), 20 mitigate against continued expansion of the few adult stem cells, causing a dilution of their frequency upon passages. On the other hand, if tumors contain both cancer stem cells and cancer nonstem cells, the existence of cancer stem cells that resist apoptosis and terminal differentiation would be able to sustain the growth of the culture. These cultures would always be heterogeneous, because there would be partial differentiation of these cancer stem cells, as was shown with Oct-4–positive and Oct-4–negative cells in both HeLa and MCF-7 cancer cell lines. 99 Therefore, if cancer stem cells are relatively rare in breast tumors, it might be expected that starting breast cancer cell lines with immortal growth in vitro would be difficult. 84
Is Oct-4 a Marker for Embryonic, Adult, and Cancer Stem Cells?
The use of Oct-4 as a marker for stemness of embryonic stem cells seems very solid, as those early studies of its absolute requirement seem to demonstrate. 74, 75 Oct-4 has been demonstrated in several adult normal and cancer tissues, in adult human and canine tissues, in human cancer cell lines, and in normal adult human stem cells. A serious challenge has been made that questions whether Oct-4 can be a marker for cancer stem cells. 7, 13, 50, 54 Multiple Oct-4 pseudogenes and multiple isoforms of the normal Oct-4 gene have also been reported. The reports of negative identification of the existence of the expression of Oct-4 in many tumors have created a dilemma in the current phase of the science to resolve the issue of a cancer stem cell marker.
The critical point here is not that adult stem cells do not exist. They have been identified in both animal and human somatic tissues. In addition, it is not that these adult stem cells can be targets for carcinogenesis, because it has been dramatically demonstrated that normal adult human breast stem cells can be neoplastically transformed. 14 In fact, the negative results and criticisms against Oct-4 as a marker for adult stem cells or cancer stem cells do not seem strictly relevant to those studies on adult stem cells where Oct-4 has been detected. In those cases, unlike all the negative studies, Oct-4 was expressed in cells that were isolated from adult tissues. These cells were characterized as being stem cells because they could divide both symmetrically and asymmetrically and could differentiate into the cell types of the tissues from which they were derived. In fact, they were first characterized by another marker, that is, the absence of the gap junction gene and GJIC function. 111 It was only after these cells were shown to have stem cell characteristics that they were characterized for their expression of Oct-4.
Therefore, although it remains that a rational explanation must be demonstrated for these apparent discrepant results, the fundamental issue at hand is whether adult stem cells, which do exist in most, if not all, somatic tissues, are target cells for cancers and the cancer stem cells. Alternatively, might somatic differentiated cells dedifferentiate or be reprogrammed to the immortalizing state of an embryonic-like stem cell to initiate the carcinogenic process?
This last point seems to provide a clue (at least from the standpoint of using “Occam's razor” to decide which of multiple hypotheses might be scientifically the one to choose) as to the explanation of the multistage hypothesis of carcinogenesis. If a somatic differentiated cell is the target cell for the initiation phase, one must invoke an extra mechanistic step to explain the initiation step. That extra step would be needed to reprogram a somatic differentiated cell, destined to senesce via the Hayflick phenomenon, 36 so that it now regains the gene expression needed for immortality. Before this cell can start the initiation phase, it must be blocked in its ability to terminally differentiate to divide asymmetrically. This, of course, is exactly the prevailing paradigm of the cancer research field, namely, one must first immortalize the mortal, normal somatic cell before it can become neoplastically transformed. 49
On the other hand, if that somatic adult stem cell, which is naturally immortal, is now blocked from mortalizing by the initiator, this hypothesis eliminates one additional step to start the carcinogenic process. Occam's razor would favor this later hypothesis.
Other Stem Cell Markers That Might Be Related to the Practical Problem of Cancer Treatment for Both Animal and Human Tumors
If we accept, for the moment, that the concept of cancer stem cells is real and related to those cells within a tumor that are responsible for the perpetuation of the growth of the tumor, then the current explanation of the relative nonsuccessful treatment of many tumors is that the current treatments were never designed to knock out the cancer stem cells. 24, 69, 128 More attention has been paid to the targeting the cancer stem cells. Through use of the analogy of a beehive, the most efficacious manner to destroy a beehive would be to kill the queen bee. The most efficacious manner to knock out a tumor would be to target the cancer stem cell, the queen bee of a tumor.
One of the interesting historical reasons for the failure of many cancer chemotherapeutic drugs was that they damaged DNA, induced mutations, and induced the multidrug resistance of the tumor cells that survived the chemotherapy. A newer interpretation of multidrug resistance of tumors is that the cancer stem cell is naturally multidrug resistant. More evidence linking stem cell biology, evolution, cancer stem cells, and multidrug resistance comes from the observations that if cells of a tumor are treated with a fluorescent toxicant and then passed through a cell sorter, 2 populations are found. The majority population consists of fluorescent tumor cells and a small “side population” of nonfluorescing cells. 32, 40, 44, 121, 127 These side population cells have been shown in many cases to have “stem-like” properties.
