
Abstract
Tissues and fecal material were collected from 14 North American bison (Bison bison) that were suspected of having Johne's disease and analyzed for the presence of Mycobacterium avium subsp. paratuberculosis (M. paratuberculosis). Sections of ileum, ileal-cecal lymph node, and three sequential sections of jejunum with their associated mesenteric lymph nodes were taken from each animal. Fecal culture indicated that 5 of 14 (35.7%) animals were infected, whereas cultures from tissues detected 12 of 14 (85.7%) animals as infected and 59 of 111 (53.2%) of the tissues as positive for M. paratuberculosis. Polymerase chain reaction analysis identified infection in 14 of 14 (100%) animals and in 91 of 112 (81.2%) tissues. In addition, tissues were processed for Ziehl-Neelsen acid-fast staining, auramine O/acridine orange fluorescent staining, and immunohistochemical staining. Ziehl-Neelsen and auramine O staining identified 7 of 14 (50%) and 5 of 14 (35.7%) animals as infected and 24 of 112 (21.4%) and 28 of 112 (25%) tissues as positive, respectively. Immunohistochemical analyses of bison tissues, using antisera collected from rabbits immunized with four different preparations of M. paratuberculosis, identified a greater percentage of infected animals (ranging from 57 to 93%) and positive tissues (ranging from 28 to 46%). Collectively, these data indicate that DNA-based detection of M. paratuberculosis was more sensitive than bacterial culture or staining, identified infection in all the bison, and detected the greatest number of positive tissues within each animal.
Paratuberculosis (Johne's disease) is a chronic inflammatory disease of ruminant animals caused by Mycobacterium avium subsp. paratuberculosis (M. paratuberculosis). Clinical disease in cattle is characterized by weight loss, diarrhea, decreased milk production, and ultimately death. Animals are most likely infected before 6 months of age by ingestion of contaminated food or milk. Because of the slow progression of the disease, clinical signs are often not observed until the animal is at least 3 years of age. 4 During the preclinical incubation stage, bacteria are shed through the feces at varying levels, serving as a potential source of infection for other animals. Although paratuberculosis has been primarily described in cattle, sheep, and goats, infection of wild ruminant animals such as white-tailed deer (Odocoileus virginianus), 5 , 16 Key deer (Odocoileus virginianus clavium), 24 fallow deer (Dama dama), 18 saiga antelope (Saiga tatarica), 10 tule elk (Cervus eleaphus nannodes), 7 , 15 , 17 bighorn sheep (Ovis canadensis), Rocky Mountain goats (Oreamnos americanus), 36 , 35 and bison (Bison bison) 2 , 3 , 31 , 32 has also been reported.
Diagnosis and culling of infected animals remains the most effective disease control measure because currently available vaccines do not prevent disease or bacterial shedding. Definitive diagnosis is complicated by the lack of a true “gold standard” because there is debate over which diagnostic test is the most sensitive and specific for paratuberculosis. Isolation and culturing of M. paratuberculosis from feces or tissue remains the most definitive detection method yet can result in false-negative results based on a low level of colonization in the tissues or negligible and intermittent shedding of the microorganisms in the feces. In addition, culturing with conventional solid agar media requires 8–16 weeks of incubation before colony growth occurs, 27 resulting in a prolonged period of time until an appropriate diagnosis can be made. Serodiagnostic tests are commercially available, but the paradoxical host immune response during M. paratuberculosis infection reduces the efficacy of detection. 28 Two of the most commonly used staining procedures for detecting mycobacteria in formalin-fixed tissues are Ziehl-Neelsen staining, which identifies acid-fast organisms, 25 and auramine O fluorescent staining, which has been reported to be specific for mycolic acids. 23 However, these two staining methods are not able to distinguish among different mycobacterial species. Immunohistochemical staining of suspect tissues using antibodies produced against M. paratuberculosis provides a more specific means of detection than chemical staining and can discriminate tissue samples containing M. paratuberculosis from those containing other pathogens. 6 , 29 , 30 In addition, immunohistochemical staining of fixed tissues has been reported to be more sensitive than acid-fast staining. 6 , 19 , 34 Finally, direct polymerase chain reaction (PCR)-based detection of the M. paratuberculosis insertion sequence, IS900, from tissues 12 , 21 , 33 and feces 13 has demonstrated that PCR is highly sensitive and specific. As proof of this specificity, a PCR analysis was performed in our laboratory, which showed that IS900 was only amplified from M. paratuberculosis samples but not from over 100 M. avium, M. intracellulare, or other M. avium subspecies. 11 PCR has been used to positively identify samples that were culture negative 21 as well as detect fentogram (less than two genome copies) amounts of DNA. 9 , 22
It was previously reported that bison with gross and histopathologic changes consistent with Johne's disease, as well as tissue samples that were positive for acid-fast organisms, were negative by agar gel immunodiffusion, enzyme-linked immunosorbent assay, fecal culture, and tissue culture. 3 Those results demonstrated that many of the testing methods commonly used for cattle are not as sensitive or specific for bison. The aim of this study was to compare the sensitivity of routinely used bovine laboratory diagnostic tests for the detection of M. paratuberculosis from the tissues of 14 bison. Six tests, including fecal culture, tissue culture, Ziehl-Neelsen acid-fast staining, auramine O fluorescent staining, immunohistochemical staining with four polyclonal antibodies, and IS900 PCR detection, were evaluated in this study.
