
Abstract
Hepatic fibrosis with bile duct ectasia and hyperplasia associated with polycystic kidney disease, analogous to Caroli syndrome in humans, was observed in a rat used as a control in a subchronic toxicity study. Light microscopy of liver sections showed multiple cystic and segmental saccular dilatations and hyperplasia of the intrahepatic bile ducts associated with overgrowth of portal connective tissue; the kidneys had diffuse cystic dilatation of cortical renal tubules. The lesions resembled those of human cases of the fibropolycystic disease termed as Caroli syndrome, which is thought to be the result of a pathologic developmental process known as ductal plate malformation. Recently, an animal model of Caroli syndrome has been described in mutant rats from a colony that constantly showed renal and hepatic cysts and an autosomal recessive mode of inheritance. The finding in our case of identical hepatorenal lesions suggests that the same mutation has occurred incidentally in a standard colony.
A 5-month-old male Sprague-Dawley rat, included as a control case in a subchronic toxicity study to test plastic material for vascular prosthesis, was euthanatized with CO2 at the end of the study. At necropsy, moderate enlargement of the liver in association with an irregularly lobular, gray pattern and increased firmness of the hepatic parenchyma was noted (Fig. 1); furthermore, both kidneys were moderately enlarged and bilaterally presented multiple cortical cysts ranging in size from 3 to 5 mm in diameter (Fig. 2). Tissue samples were fixed in 10% neutral buffered formalin, embedded in paraffin, sectioned at 4 µm, and