
Abstract
Thyroid carcinomas are the most common endocrine neoplasms in humans, with a globally increasing incidence. Thyroid follicular cells and neuroendocrine (parafollicular) C cells are each susceptible to neoplastic transformation, resulting in thyroid cancers of differing phenotypes with unique associated genetic mutations and clinical outcomes. Over the past 15 years, several sophisticated genetically engineered mouse models of thyroid cancer have been created to further our understanding of the genetic events leading to thyroid carcinogenesis in vivo. The most significant mouse models of papillary, follicular, anaplastic, and medullary thyroid carcinoma are highlighted, with particular emphasis on the relationship between the relevant oncogenes in these models and genetic events in the naturally occurring human disease. Limitations of each model are presented, and the need for additional models to better recapitulate certain aspects of the human disease is discussed.
Thyroid carcinomas are the most common endocrine neoplasms in humans, affecting approximately 1% of the population. 77 Roughly 95% of all thyroid tumors are of thyroid follicular epithelial cell origin, including papillary, follicular, and anaplastic thyroid carcinomas. The remaining 5% are medullary thyroid carcinomas of C cell origin. 17, 77 The subclassification of thyroid cancers into these 4 categories is clinically significant. Papillary thyroid carcinomas metastasize via lymphatics to local lymph nodes in an estimated 50% of cases but have the most favorable prognosis, with a 98% 10-year survival rate. Follicular and tall cell variants of papillary thyroid carcinoma are associated with poorer prognoses. Follicular thyroid carcinomas are more prevalent in areas of dietary iodine deficiency, metastasize hematogenously, and are less likely than papillary thyroid carcinomas to take up radioactive iodide for imaging and therapeutic ablation. However, the 10-year survival rate for follicular thyroid carcinomas is still high at 92%. Anaplastic thyroid carcinomas are almost invariably fatal as a result of rapid invasion of critical structures in the neck, distant metastases, and a failure to take up radioactive iodide. In many cases, anaplastic thyroid carcinomas arise from dedifferentiation of follicular or papillary carcinomas or in patients with a history of multinodular goiter. 17
Medullary thyroid carcinomas, also commonly known as C-cell carcinomas, arise from calcitonin-secreting C cells and are associated with inherited syndromes, such as multiple endocrine neoplasia (MEN), in approximately 20%–25% of cases. 17, 77 Medullary carcinomas frequently metastasize via the bloodstream, in addition to lymphatic spread, and are treated with surgical resection and/or external beam radiation. In contrast to follicular-origin thyroid tumors, C cells and tumors arising from them do not have the ability to take up radioactive iodide. The 5-year survival rate for medullary thyroid carcinomas is approximately 50%. According to the National Cancer Institute Surveillance Epidemiology and End Results (SEER) data, as well as other studies, thyroid cancer is one of the few cancer types with increasing incidence in the United States and around the globe, particularly among women. 76
Genetic Events in Thyroid Tumors of Follicular Cell Origin
Thyroid follicular cells are responsible for iodide uptake and thyroid hormone synthesis and can undergo neoplastic transformation to carcinomas of 3 histotypes: papillary, follicular, and anaplastic. It is well-known that papillary thyroid carcinomas can occur secondary to ionizing radiation exposure, particularly in children.
6, 53, 77
After the Chernobyl nuclear reactor accident in 1986, the incidence of thyroid carcinomas in children in affected areas of Belarus increased from less than 1 per million to more than 90 per million.
10
The primary known molecular mechanism of radiation-induced papillary carcinoma development is through the Ret/PTC group of oncogenes.
Sporadic papillary thyroid carcinomas unrelated to radiation exposure make up more than two thirds of all cases, and several genetic events have been identified as important in their tumorigenesis. 54 A form of the B-type Raf kinase, or BRAF, with a point mutation resulting in V600E has been identified in approximately 45% of sporadic papillary carcinomas, particularly the tall-cell variant. 92 BRAF expression has been associated with dedifferentiation and disease progression. NTRK1 is a receptor tyrosine kinase normally involved in nerve growth factor signaling. 83 Like Ret/PTC rearrangements, NTRK1 can recombine with the 5′ end of other heterologous genes and form a constitutively active oncogene, such as TRK-T1, leading to papillary carcinomas. However, NTRK1 rearrangements are less frequent than Ret/PTC rearrangements and are not associated with radiation exposure. Finally, chromosomal rearrangement of the PTEN tumor-suppressor gene leading to its loss of function has been recently associated with papillary carcinoma formation. 60
Ras mutations have been implicated in the development of follicular thyroid carcinomas, particularly N-ras activating mutations at codon 61. 54, 86,89 Expression of a Pax8-PPARγ fusion protein has been detected in a small number of follicular carcinomas and follicular variants of papillary carcinomas. 42, 43 While controversy exists over the ability of chronic thyroid stimulating hormone (TSH) stimulation to result in thyroid tumorigenesis, follicular carcinomas are most prevalent in iodine-deficient regions, suggesting that TSH stimulation may play a role. 24, 45, 68, 89 An elevation in the risk of thyroid tumorigenesis in patients with certain types of goiter has been reported, 24, 50, 67, 85 although there is a lack of consensus regarding this association.
Loss of p53 function is the primary genetic alteration identified in the development of anaplastic thyroid carcinomas, 30, 54, 63 although BRAF and Ret/PTC3 mutations have been associated with some degree of anaplasia in papillary carcinomas. 54, 92 β-catenin activating mutations have also been detected in anaplastic thyroid carcinomas, where the protein likely mediates loss of cell-cell adhesions and also acts as a transcription factor in upregulating growth-promoting genes. 25
Genetic Events in Thyroid Tumors of C Cell Origin
Medullary thyroid carcinomas, or C-cell carcinomas, arise from the calcitonin-secreting parafollicular C cells of the thyroid gland. These tumors can occur as part of an inherited syndrome, such as MEN type 2A or type 2B or familial medullary thyroid carcinoma, which together account for 20%–25% of all medullary thyroid carcinomas.
17,
61, 77
The
Animal Models of Thyroid Cancer
Thyroid cancer arises as a spontaneous disease in domestic animal species, particularly follicular adenomas in cats, solid-to-follicular carcinomas in dogs, and C-cell (ultimobranchial) carcinomas in bulls. 5, 9, 64 However, the incidence of tumorigenesis in these species is too low for feasible use as experimental models of the human disease, and the genotypic similarities to the human disease remain unclear.