Materials and Methods
Gross and histopathologic lesions consistent with M. paratuberculosis infection were reported previously from 70 North American bison. 3 A subset of this group, group C, consisted of 14 bison (numbered sequentially from 1 to 14 for this study) that were culled and necropsied at a slaughter plant in the western United States because they were suspected of having Johne's disease. Each bison was suffering from severe weight loss, failure to shed winter hair, and poor body condition. Previously, animals from this herd had been culled and were fecal and tissue culture positive for paratuberculosis. 3 , 31 Feces were collected for culture of M. paratuberculosis, and eight tissue samples were taken from each animal, including one section of midileum, one section of ileal-cecal lymph node, three consecutive sections of jejunum (proximal, middle, and distal), and three sections of the associated mesenteric lymph nodes (proximal, middle, and distal). A portion of each tissue section was frozen at −80 C and stored until it could be processed for culture of M. paratuberculosis. Additional sections were formalin fixed (not to exceed 1 week in fixative) and paraffin embedded. Blocks were stored for up to 2 years before being serially sectioned at 4 µm thickness onto Probe-On Plus positively charged glass slides (Fisher Scientific, Pittsburgh, PA). Concurrently, two 4-µm sections were aseptically placed into sterile 1.5-ml microcentrifuge tubes for DNA extraction. The paraffin sections were cut with disposable blades, and a new blade was used for each tissue block. The microtome block holder and blade holder were cleaned with xylene and ethanol between tissue blocks. In addition to the bison tissues described above, a section of ileum from a clinical M. paratuberculosis–infected cow was used as a positive control, whereas a section of lung from an M. bovis–infected cow (graciously provided by Mitchell Palmer, National Animal Disease Center, Ames, IA) and a section of ileum from a control, noninfected bison from an unrelated project (graciously provided by Steven Olsen, National Animal Disease Center) were used as negative controls.
Mycobacterial DNA was crudely extracted from paraffin-embedded tissues using a previously described protocol. 20 Microcentrifuge tubes containing sectioned tissue were centrifuged at 16,000 × g for 1 minute to pellet the sections. To each tube, 200 µl of ultrapure distilled water (GIBCO-BRL, Grand Island, NY) with sterile 0.5% Tween 20 was added. The tubes were placed in a boiling water bath for 10 minutes and then snap-frozen for 2 minutes in a dry ice–ethanol bath. The boil-freeze step was repeated three times, and then the samples were centrifuged at 3,000 × g for 20 minutes to pellet tissue fragments. Supernatant was removed from the microcentrifuge tube and transferred to a new, sterile microcentrifuge tube for PCR amplification. If the first PCR amplification did not yield an IS900 amplicon, DNA was reextracted from a different section of the same tissue block. In addition to the boil-freeze procedure listed above, this second DNA preparation was phenol-chloroform purified. In brief, an equal volume of phenol : chloroform : isoamyl alcohol 25 : 24 : 1 (Amresco, Solon, OH) was added to the DNA supernatant, mixed, centrifuged at 21,000 × g for 1 minute, and the aqueous layer was removed. To this solution, two volumes of 100% ethanol and one-tenth volume of 3 M sodium acetate (pH 5.2) were added. The solution was placed at −80C for 30 minutes and then centrifuged at 21,000 × g for 15 minutes. The supernatant was removed, and the pellet was dried and resuspended in 100 µl of ultra-pure water. Tissue samples from a clinical M. paratuberculosis cow were used as positive control samples, whereas tissue sections from an M. bovis–infected cow and a non-infected bison were used as negative control samples. DNA was extracted from control and suspect tissues in parallel to account for possible cross-contamination and false positives.
PCR amplifications were performed in 200-µl tubes and cycled in a GeneAmp PCR System 9700 thermocycler (Applied Biosystems, Foster City, CA). Each 50-µl reaction contained 5 µl of GeneAmp 10× PCR Buffer (100 mM Tris-HCl, pH 8.3, 500 mM KCl, 15 mM MgCl2, 0.01% gelatin; Applied Biosystems), 1 µl of diethylnitrophyenyl thiophosphate (dNTP) (10 mM each dNTP; Roche Diagnostics, Indianapolis, IN), 0.8 µl of IS900R 11 (5′-AATCAACTCCAGCAGCGCGGCCTCG-3′) and IS900L 11 (5′-CCGCTAATTGAGAGATGCGATTGG-3′) oligonucleotide primers (100 pmol/µl each), 2.5 U of AmpliTaq Gold DNA polymerasek (Applied Biosystems), 33 µl of ultrapure distilled water, and 10 µl of DNA extracted from tissue (described above). Samples were denatured at 95 C for 5 minutes, then 50 cycles of denaturation at 95 C for 30 seconds, annealing at 55 C for 30 seconds, and extension at 72 C for 1 minute were performed, followed by a final extension-termination of 72 C for 10 minutes and holding at 4 C. PCR-amplified products were observed on 1.2% agarose gels stained with ethidium bromide. In addition, a 100–base pair (bp) molecular weight marker (MBI Fermentas, Hanover, MD) was loaded on the gel to aid in determining the size of the PCR amplicons. Positive samples were identified by the presence of a 229-bp product.