stained with hematoxylin-eosin and Masson trichrome. In histologic sections of the liver the connective tissue in portal tracts was widened and formed thick bands that surrounded bile ducts and ductules (portal fibrosis); bile ducts were increased in number (bile duct hyperplasia) and markedly dilated (bile duct ectasia) (Fig. 3). The irregularly shaped bile ducts were lined by normal cuboidal epithelium, but there were bulbar protrusions of the duct wall into the lumen and occasional bridge formation of the duct wall across the lumen (bile duct dysplasia suggestive of ductal plate malformation) in more dilated bile ducts. The lumen of dilated bile ducts occasionally contained amorphous or globular weakly eosinophilic material. In some portal tracts, there was hypoplasia of portal vein branches in association with supernumerary hepatic artery branches. Occasionally, portal tracts showed mild lymphocytic infiltration. The hepatic parenchyma lacked regenerative features, and the lobular pattern was maintained. The kidneys showed multiple cystic dilatations of renal tubules, particularly marked at the cortical-medullary junction, that tended to spread to the cortex; glomeruli were spared, and there were no glomerulocystic lesions (Fig. 4). Some dilated tubules contained amorphous eosinophilic proteinaceous material; multiple, small, lymphoplasmacytic foci infiltrated the interstitial space. Renal papilla did not show cysts.
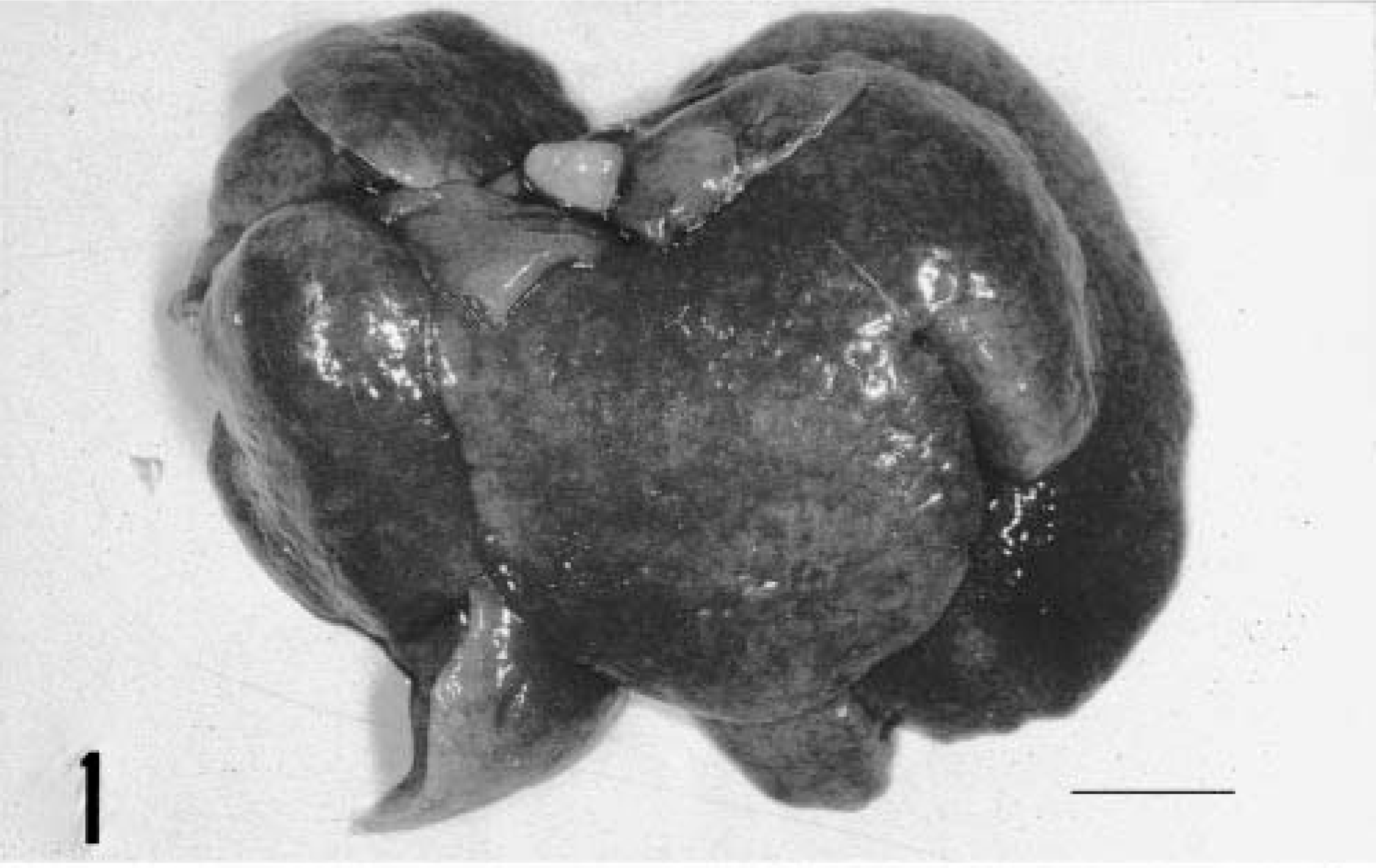
Liver; rat. Irregular surface and diffuse lobular gray pattern. Bar = 1 cm.
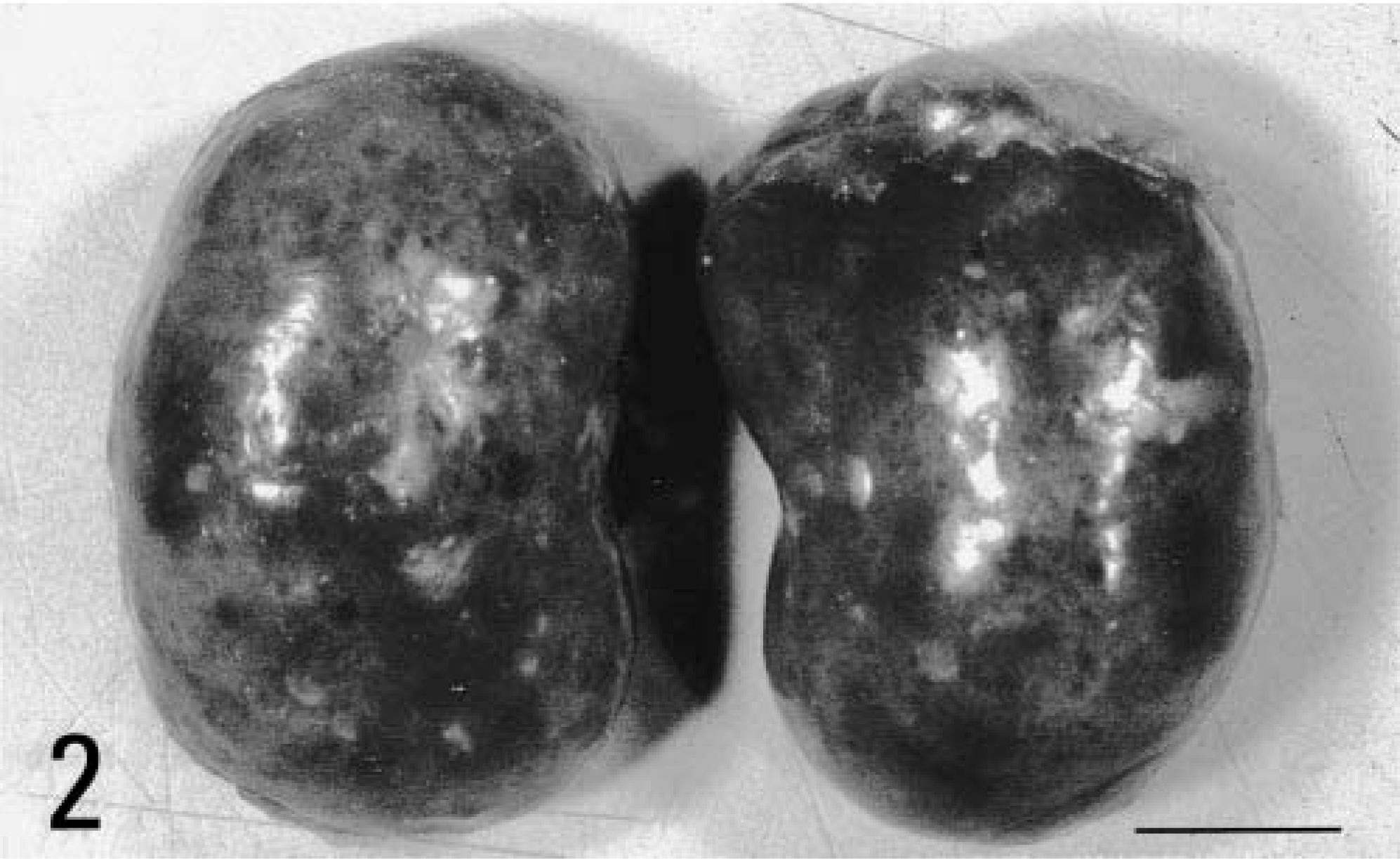
Kidneys; rat. Multiple cortical cysts. Bar = 1 cm.