Many xenobiotics result in thyroid tumors of follicular cell origin in laboratory mice and rats, generally by lowering thyroxine (T4) levels, leading to an increase in circulating TSH and thyroid follicular cell proliferation. 9 Some of the implicated compounds include hepatic Cyp2B inducers, thyroperoxidase and 5′-deiodinase inhibitors, and inhibitors of the sodium/iodide symporter. 8 This pathway of thyroid tumorigenesis is believed to be much less important in humans than in rodents, as the half-life of thyroid hormones is 5–18 times greater in humans than in rodents because of the presence of the transport protein thyroxine-binding globulin in circulation. 8 Thus, the shorter half-life of thyroid hormones in rodents results in greater ease of disruption of thyroid hormone homeostasis. In the B6C3F1 laboratory mouse strain, which is commonly used in long-term toxicity studies, thyroid tumorigenesis occurs in 1%–10% of females and 0.8% of males by age 24 months (Charles River Laboratories, www.criver.com). However, the incidence rate and distribution of tumor types make these spontaneous models insufficient for use in mechanistic studies of thyroid cancer.
The majority of published studies utilizing mouse models of thyroid cancer have involved xenografts of human thyroid tumor tissue or immortalized thyroid carcinoma cell lines into severe combined immunodeficient (SCID) mice. SCID mouse tumor xenograft models have several drawbacks, including tumor implantation into an artificial microenvironment, rare success of metastasis from a subcutaneous location, lack of normal B- and T-cell function for evaluation of immune components of disease, and frequent onset of multiple spontaneous neoplasms. 2 Attempts to address these drawbacks in thyroid models have included orthotopic implantation of a thyroid carcinoma cell line into the mouse thyroid gland, 20, 37 selection of an anaplastic thyroid carcinoma cell line for pulmonary metastatic ability by serially passaging through nude mice, 97 and implantation of human peripheral blood lymphocytes into a SCID mouse model of thyroid carcinoma to confer partial immune competence. 28 The most commonly used cell lines in SCID mouse models of thyroid cancer include papillary thyroid carcinoma cell lines and NPA; 16, 56 follicular thyroid carcinoma cell lines FTC-133, FTC-238, RTC-R2 and FRO; 26, 74, 79, 84 anaplastic thyroid carcinoma cell lines ARO, DRO, WRO, KAT-4, and Thena; 3, 11, 20, 31, 32, 57, 72, 78, 87, 88, 94, 95, 97 and medullary thyroid carcinoma cell lines TT and rMTC. 4, 21, 41, 51, 62, 80, 81, 93
Genetically Engineered Mouse Models of Thyroid Cancer
Genetically engineered mouse models of thyroid cancer facilitate analysis of the roles of specific genetic mutations in thyroid tumorigenesis. Because thyroid cancer truly encompasses several diseases with different etiologies and relevant genetic mutations, no single transgenic mouse model of thyroid cancer can fully recapitulate the full spectrum of disease. However, several models have successfully reproduced various aspects and have offered insight into genetic mutations underlying thyroid tumorigenesis. These mouse models of thyroid cancer offer examples of positive genotype-phenotype correlation.
Over the past decade, several genetically engineered mouse models of thyroid cancer have been utilized to replicate variants of the human disease (Tables 1, 2). In most cases, transgenic mice have been produced using the highly active bovine thyroglobulin (Tg) promoter to specifically target transgene expression to thyroid follicular cells or using the human or rat calcitonin/calcitonin gene-related peptide (CGRP) promoter to target transgene expression to C cells. In some cases, tumor suppressor gene knockout mice have been created and cross-bred with transgenic mice expressing thyroid-specific oncogenes, resulting in increased tumorigenesis and/or an aggressive phenotype.
Summary of genetically engineered mouse models of thyroid cancer.
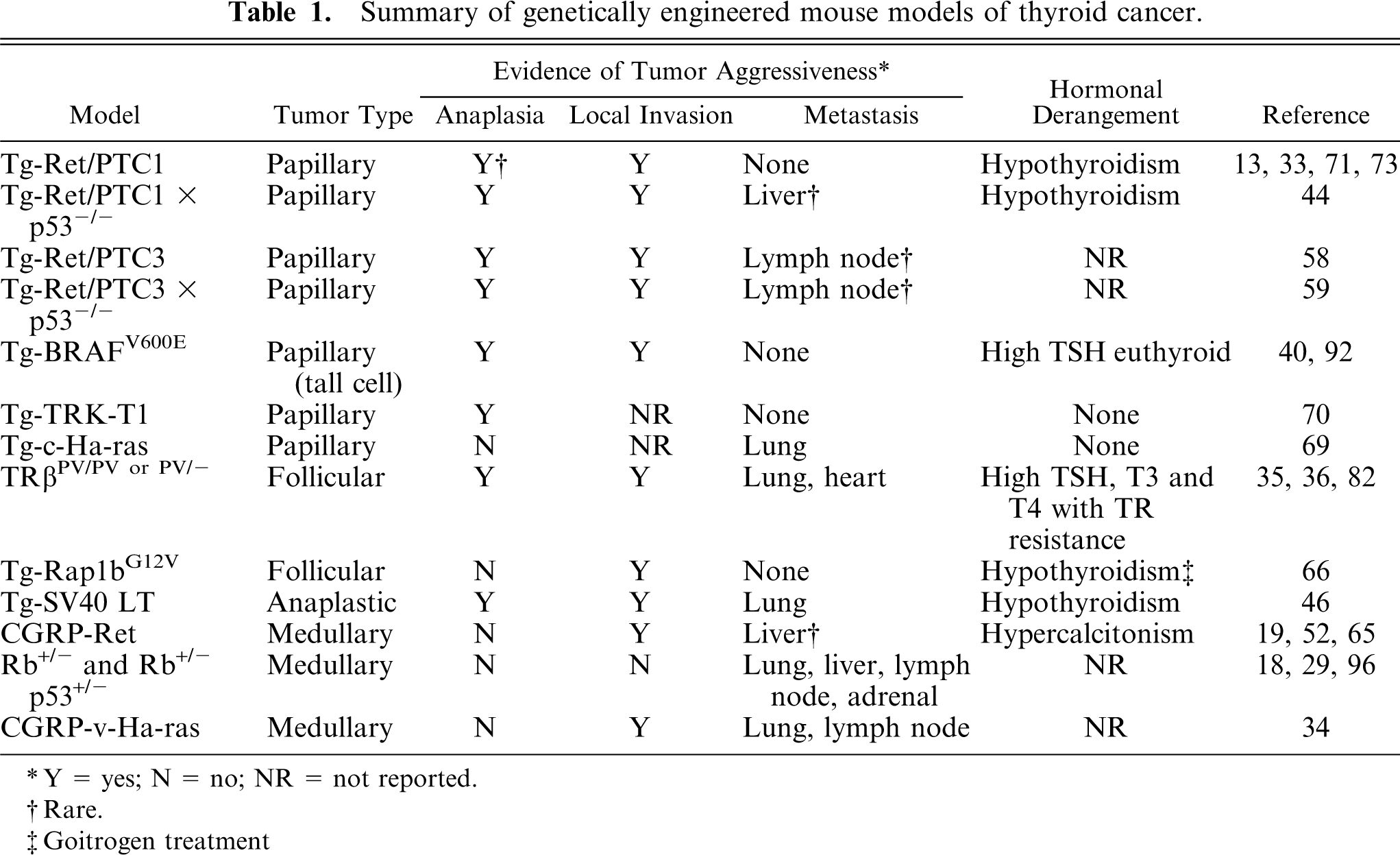

∗ Y = yes; N = no; NR = not reported.