Fecal and tissue culture for the detection of M. paratuberculosis was performed at the University of Pennsylvania by R. H. Whitlock as described previously. 31
For Ziehl-Neelsen staining, tissue sections were deparaffinized and hydrated by three washes in xylene for 5 minutes each, two washes in 100% ethanol for 1 minute each, two washes in 95% ethanol for 1 minute each, and one wash in distilled water for 5 minutes. The tissue sections were stained for 1 hour with TB carbol fuchsin Ziehl-Neelsen acid-fast stain (Becton Dickinson, Sparks, MD). The sections were washed for 2 minutes in tap water, decolorized in two brief washes of acid alcohol (1% hydrochloric acid in 70% ethanol), washed for 2 minutes in tap water, and briefly counterstained with methylene blue. The sections were dehydrated by two brief washes in 95% ethanol, two brief washes in 100% ethanol, and two brief washes in xylene before being coverslipped. All tissue sections were examined by two people, with multiple fields being evaluated.
Auramine O/acridine orange staining was performed on deparaffinized and hydrated tissue sections by two washes in xylene for 5 minutes each, two washes in 100% ethanol for 1 minute each, and one wash in distilled water for 2 minutes. The tissue sections were stained in auramine O solution (9.3 mM auramine O [Sigma Chemical Co., St. Louis, MO], 7% glycerol, and 3% liquid phenol) for 10 minutes and then rinsed in tap water for 1 minute. The slides were treated with 10% ferric chloride solution for 5 minutes, washed in tap water for 1 minute, and counterstained with 0.67 mM acridine orange solution (Sigma Chemical Co.) for 2.5 minutes. The slides were then washed in tap water for 1 minute, in 70% ethanol for 1 minute, in 95% ethanol for 1 minute, in 100% ethanol for 1 minute, and in two changes of xylene for 5 minutes and coverslipped.
For immunohistochemical staining, tissue sections were deparaffinized and hydrated by three washes in xylene for 5 minutes each, one wash in 100% ethanol for 2 minutes, one wash in 95% ethanol for 1 minute, one wash in 70% ethanol for 1 minute, and one wash in distilled water for 5 minutes. The tissues were rinsed in 1% bovine serum albumin in Trisbuffered saline (TBS; 50 mM Tris-HCl, 150 mM NaCl, 0.03% Tween 20; pH 7.6) for 5 minutes and treated with a 0.1% trypsin–calcium chloride enzyme solution for 20 minutes at 37 C. The samples were rinsed in TBS for 5 minutes and then treated with hot hydrochloric acid (2 N) for 4 minutes. The slides were rinsed with TBS for 5 minutes and then stained with the streptavidin–alkaline phosphatase detection kit (HistoMark Red, Kirkegaard and Perry Laboratories, Gaithersburg, MD) according to manufacturer's instructions. In brief, the samples were blocked with 10% normal goat serum for 30 minutes and then incubated with primary antibody (diluted 1 : 1,000 in TBS) for 1.5 hours at 37 C. Four primary rabbit antibodies were tested in this study: a commercially available antibody made against a sonicated preparation of M. paratuberculosis strain 2E (Dako Corporation, Carpinteria, CA); anti-a362 serum was made against recombinant polypeptide a362, the carboxyl-terminal portion of a 34-kd M. paratuberculosis antigen 6 (kindly provided by Claus Buergelt, Department of Pathobiology, University of Florida, Gainesville, FL); antibody NADC 272 was generated against M. paratuberculosis cell wall proteins; and antibody NADC 275 was generated against heat-killed, whole-cell M. paratuberculosis. Both NADC 272 and NADC 275 were produced at the National Animal Disease Center, Ames, Iowa, as reported previously. 29 After incubation with primary antibody, the samples were washed in TBS for 5 minutes, the biotinylated secondary antibody was added and incubated for 30 minutes at room temperature, the samples were washed in TBS for 5 minutes, strepavidin–alkaline phosphatase was added and incubated for 30 minutes at room temperature, and the samples were washed in TBS for 5 minutes. The red chromogen solution was added to each sample and incubated for 10 minutes at room temperature, the samples were washed in distilled water for 5 minutes, counterstained in three brief washes of hematoxylin solution Gill No. 2 (Sigma Chemical Co.), and decolorized in three brief washes of blueing water (0.11% ammonium hydroxide). The samples were dehydrated by one wash in 95% ethanol for 1 minute, three washes in 100% ethanol for 1 minute, and three washes in propar for 1 minute and coverslipped. Sections of lung from an M. bovis–infected cow (supplied by Mitchell Palmer, National Animal Disease Center, Ames, IA) were used as control samples to evaluate cross-reactivity of the polyclonal antibodies. Antibodies anti-a362 and NADC 272 performed optimally using the standard protocol described above. However, for the commercial antibody and NADC 275, slight variations in the immunohistochemical staining protocol provided optimal staining with no cross-reactivity to M. bovis–infected tissues. In particular, the commercial antibody stained most specifically when tissues were not treated with hot hydrochloric acid. In addition, antibody NADC 275 performed best when tissues were not treated with enzyme. If a test result for acid-fast staining (Ziehl-Neelsen or Auramine O) or immunohistochemical staining was negative, additional tissue sections were cut and processed for restaining to preclude false negatives.
Results
Fourteen bison were culled from a ranch in the northern United States because of weight loss, failure to shed winter hair, and poor body condition. Gross and microscopic lesions were previously described 3 and are summarized in Table 1. In this study, a total of 112 tissues from 14 bison were analyzed by six routinely used paratuberculosis diagnostic tests. The testing results for all tissues are shown in Fig. 1, with the data summarized in Table 2. Amplification of IS900 from bison tissues identified infection in 100% (14 of 14) of the animals and indicated that 81.2% (91 of 112) of the tissues contained M. paratuberculosis. On the basis of PCR analysis, no single tissue could be relied on to consistently determine infection status (Fig. 2). Sections of lesioned ileum from a cow with clinical signs of M. paratuberculosis infection were positive for IS900. Conversely, sections of lesioned lung from an M. bovis–infected cow and ileum from a control, noninfected bison were negative.