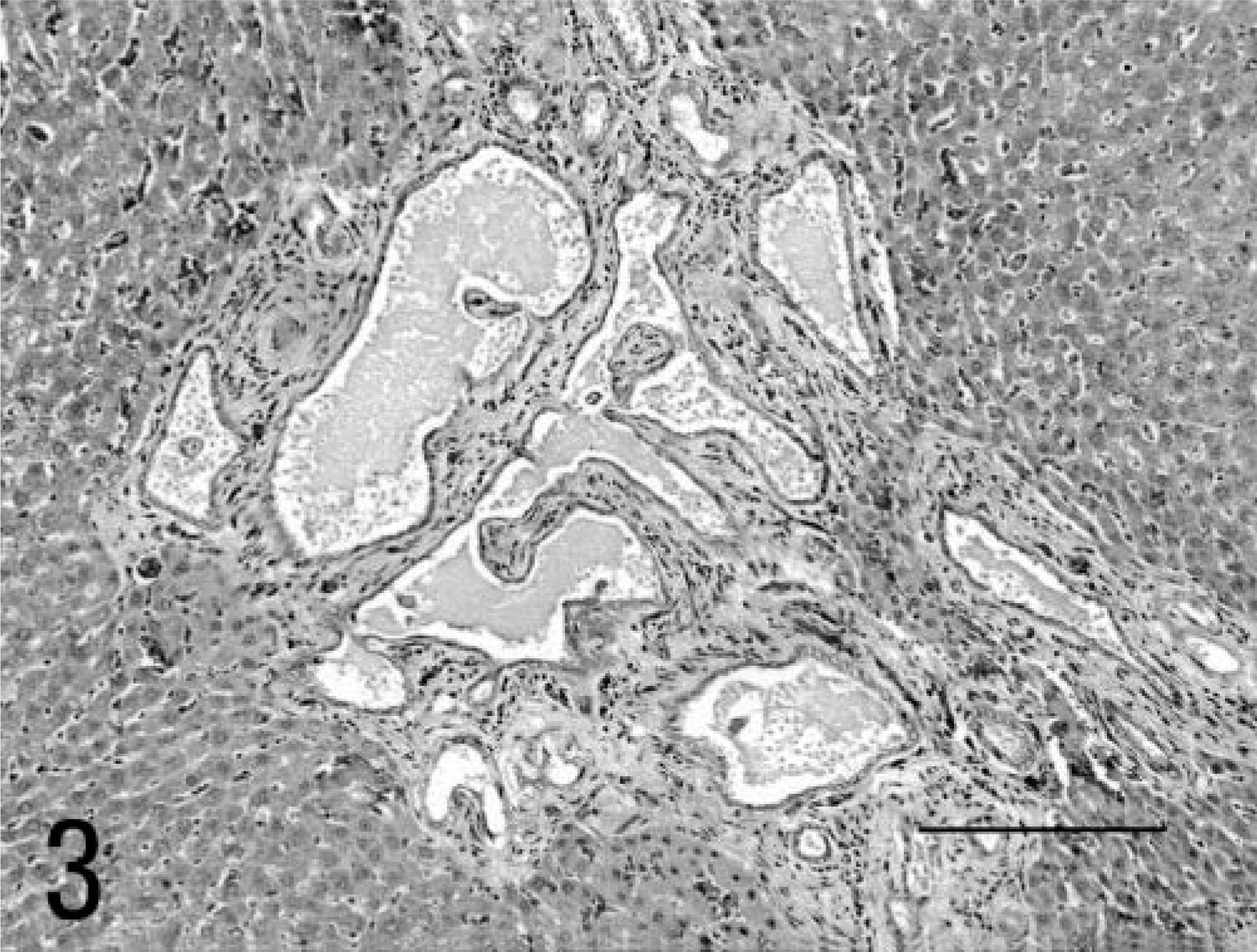
Liver; rat. Dilatation and hyperplasia of bile ducts in a fibrotic portal space. HE. Bar = 200 µm.