† Rare.
‡ Goitrogen treatment
Comparison of human thyroid tumors and genetically engineered mouse models of thyroid cancer with similar genetic mutations.

PTC = papillary thyroid carcinoma; FTC = follicular thyroid carcinoma; ATC = anaplastic thyroid carcinoma; MTC = medullary thyroid carcinoma.
NR = not reported.
hereditary cases (e.g., MEN 2A).
Mouse Models of Papillary Thyroid Carcinoma
Tg-Ret/PTC1
FVB/N strain transgenic mice expressing thyroid-targeted Ret/PTC1 tyrosine kinase consistently developed bilateral papillary thyroid carcinomas with similar histopathologic features to the human disease, including papillary folds, ground-glass nuclei, and nuclear invaginations (Figs. 1–3). 33 Tumorigenesis was abolished when all 3 critical phosphotyrosine residues involved in signaling pathways mediated by Ret/PTC1 were mutated, but not when single residues were mutated, although tumor incidence was decreased. 7 Thus, signaling pathways mediated by each single phosphotyrosine residue were not solely essential for tumor development, but all 3 worked in concert to induce tumorigenesis. Like the human disease, these tumors accumulated less radioactive iodide than did normal thyroid tissue. The severity of disease varied markedly between high- and low-copy founder lines. High-copy lines had dysplastic thyroid glands at birth and developed carcinomas as young as 4 days of age, as compared with 1–6 months of age in the low-copy line. 13, 71
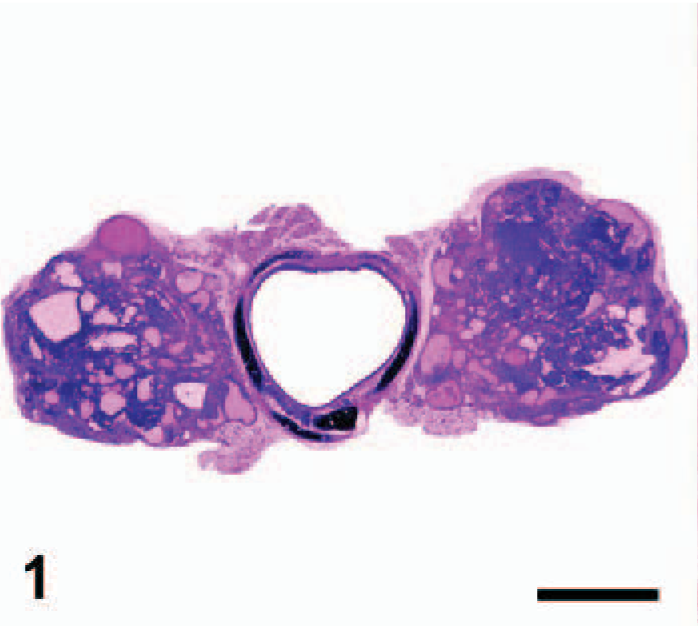
Thyroid gland; Ret/PTC1 transgenic mouse. Well-demarcated bilateral papillary thyroid carcinomas are present. Remaining non-neoplastic thyroid gland has large, quiescent follicles due to dietary L-thyroxine supplementation. HE; Bar = 1 mm. Tissue courtesy of Dr. Je-Yoel Cho.
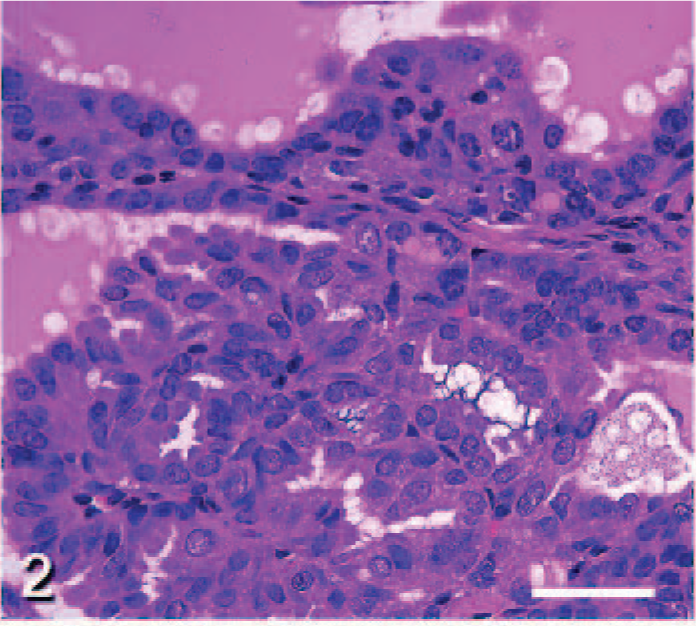
Thyroid gland; Ret/PTC1 transgenic mouse. Higher magnification of a well-differentiated papillary thyroid carcinoma, illustrating the fronds of neoplastic cells extending into colloid-filled follicles. HE; Bar = 50 μm. Tissue courtesy of Dr. Je-Yoel Cho.