Summary of gross pathology and histopathology from 14 bison as previously described.3

∗1 = mucosal corrugation; 2 = prominent lymphatics; 3 = lymphadenopathy; 4 = weight loss; NS = none seen.
† The extent of granulomatous infiltration: 0 = none; 1 = mild; 2 = moderate; 3 = severe.
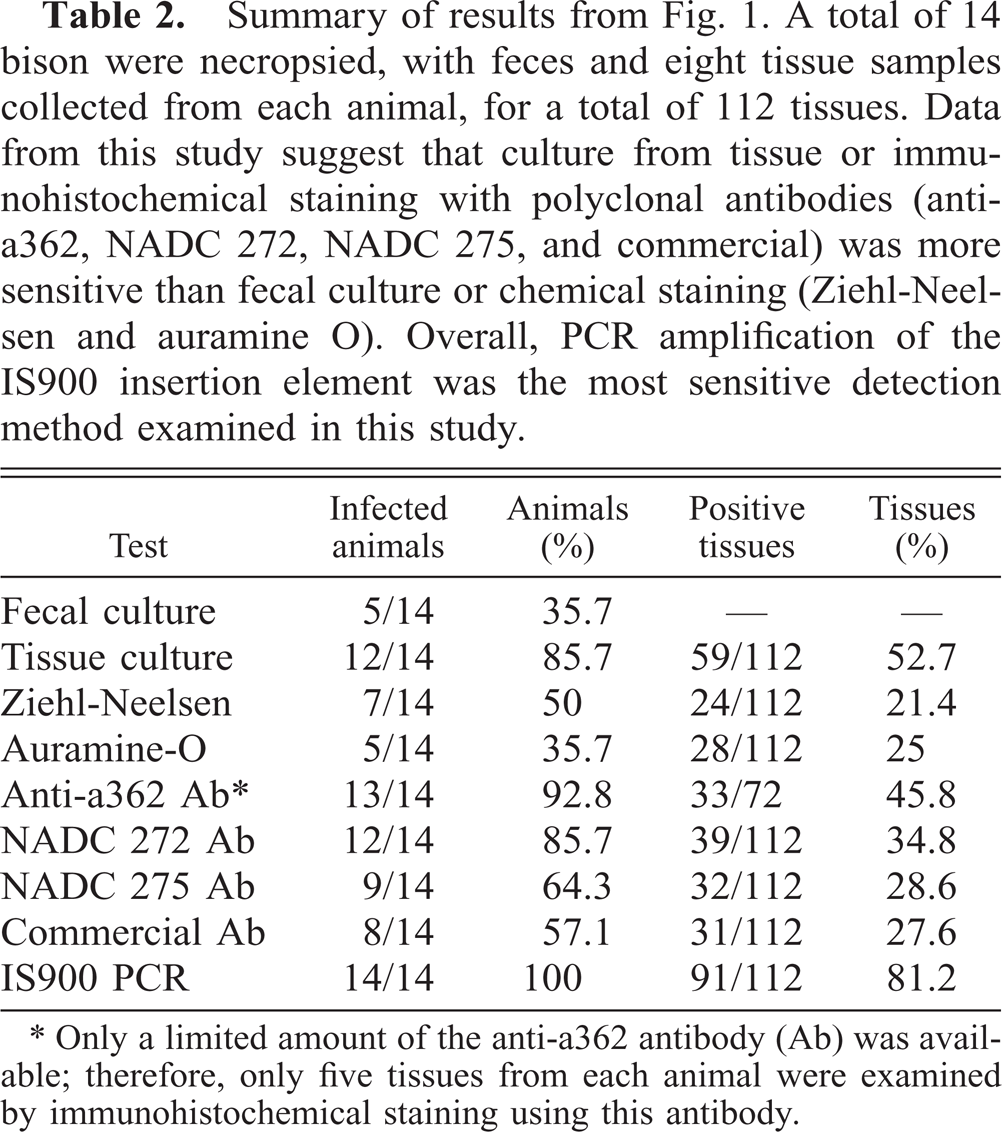
Summary of results from Fig. 1. A total of 14 bison were necropsied, with feces and eight tissue samples collected from each animal, for a total of 112 tissues. Data from this study suggest that culture from tissue or immunohistochemical staining with polyclonal antibodies (anti-a362, NADC 272, NADC 275, and commercial) was more sensitive than fecal culture or chemical staining (Ziehl-Neelsen and auramine O). Overall, PCR amplification of the IS900 insertion element was the most sensitive detection method examined in this study.
∗ Only a limited amount of the anti-a362 antibody (Ab) was available; therefore, only five tissues from each animal were examined by immunohistochemical staining using this antibody.