An evolutionary explanation for this association might have resulted from the early need for a stem cell to be less sensitive to an environmental toxicant than the differentiated derivatives of this stem cell. If both the adult stem cell and their differentiated daughters within an organ were equally sensitive to a toxicant, the organ would not survive, jeopardizing the survival of the organism. Therefore, the normal stem cell probably expresses these drug-resistant genes. If these normal adult stem cells, which express Oct-4 for stemness, do not express functional gap junctions or connexin genes to prevent differentiation but do express drug resistance (drug transporter genes resistant to toxicants), then the initiation of these adult stem cells would be expected to maintain these gene patterns. They might be expected to be maintained in the precancer stem cell and the cancer stem cells. As these cancer stem cells proliferate, they change the tumor microenvironment, allowing some of these Oct-4–positive, connexin-negative, and drug transporter–positive cells to partially differentiate to the Oct-4–negative, connexin gene–expressing, and drug transporter–negative cancer nonstem cells. It would be these cells that might respond to some toxic chemotherapeutic drug.
It should be noted that although tumor cells are characterized by their inability to have functional GJIC, 2 types of tumors exist. One type does not express any connexins, whereas the other non-GJIC tumor cell does express their connexin genes and have proteins that are rendered nonfunctional by oncogenes. 106
It should also be stressed that single-cell organisms do not contain the connexin genes. However, the early metazoan, a social collection of cells, adapted new phenotypes to survive via capacities other than cell proliferation. These new phenotypes included 1) growth control, 2) cell differentiation to produce specified survival-related cell types (e.g., nerves, muscles, optical), 3) apoptosis to remove cells needed at one stage of development but not at others, and 4) germ cells to maintain the genome of the species. The connexin genes are a highly evolutionary conserved family of genes that code for a membrane-associated protein channel to allow ions and small–molecular weight molecules that could be transported, selectively and directly, from the cytoplasm of one cell to the surrounding coupled cell. 26 Interestingly, cancer cells, which have no growth control and cannot terminally differentiate or apoptose under normal conditions, do not have functional GJIC. 112 Stem cells must maintain their nondifferentiated and unlimited proliferated or immortal status. In those adult stem cells that have been analyzed, no GJIC was detected. 111
It has also been demonstrated that tumor-promoting agents and conditions can reversibly block GJIC in progenitor cells (inflammatory microorganisms, cytokines, chemical tumor promoters, wound healing, cell removal or cell death, growth factors, and certain hormones). 114 On the other hand, agents such as dietary antioxidants and other factors that increase GJIC or prevent downregulation by tumor-promoting agents seem to be chemopreventing agents. 97, 108, 113 There is now even evidence that some chemotherapeutic agents can either transcriptionally turn on connexin genes in cancer cells or inhibit oncogene-inhibited connexin proteins in various cancer cells. 108, 113
Therefore, if this hypothesis that cancer stem cells express Oct-4, drug-resistant transporter genes and do not express connexins is correct, then strategies 1) to turn off, transcriptionally, the Oct-4 gene; 2) to turn on, transcriptionally, the connexin genes; and 3) to inhibit the drug transporter proteins might work to cause the cancer stem cells to differentiate or apoptose. On the other hand, the current strategy to target, specifically, the oncogene product might render the cancer nonstem cell to re-establish its ability to have functional GJIC and to regain growth control and differentiate.
The strategy of chemoprevention would have to take into account the 2 types of initiated cells, namely, those that have not expressed their connexin genes and those that have but that still express the Oct-4 gene. Any chemopreventive agent that might induce transcriptional suppression of the Oct-4 gene or connexin transcription in the initiated premalignant stem cell would stop its growth. On the other hand, prevention of either secreted tumor promoters or inhibitors of GJIC of initiation stem cells might restrict the growth of these cells. 106 Of course, the exact same potential theoretical problem exists for this strategy as it does for current cancer therapies, namely, any treatment that might cause premalignant and malignant adult stem cells would be expected to affect the normal stem cells. Therefore, the problem of targeting only the premalignant and malignant stem cell must be solved.
Therefore, at this stage of our understanding of carcinogenesis, the scientific information does not yet exist to resolve the issue as to what is (are) the target cell(s) that give rise to cancer and the cancer stem cell in either animal or human carcinogenesis. It seems that comparative analyses of animal and human carcinogenesis, from classic experimental cancer bioassays to human epidemiology, as well as cross-disciplinary studies, using common molecular tools and concepts, could set the stage for a new approach to cancer prevention and treatment.
Footnotes
Acknowledgements
I was not supported by any research grant during the writing of the review. In addition, I have no financial holdings or obligations to any pharmaceutical or chemical chemopreventive/chemotherapeutic agents mentioned in this review. I acknowledge the academic input over the years of all my students, postdoctoral fellows, visiting scholars, colleagues, and coworkers who have, in multiple ways, contributed to both the scientific research findings and the generation of ideas that found their way into this review. In addition, I wish to acknowledge my late mentor's (Dr. Van R. Potter) contribution to having me think in a more “systems” fashion, rather than in the reductionist's way that I received during my formal scientific training. Lastly, I thank my good friend, Dr. Jay Goodman, who, for decades, challenged my thinking with our friendly discourses during our lunches.