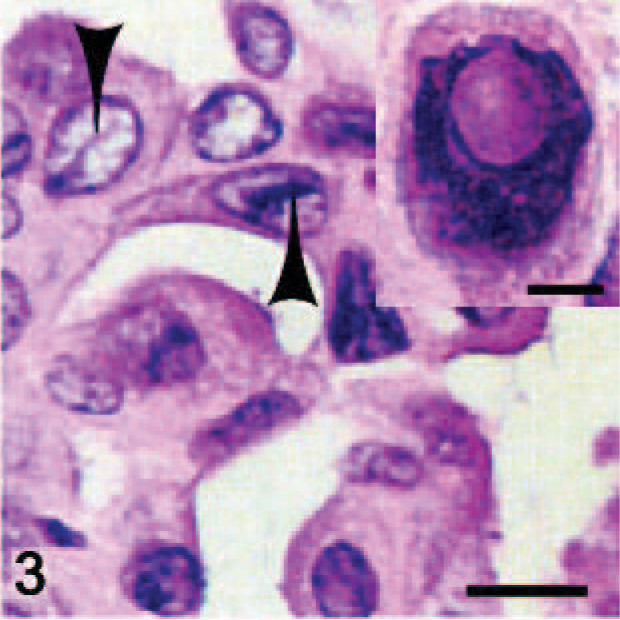
Thyroid gland; Ret/PTC1 transgenic mouse. High-magnification thin section of a papillary thyroid carcinoma demonstrating the nuclear invaginations (arrowheads) and ground-glass nuclei (
Mice from the high-copy line were profoundly hypothyroid, as indicated by low serum T4 and triiodothyronine (T3) levels, dwarfism, infertility, paraphimosis, and hyperplasia of TSH-secreting pituitary thyrotrophs. Hypothyroidism likely resulted from either a dedifferentiating effect of the Ret/PTC1 oncogene on thyroid follicular cells or reduced thyroglobulin synthesis due to competition for transcription factors between the endogenous Tg promoter and the highly active bovine Tg promoter used to drive transgene expression. Maintaining the mice on a low iodide diet resulted in persistent elevation of TSH levels and enhanced tumorigenesis. 71 The elevation of TSH levels and TSH responsiveness of the Tg-Ret/PTC1 transgene is a drawback to this model, as chronic TSH stimulation is not believed to be involved in papillary carcinoma development in humans. 90 The lack of any metastases is also a limitation to this model, although cross-breeding Tg-Ret/PTC1 mice with p53-/- mice did result in rare metastases, in addition to a more anaplastic phenotype, larger primary tumor size, and enhanced local invasion (Figs. 4–6). 44 When a second research group independently created Ret/PTC1-expressing mice using the less active rat thyroglobulin promoter and a C57BL/6J background strain, less than half of the mice developed tumors after a long latent period (8–16 months). 73
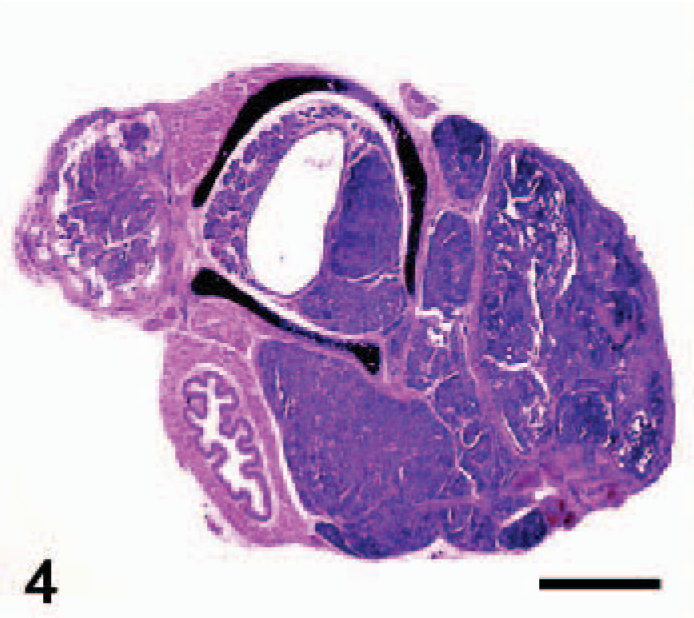
Thyroid gland; Ret/PTC1 transgenic × p53-/- mouse. Bilateral papillary thyroid carcinomas of large size have replaced all normal thyroid tissue and invaded the adjacent trachea. HE; Bar = 1 mm. Photo reprinted from La Perle et al: Am J Pathol
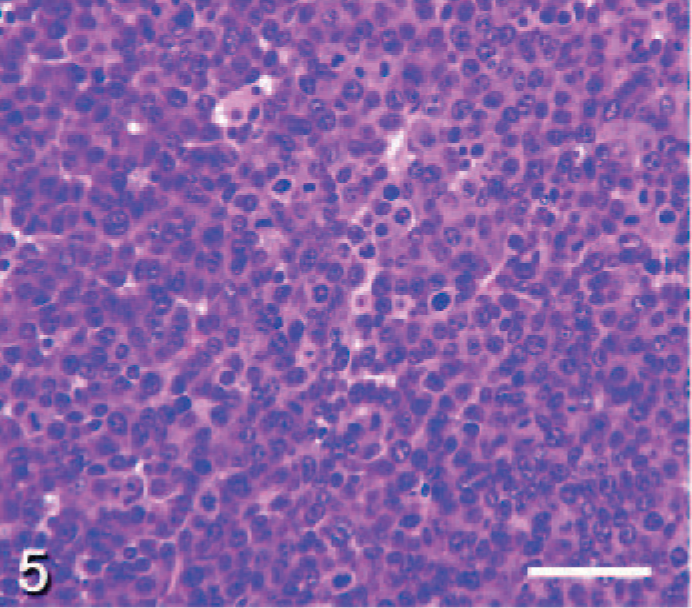
Thyroid gland; Ret/PTC1 transgenic × p53-/- mouse. Higher magnification of a poorly differentiated papillary thyroid carcinoma with multiple mitotic figures. HE; Bar = 50 μm. Photo reprinted from La Perle et al: Am J Pathol
Liver; Ret/PTC1 transgenic × p53-/- mouse. Two metastatic papillary thyroid carcinomas are present within the hepatic parenchyma (
Tg-Ret/PTC3
Thyroid-targeted Ret/PTC3 tyrosine kinase expression led to the follicular cell hyperplasia and development of a solid variant of papillary thyroid carcinoma in almost all C3H/He strain transgenic mice by the age of 6 months. 58 Metastases to cervical lymph nodes did occur in this model, albeit rarely (less than 10%). Ret/PTC3–induced thyroid tumors did not dedifferentiate into the more aggressive phenotype often seen in human patients with Ret/PTC3 chromosomal rearrangements. Cross-breeding Tg-Ret/PTC3 transgenic mice with p53-/- mice resulted in earlier onset of tumorigenesis, with microcarcinoma formation by the age of 3 months and the development of primary tumors up to 285 times the mass of wild-type mouse thyroid glands. 59 However, metastases were still rare in the p53-/- crosses, and progression did not occur to an anaplastic phenotype.