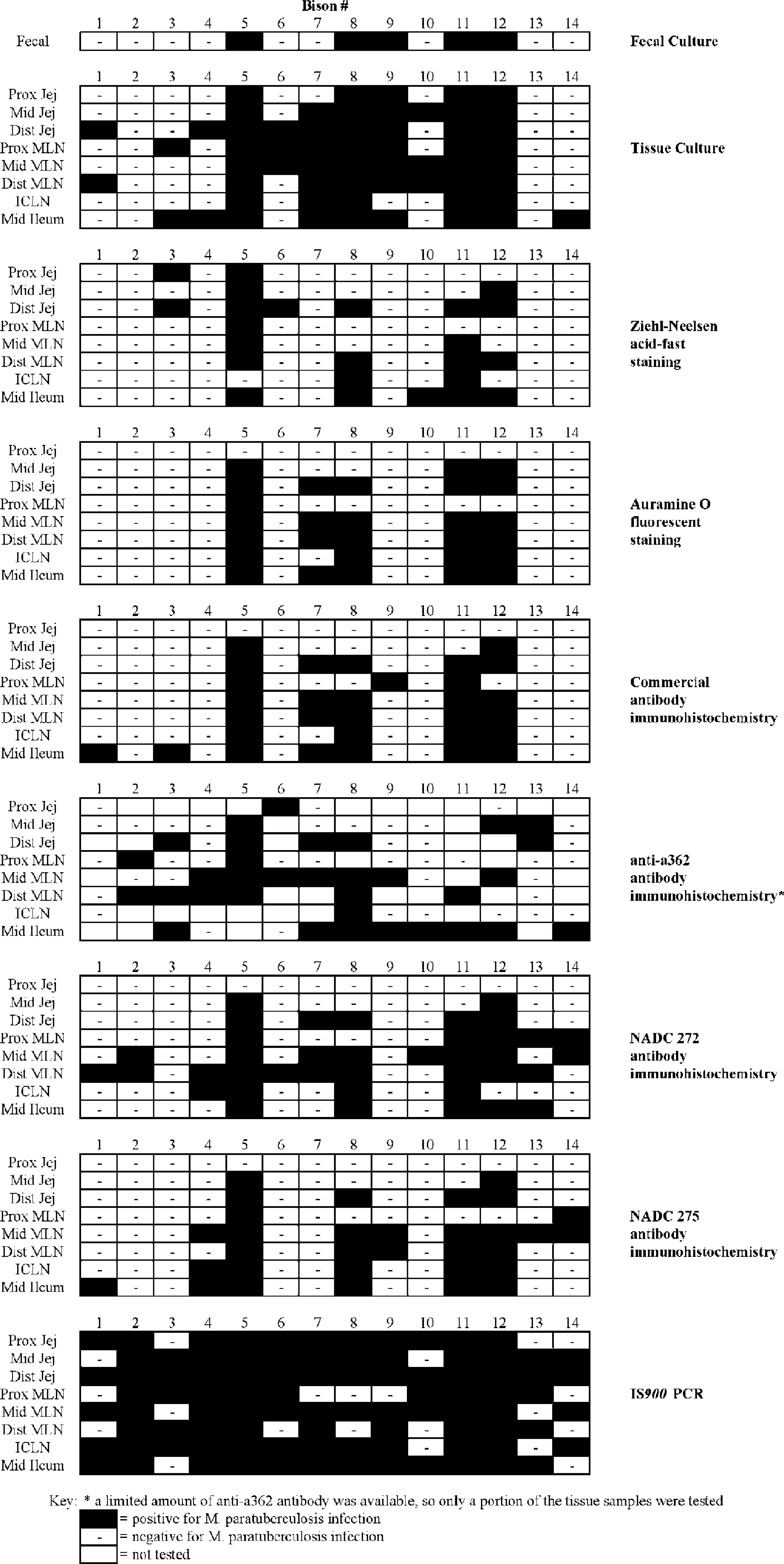
Detection of M. paratuberculosis from bison tissues by fecal culture, tissue culture, acid-fast staining (Ziehl-Neelsen and auramine O), immunohistochemical staining (commercial antibody, anti-a362 antibody, NADC 272 antibody, and NADC 275 antibody), and PCR amplification of M. paratuberculosis–specific IS900 sequence. Fourteen bison (numbered sequentially) were necropsied, with feces and eight tissues taken from each animal as described in Materials and Methods.
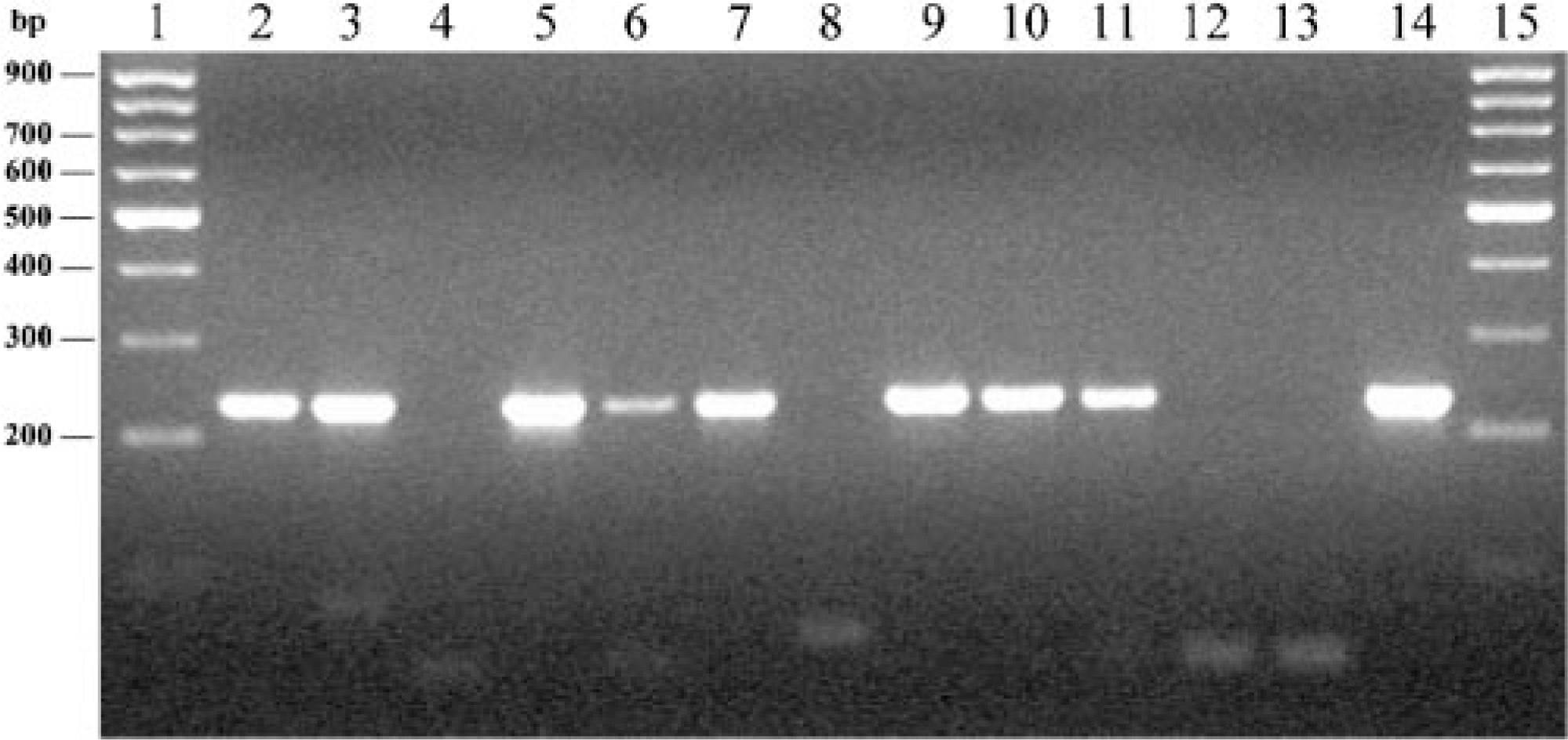
Identification of M. paratuberculosis–infected bison tissues by PCR amplification of the IS900 insertion element. Lane 1, 100 base pair ladder; lane 2, djej bison No. 1; lane 3, icln bison No. 2; lane 4, pjej bison No. 3; lane 5, dmln bison No. 5; lane 6, mil bison No. 7; lane 7, mjej bison No. 8; lane 8, pmln bison No. 9; lane 9, djej bison No. 11; lane 10, dmln bison No. 12; lane 11, mjej bison No. 13; lane 12, uninfected control bison; lane 13, M. bovis–infected bovine lung; lane 14, M. paratuberculosis–infected bovine ileum; lane 15, 100 base pair ladder. Abbreviations: djej = distal jejunum; icln = ileal-cecal lymph node; pjej = proximal jejunum; dmln = distal mesenteric lymph node; mil = midileum; mjej = middle jejunum; pmln = proximal mesenteric lymph node.