Tg-BRAFV600E
Transgenic mice of the FVB/N strain with thyroid targeted BRAFV600E serine/threonine kinase expression developed multifocal bilateral papillary thyroid carcinomas between the ages of 12 and 22 weeks. 40, 92 Features of the human tall cell variant, which is characterized by tall columnar cells with intensely eosinophilic cytoplasm, 17 were present in approximately half of the tumors. Foci of anaplasia were noted arising within and adjacent to well-differentiated papillary tumors, although there was no evidence of the necrosis commonly seen in human anaplastic carcinomas. In a high-expression transgenic line, tumors frequently invaded the surrounding fibrovascular tissue, sometimes leading to tracheal compression, but did not metastasize. Similar to the high-copy Tg-Ret/PTC1 mice, high-expression Tg-BRAF mice developed thyroid hormone derangements at a young age. However, in contrast to the Tg-Ret/PTC1 model, Tg-BRAF mice remained euthyroid and developed large goiters as a compensatory response to the high circulating TSH levels.
Tg-TRK-T1
More than 50% of transgenic mice of the B6C3F1 strain with thyroid-targeted TRK-T1 tyrosine kinase expression developed follicular cell hyperplasia, which often progressed to papillary thyroid carcinomas in animals over 7 months of age. 70 While tumors were composed of the papillae found in the human tumor, they lacked the classic nuclear clearing typical of papillary carcinomas. Few thyroid tumors in TRK-T1 mice developed the less-differentiated solid regions, and none of the tumors metastasized. It is likely that while TRK-T1 predisposed mice to develop papillary thyroid carcinomas, additional genetic mutations were required for transformation based upon the variability of tumor formation even within the same founder line.
Tg-c-Ha-ras
Transgenic mice expressing thyroid-targeted c-Ha-ras had a predisposition to developing papillary thyroid carcinomas. 23, 69 Two founder mice developed the disease at 10–12 months of age but did not transmit the transgene to offspring. A third founder mouse and 90% of progeny developed follicular cell hyperplasia, but not neoplasia. The fourth founder, which carried 20–30 copies of the ras transgene, developed papillary thyroid carcinoma with lung metastases at 9 months of age, although a thyroid origin of the lung tumors was not definitively established. When this founder, a C57BL/6J.DBA/2J strain, was backcrossed with DBA2J mice, all of the offspring had thyroid dysgenesis and early lethality. Backcrossing with C57BL/6J mice resulted in a normal phenotype. Thus, while the founder mice demonstrated papillary thyroid carcinomas, this phenotype could not be maintained in further generations.
Other models
Tumors with pathologic features of papillary thyroid carcinoma have been described in transgenic mice expressing thyroid-targeted human papillomavirus type 16 E7 oncogene, which inhibits retinoblastoma (Rb) function, 47 or the adenosine A2β receptor, which stimulates cAMP signaling. 14, 15, 48 Coexpression of both transgenes enhanced tumorigenesis. In a mouse model of Carney complex tumor syndrome, a disease involving multiple endocrine and non-endocrine neoplasms, heterozygous knockout of the R1A regulatory subunit of protein kinase A led to papillary thyroid carcinoma development in 5 out of 44 mice. 38
Mouse Models of Follicular Thyroid Carcinoma
TRβPV/PV
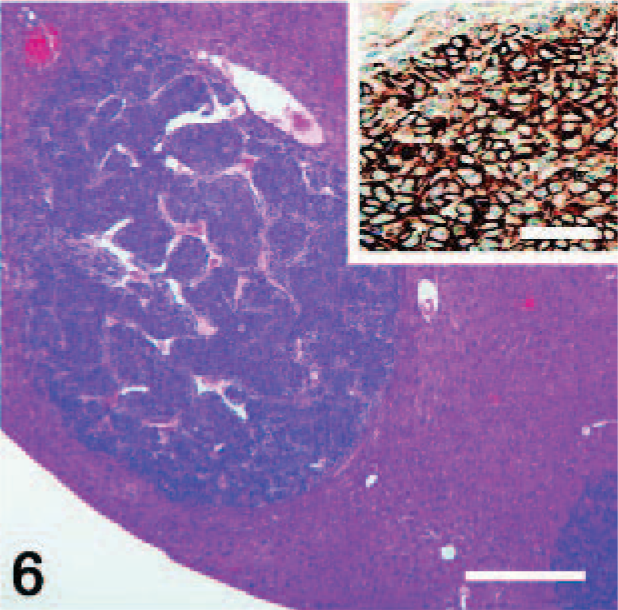
A genetically engineered mouse model of follicular thyroid carcinoma was created by introduction of a dominant-negative mutation, named PV for the patient first identified with the mutation, into the thyroid hormone receptor β (TRβ) locus by homologous recombination in a 129/Sv × C57BL/6J background strain. 35 The PV mutation results in loss of TRβ binding ability to active T3 hormone, abolishing transcriptional activation. The lack of negative feedback by T3 on the hypothalamus and pituitary gland led to persistent upregulation of TSH and follicular cell hyperplasia in these mice, which often caused dyspnea due to tracheal compression. 82 TRβPV/PV homozygotes developed follicular cell hyperplasia by 3 weeks of age and invasive follicular thyroid carcinomas by 4–5 months of age. Hematogenous metastases to the lungs and heart were noted in the majority of mice over 5 months of age, and the metastases often had foci of anaplasia consisting of spindle-shaped cells. A similar phenotype was evident when TRβPV/- mice were created by cross-breeding TRβPV/PV mice with TRβ knockout mice (Figs. 7–9). 36 While this mouse model offers a reasonably good recapitulation of human follicular carcinoma arising from hyperplastic goiter, it differs in that these mice have elevated circulating T3 and T4 levels due to production by the tumor cells, while human follicular thyroid carcinomas are infrequently functional. 17
Thyroid gland; TRβPV/- mouse. A follicular thyroid carcinoma invades the thyroid capsule (arrow). HE; Bar = 50 μm. Photo reproduced from Kato et al: Endocrinology
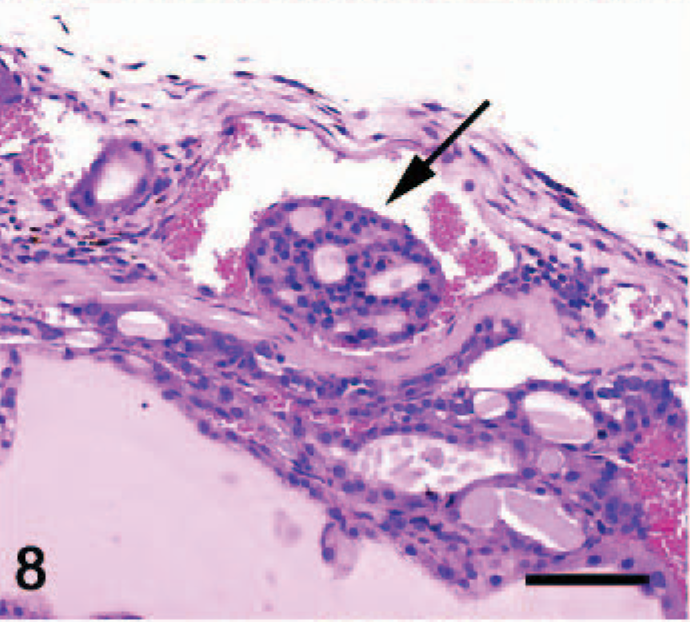
Thyroid gland; TRβPV/- mouse. Vascular invasion is characteristic of follicular thyroid carcinomas (arrow). HE; Bar = 50 μm. Photo reproduced from Kato et al: Endocrinology
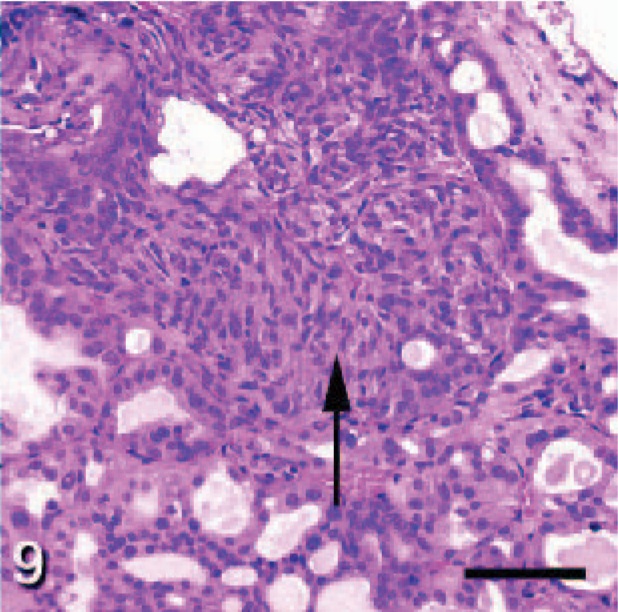
Thyroid gland; TRβPV/- mouse. Foci of anaplasia are noted in regions of follicular hyperplasia and carcinoma (arrow). HE; Bar = 50 μm. Photo reproduced from Kato et al: Endocrinology
Tg-Rap1bG12V
Transgenic mice overexpressing a constitutively active form of Rap1 that lacks GTPase activity, Rap1bG12V, developed follicular cell hyperplasia and adenomas within 6 months of goitrogen (methimazole and perchlorate) treatment and follicular carcinomas after 1 year. 66 Hyperplasia and adenomas were reversible upon 2 months of goitrogen removal provided that progression to carcinoma was absent, indicating that the early adenomas were not truly autonomous. While follicular carcinomas were locally invasive into the thyroid capsule, local blood vessels and perithyroidal soft-tissue metastases were not detected. The constitutively active Rap1bG12V cassette used to create this mouse model was floxed and followed by a dominant negative Rap1bS17N such that upon cross-breeding with a tamoxifen-inducible Cre mouse and administration of tamoxifen, the constitutively active Rap1bG12V was removed and the dominant negative Rap1bS17N was expressed in its place. When the Rap1b “switch” was made from constitutively active Rap1bG12V to dominant negative Rap1bS17N, thyroid gland size was reduced by 50% in the face of continued goitrogen treatment. Ki67, BrdU, and PCNA labeling indices and terminal deoxynucleotidyltransferase-mediated dUTP nick end-labeling indicated that decreased cellular proliferation, rather than increased apoptosis, accounted for the decrease in thyroid hyperplasia. This study suggests an important role for Rap1b in TSH-induced thyroid hyperplasia and tumorigenesis.
Other models
Follicular thyroid carcinomas have been described in mice expressing a mutated form of the α1B adrenergic receptor under the control of the Tg promoter, which leads to upregulated cAMP and IP3-Ca++ signaling in the thyroid gland. 15, 48, 49 Tg-ki-ras transgenic mice also developed well-differentiated follicular thyroid carcinomas at a low rate with long latency upon treatment with TSH or goitrogens, suggesting that while ras mutation alone is not sufficient to induce follicular thyroid carcinomas, it acts as a predisposing factor. 12
Mouse Models of Anaplastic Thyroid Carcinoma
Tg-SV40 LT
Anaplastic thyroid carcinomas developed in transgenic mice expressing thyroid-targeted Simian virus 40 large T antigen on a C57B/6J × DBA/2J background, which is the only published transgenic mouse model of this type of thyroid cancer. 46 Mice were profoundly hypothyroid and required T4 supplementation in order to survive to an age where they could reproduce. By the age of 4–6 weeks and as early as 9 days of age, most mice developed solid, anaplastic carcinomas with hemorrhage, necrosis, and few, if any, residual thyroid follicles. A few mice survived long enough to develop lung metastases, while invasion of the trachea led to dyspnea and death in 90% of the transgenic mice by 22 weeks of age.