Fecal culture identified M. paratuberculosis infection in 35.7% (5 of 14) of the bison. Tissue culture identified 85.7% (12 of 14) of the bison as infected or 52.7% (59 of 112) of the tissues as positive for M. paratuberculosis. Specific tissues and animals that tested positive by fecal and tissue culture are listed in Fig. 1.
Using the Ziehl-Neelsen acid-fast staining technique, M. paratuberculosis–infected tissue samples were identified by the red staining of bacteria (Fig. 3A). In contrast, auramine O detected infected tissues by green fluorescent labeling of bacteria (Fig. 3B). Data from the acid-fast and fluorescent staining of all tissues are listed in Fig. 1 and summarized in Table 2. Ziehl-Neelsen staining identified 50% (7 of 14) of the animals as infected, with 21.4% (24 of 112) of the tissue samples positive for acid-fast organisms. Tissue samples from the distal jejunum, distal mesenteric lymph node, and midileum were found to contain M. paratuberculosis most frequently. Auramine O fluorescent staining of organisms identified 35.7% (5 of 14) of the animals as M. paratuberculosis infected and 25% (28 of 112) of the tissues as positive. The same types of tissues were found to be positive for M. paratuberculosis infection with both the auramine O and Ziehl-Neelsen staining, with the exception that auramine O was better at identifying organisms from the midmesenteric lymph nodes. Control tissues from an M. paratuberculosis–infected cow and the lung section from an M. bovis–infected cow were positive by both Ziehl-Neelsen and auramine O staining.