Mouse Models of Medullary Thyroid Carcinoma
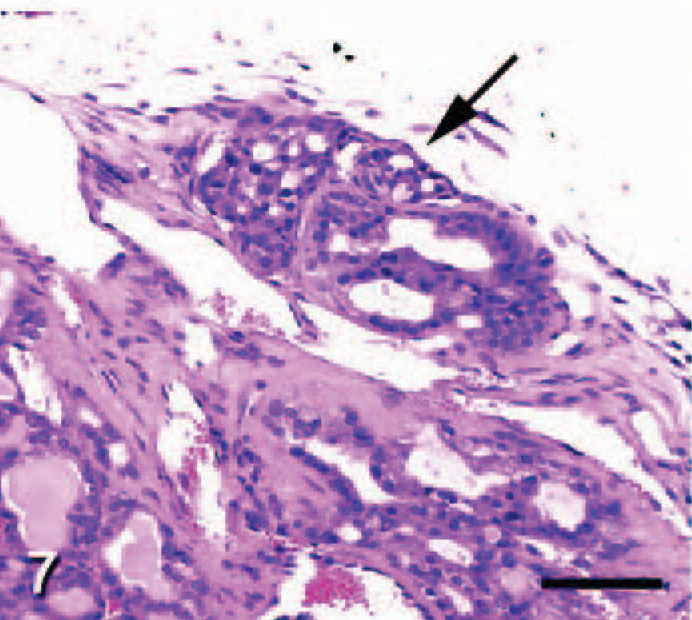
CGRP-RetC634R
It is well known that in humans, point mutation of the
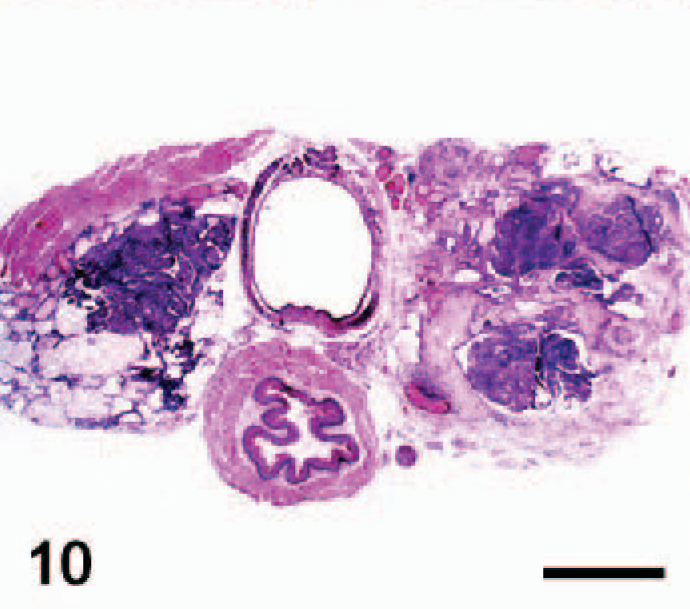
Thyroid gland; CGRP-RetC634R transgenic mouse. A large, invasive C-cell carcinoma is present within the right lobe of the thyroid gland (arrow), while the opposite lobe contains a papillary thyroid carcinoma. HE; Bar = 1 mm. Photo reprinted from Reynolds et al: Oncogene
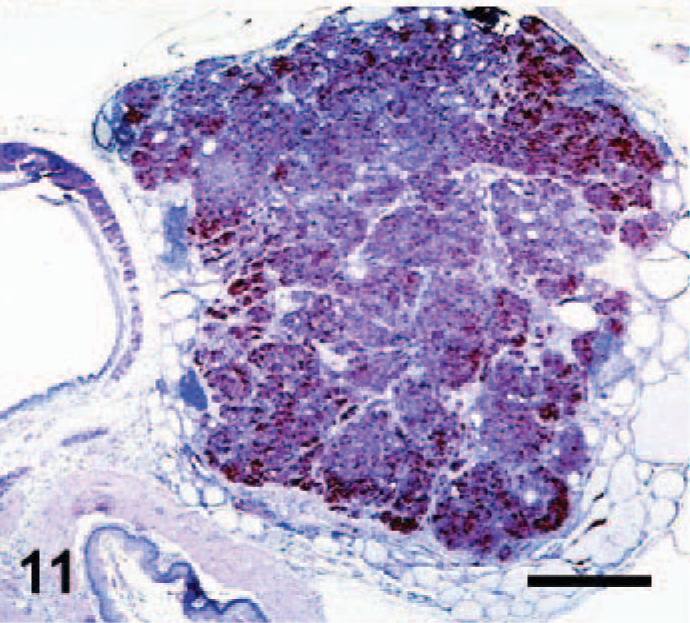
Thyroid gland; CGRP-RetC634R transgenic mouse. Higher magnification of a C-cell carcinoma reveals characteristic neuroendocrine packeting of neoplastic cells. Calcitonin immunostain with hematoxylin counterstain; Bar = 500 μm. Photo reprinted from Reynolds et al: Oncogene
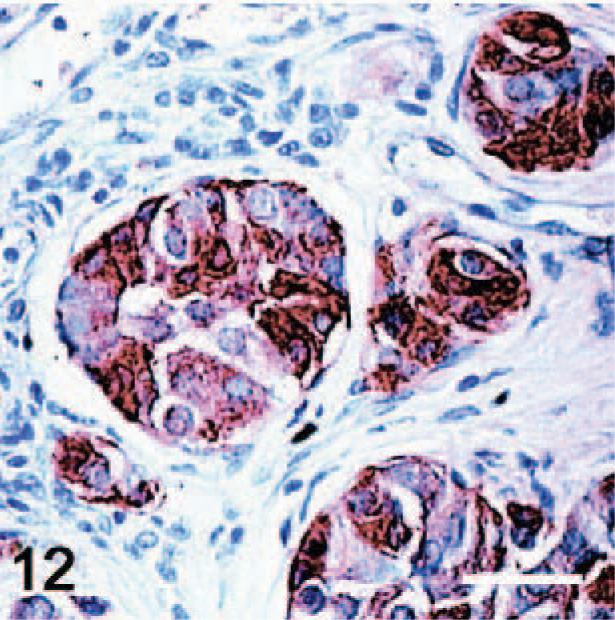
Thyroid gland; CGRP-RetC634R transgenic mouse. High magnification of a C-cell carcinoma demonstrates the fine fibrovascular stroma dividing lobules of polyhedral neoplastic cells. Ret immunostain with hematoxylin counterstain; Bar = 30 μm. Photo reprinted from Reynolds et al: Oncogene
Rb and p53 deletional mutation
Approximately 40% of mice with simultaneous heterozygous deletional mutation of Rb and p53 (Rb+/- × p53+/-) on a 129 strain background developed C-cell hyperplasia and medullary thyroid carcinomas at approximately 7 months of age. 29 Tumors were generally well differentiated and nonmetastatic. Interestingly, in one recent study using an Rb+/- model with normal p53 status on a 129/Sv × C57BL/6 background, 56% of mice developed medullary thyroid carcinomas, and there were rare metastases to liver, lung, lymph node, and adrenal gland. 96 Thus, loss of 1 allele of Rb was sufficient to predispose mice to medullary thyroid carcinomas, depending on the background strain. Naturally, Rb+/- and p53+/- mice also developed many nonthyroidal neoplasms and had shortened life spans. Spontaneous activating mutations of Ret were detected in 4 out of 8 medullary thyroid carcinomas from Rb+/- × p53+/- mice. 18 While Rb loss has been detected immunohistochemically in human thyroid malignancies of many types, 1 its precise role is unclear in the development of spontaneous medullary thyroid carcinoma in humans.