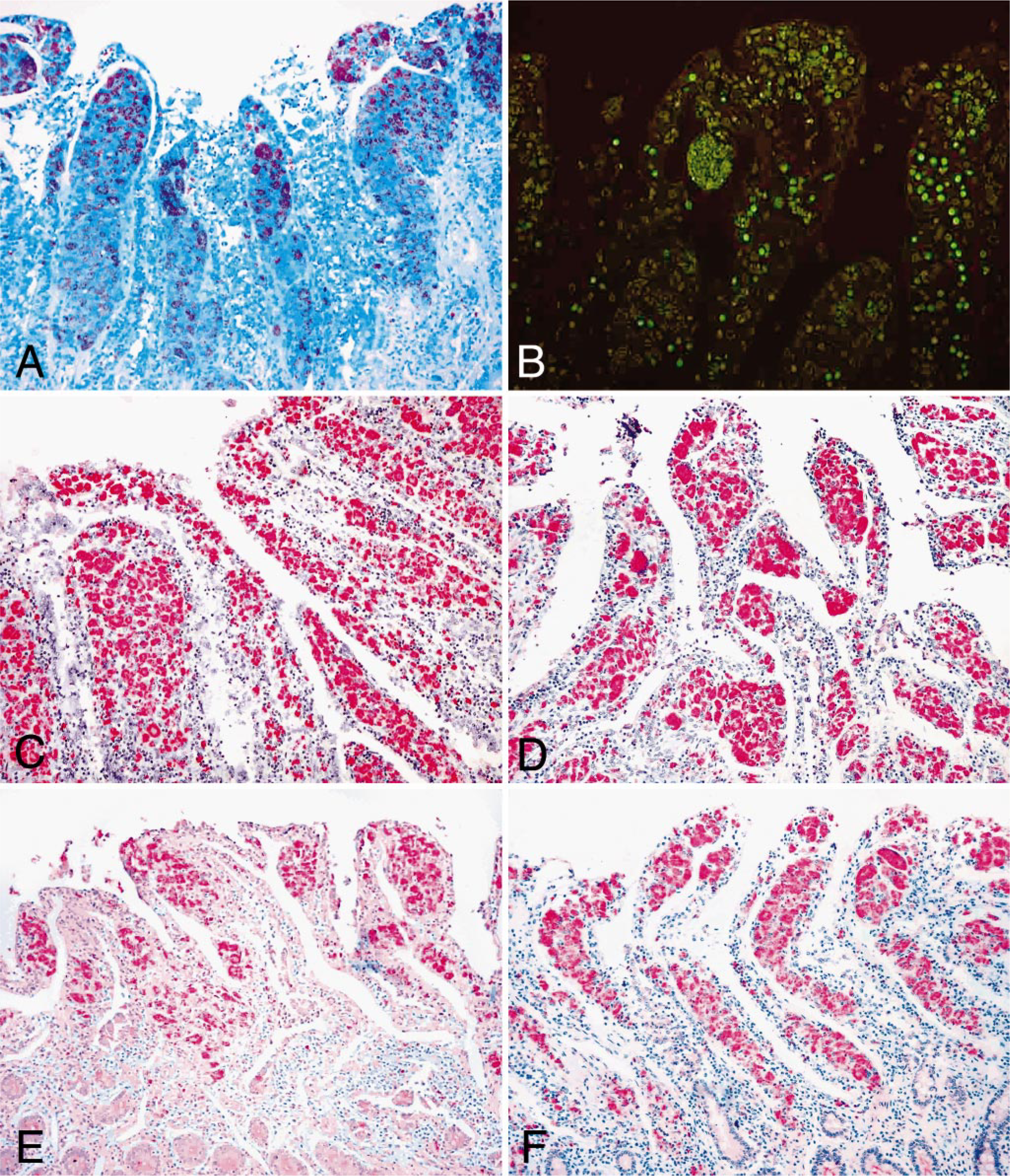
Staining of distal jejunum; bison No. 5.
Figure 1 shows information on the number of M. paratuberculosis–positive animals and tissues as detected by immunohistochemical staining. These results are summarized in Table 2. Examples of immunohistochemical staining patterns after the use of four polyclonal antibodies are shown in Fig. 3C–F. Staining of tissue sections using immunohistochemistry identified a greater number of M. paratuberculosis–infected bison and M. paratuberculosis–positive tissues as compared to acid-fast or fluorescent staining (Fig. 3A, B). The anti-a362 antibody identified M. paratuberculosis in 92.8% (13 of 14) of the animals, whereas NADC 272 antiserum identified 85.7% (12 of 14), NADC 275 antiserum identified 64.3% (9 of 14), and the commercially available antibody identified 57.1% (8 of 14) of the animals as paratuberculosis positive. With respect to staining of tissues, anti-a362 identified 45.8% (33 of 72) of the tissues examined, NADC 272 identified 34.8% (39 of 112), NADC 275 identified 28.6% (32 of 112), and the commercial antibody identified 27.6% (31 of 112) of the tissues as positive for M. paratuberculosis. The midileum and distal jejunum were found to contain M. paratuberculosis most frequently by immunohistochemical staining. All antibodies selectively stained sections of ileum from a cow with clinical signs of M. paratuberculosis infection. Sections of lesioned lung from an M. bovis–infected cow and sections of ileum from a control, noninfected bison were unstained.
Discussion
Among other factors, particularly testing costs, turnaround time, and reproducibility, a practical diagnostic test for paratuberculosis must be sensitive and specific. Although a number of diagnostic tests are available, debate exists over which is the most effective. To date, culturing of M. paratuberculosis from feces or tissue remains the most definitive detection method and is considered the gold standard of diagnostic tests for paratuberculosis. However, a low level of colonization in the tissues may preclude accurate diagnosis, particularly in subclinically infected animals. In addition, the long period of incubation required for primary culture of the microorganism (8–16 weeks) impedes management decisions to cull infected animals and control the spread of the disease. With respect to detection of M. paratuberculosis from wild ruminants such as bison, a previous study reported that bison isolates of M. paratuberculosis were more difficult to culture from feces and tissues and may require specialized media for optimal recovery of the microorganism. 31 Diagnosis of infection in bison has been reported to be further complicated by conflicting test results because the same animal can test positive and negative with different tests. 3 Taken together, those results demonstrated that the testing methods commonly used for cattle are not as sensitive or specific for bison.
In this study, we evaluated six routinely used diagnostic tests for the detection of M. paratuberculosis from suspect bison to identify the most sensitive test. Our results demonstrated that PCR amplification of M. paratuberculosis DNA was the most reliable technique for the identification of infected animals, although immunohistochemical staining and tissue culture performed nearly as well (Table 2). Comparatively, fecal culture and auramine O staining detected the least number of infected animals. In terms of the number of positive tissues, PCR was far superior to other techniques at detecting M. paratuberculosis infection, whereas staining with Ziehl-Neelsen or auramine O revealed few infected tissues.
The use of PCR as a diagnostic tool requires that an M. paratuberculosis–specific sequence be amplified. To date, a number of paratuberculosis-specific sequences and genes have been reported. 1 , 14 The insertion element, IS900, is the most routinely exploited sequence for PCR-based detection of suspect samples. 14 IS900 PCR has been used to detect minute amounts of M. paratuberculosis DNA in tissue 12 , 33 and feces, 13 including from tissues that were culture negative. 21 One study noted the presence of IS900 in environmental nonparatuberculosis mycobacteria, which has sparked debate about the specificity of the IS900 insertion element. 8 However, a PCR analysis previously performed by our laboratory demonstrated that IS900 was specific for M. paratuberculosis and could not be amplified from over 100 M. avium subspecies isolates. 11 The primers used to amplify IS900 differed between the two studies, 8 , 11 and thus, probably account for the reported differences in PCR specificity. In this study, we chose to amplify the IS900 sequence, as described to be specific for M. paratuberculosis, 11 from bison tissues because of its current use in many diagnostic laboratories. Amplification of the IS900 insertion element was the most specific and sensitive diagnostic detection method analyzed in this study. This protocol required little optimization, was rapid, and the results were easy to interpret. The reported specificity of IS900, taken together with our parallel DNA extraction from both control and suspect tissues, makes it extremely unlikely that false positives occurred during our PCR analysis.
Acid-fast staining of suspect tissues is rapid and requires little optimization. However, Ziehl-Neelsen staining has been reported to falsely identify Nocardia and Corynebacteria 26 and cannot differentiate among the various mycobacterial species. Similar specificity problems exist for the auramine O fluorescent stain. Previous reports have determined the sensitivity of Ziehl-Neelsen to be 36.4%. 37 Data from this study suggest that Ziehl-Neelsen staining may be slightly more sensitive at detecting M. paratuberculosis than auramine O because it detected two additional bison as positive (Table 2).
When M. paratuberculosis–specific antibodies are used, immunohistochemical staining has been reported to be more specific 6 , 29 , 30 and more sensitive 6 , 19 , 34 than other mycobacterial staining methods. Antibody anti-a362 was selected for this study because it was previously shown to distinguish M. paratuberculosis–infected intestinal and lymph node tissues from uninfected or M. bovis–infected bovine tissues. 6 Similarly, antibodies NADC 272 and NADC 275 were previously shown to identify M. paratuberculosis–infected ileum and mesenteric lymph node samples from cattle but not tissues from M. bovis–infected animals. 29 In this study, each of the four polyclonal antibodies was more sensitive at detecting infected tissues than either the Ziehl-Neelsen or auramine O staining techniques. However, some optimization was required for each antibody and tissue type to obtain optimal positive staining with minimal background. The use of antibodies anti-a362 and NADC 272 as immunohistochemical reagents demonstrated that when multiple tissue samples are taken from an animal for postmortem diagnosis of suspect animals, immunohistochemistry can be very effective (Table 2). In addition, further analysis of the staining results revealed that detection through staining was more likely in distal portions of the small intestine (jejunum and mesenteric lymph node) than proximal portions. Our results suggest that if only one tissue is collected from a suspect bison, the best tissue appears to be the middle ileum. Under the optimized conditions described for each antibody, we saw no cross-reactivity with M. bovis–infected tissues, further validating the specificity of immunohistochemical detection. Finally, the results of this study suggest that the panel of polyclonal antibodies, previously tested on tissues from cattle, is able to detect M. paratuberculosis infection in bison.
Data from this study are compiled in Fig. 1 and summarized in Table 2, demonstrating the detection of M. paratuberculosis from multiple tissues of 14 bison with different diagnostic tests. Collectively, the data indicate that animal Nos. 5, 8, 11, and 12 were the most heavily infected, based on the total number of positive tissues among all tests examined in this study. This finding agrees with the previously published gross pathology and histopathology results because bison Nos. 8, 11, and 12 had histopathologic evidence of moderate to severe granulomas (Table 1). However, no gross lesions were reported from bison No. 12, despite a high number of positive tissue samples in this study. Conversely, in this study, M. paratuberculosis infection was difficult to detect in animal Nos. 1 and 14, regardless of the diagnostic test or tissue sample evaluated. Previously, both bison Nos. 1 and 14 had gross lesions, but only bison No. 14 had severe granulomas. Given the extremely nonspecific and subjective nature of gross pathology and histopathology, these differences are difficult to interpret.
The results of this study suggest that PCR amplification of the species-specific sequence, IS900, was more sensitive than any of the staining techniques used in this study, as well as culture of feces or tissues. The ease of DNA sample preparation and rapid detection make PCR the superior diagnostic method. The conclusion of this study is that PCR examination of suspect tissues is the best way to confirm a diagnosis of M. paratuberculosis infection in wildlife species.
Footnotes
Acknowledgements
We thank Jennifer Greiner for her excellent technical assistance.