CGRP-v-Ha-ras
Transgenic mice of a C57BL/6 × SJL strain expressing v-Ha-ras in C cells under control of the CGRP promoter developed C-cell hyperplasia progressing to medullary thyroid carcinoma in 85%–93% of animals (depending on founder line) between the ages of 6–12 months. 34 Tumors were functional in secreting calcitonin, as is common in the human disease. Metastasis occurred rarely to the lungs and cervical lymph nodes. Some tumors had amyloid deposition, cystic cavitation, and vascular invasion and hemorrhage, characteristic features of human medullary thyroid carcinomas. Ras mutations have not been detected in human cases of medullary thyroid carcinoma. Thus, this mouse model offers a better phenotypic than genotypic representation of the human disease.
Other models
All transgenic mice coexpressing a truncated form of the polyomavirus (Py) middle-T antigen and the full-length Py small-T antigen under control of a composed Py early promoter/enhancer developed medullary thyroid carcinomas by 3–7 months of age. 22 Tumors did not metastasize, and the animals had an unexpected waviness of hair and whiskers due to disorganized hair follicles in the dermal layer. In one of the earliest mouse models of medullary carcinomas, transgenic mice carrying the c-mos proto-oncogene, under control of the Moloney murine sarcoma virus long-terminal repeat, developed a syndrome similar to MEN 2A, consisting of medullary carcinomas and adrenal medullary pheochromocytomas, after a long latency. 75 In this model, like the CGRP-RetC634R model, 19 tumors were more prevalent on a BALB/c background than an FVB/N background.
The Future of Thyroid Cancer Research Using Genetically Engineered Mouse Models
Transgenic mice expressing Ret/PTC1, Ret/PTC3 Trk-T1, BRAF, or ras offer a reasonable approximation of the features of papillary thyroid carcinomas in humans. However, even with a p53 knockout, no model has been able to demonstrate significant tumor dedifferentiation or metastasis. One limitation has been the fact that p53 knockout mice develop extrathyroidal neoplasms at a high rate, resulting in a shortened life span. A tissue-specific p53 knockout would help to eliminate this problem and allow the mice to live long enough to potentially develop more advanced thyroid cancer.
Follicular thyroid carcinoma has been primarily investigated using a single mouse model, TRβPV/PV. The limitation of this model is the presence of hyperthyroidism and thyroid hormone resistance, which is not typical of the human disease. Because follicular thyroid carcinomas are more common in areas of dietary iodide deficiency, it is interesting that chronic TSH stimulation in the TRβPV/PV mice resulted in this type of neoplasm. However, rodents are notoriously susceptible to thyroid neoplasia because of perturbations of the pituitary-thyroid axis. 8, 9
There currently is a paucity of genetically engineered mouse models of anaplastic thyroid carcinoma, although in mice, SCID xenograft models are abundant. As previously mentioned, few papillary and follicular thyroid carcinomas undergo significant dedifferentiation in transgenic mouse models. The only published transgenic model of anaplastic carcinoma, Tg-SV40 LT, rapidly developed severe hypothyroidism and fatal thyroid tumors as a result of tracheal invasion. An inducible model would facilitate maintenance and breeding of these mice and would better reproduce anaplastic carcinoma in the adult animal, which is more typical in human disease.
Many of the mouse models of thyroid follicular cell neoplasia fail to reproduce the normal hormonal milieu present in humans with thyroid cancer. For example, derangement of thyroid function in mouse models remains a problem. Rapid onset of dysplasia or neoplasia replacing the majority of the normal thyroid tissue, especially in neonatal and juvenile mice, often results in significant hypothyroidism unless thyroid hormone supplementation is instituted. In addition, human thyroid neoplasms are more than twofold more common in women than in men, suggesting that estrogen plays a role in tumorigenesis, while no sex predilection has been achieved in the current mouse models.
Reproduction of the MEN 2A syndrome using CGRP-RetC634R has been quite successful. However, in this model, as well as the Rb+/- models, there appears to be a significant contribution of background strain to the development of medullary thyroid carcinomas. In addition, these models develop several extrathyroidal neoplasms common to the MEN 2A syndrome, while only the CGRP-v-Ha-ras model mimics the presence of medullary thyroid carcinoma alone, which is the most common form of the disease in humans.
Overall, there is a need for the development of inducible mouse models of thyroid cancer. All of the available mouse models have genetic mutations from the earliest stages of development onward, while many naturally occurring human cases of thyroid cancer result from somatic mutations during childhood or beyond. Additionally, little research has been performed to test novel therapies in vivo. For example, while a great deal of recent research has focused upon signal transduction inhibitors directed toward Ret, ras, raf, and ERK in tumors of follicular and C-cell origin in vitro, 91 these compounds have only been tested in a few SCID mouse xenograft models of thyroid cancer. Pharmacologic therapeutic intervention using genetically engineered mouse models of thyroid cancer would provide more useful information as to the feasibility, efficacy, and safety of signal transduction inhibitors in a relatively realistic in vivo setting. The continually increasing incidence of thyroid cancer in humans and lack of successful treatment options for dedifferentiated, invasive, or metastatic lesions makes mouse models invaluable in future therapeutic investigations.
Footnotes
Acknowledgement
We would like to thank Tim Vojt in the Ohio State University College of Veterinary Medicine for technical assistance with figure preparation. This work was generously supported by Schering-Plough Research Institute.