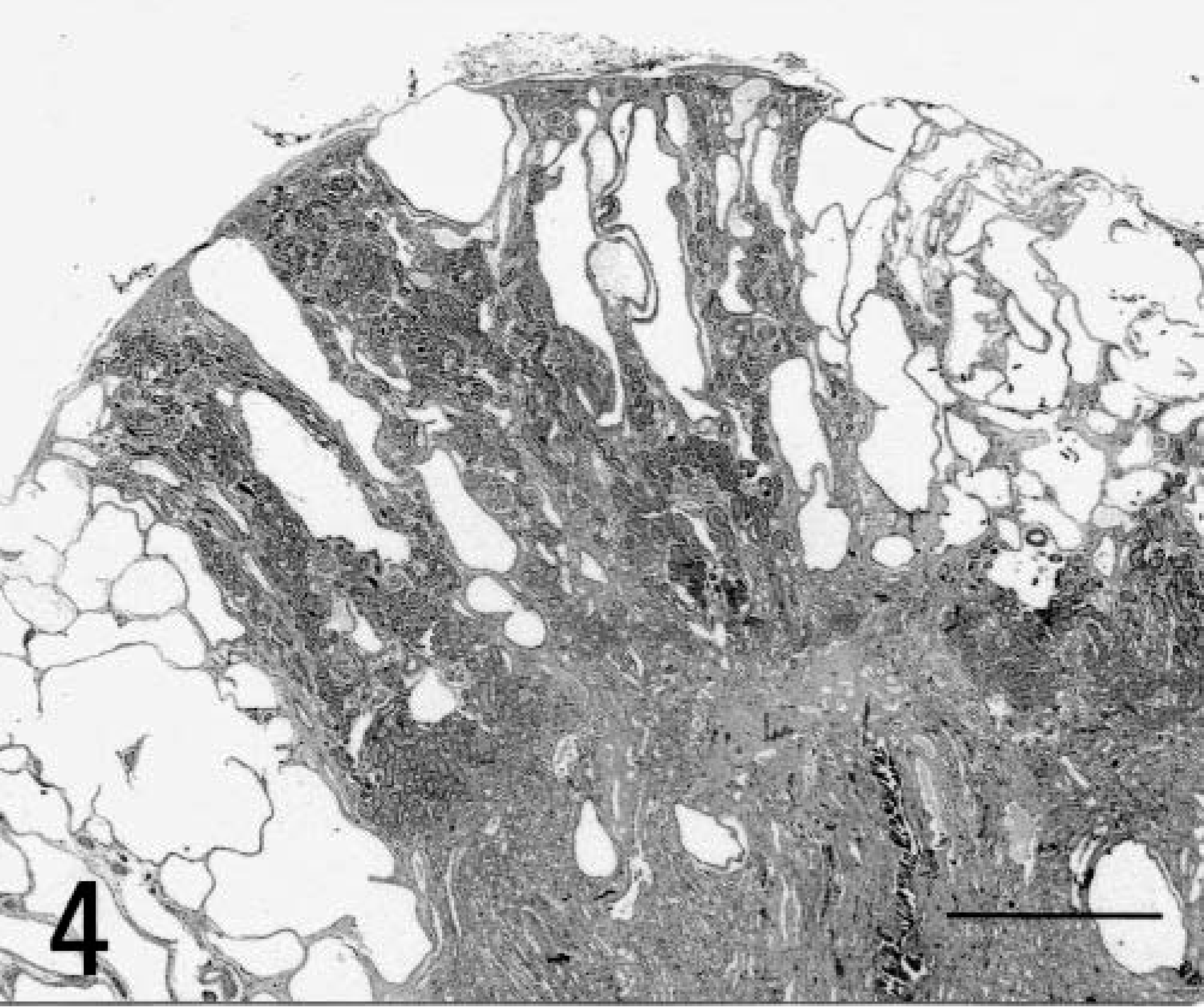
Kidneys; rat. Cystic dilatation of renal tubules. HE. Bar = 500 µm.
The morphologic diagnosis was hepatic fibrosis with bile duct ectasia and hyperplasia associated with polycystic kidney disease that resembled cases of the human fibropolycystic disease termed as Caroli syndrome.
Human hepatic fibropolycystic diseases are a heterogeneous group of congenital lesions (von Meyenburg complexes, polycystic liver disease, congenital hepatic fibrosis, Caroli disease) in which the primary abnormality is altered architecture of the intrahepatic biliary tree. 1 The precursor of the intrahepatic biliary tree is a double-layered sleeve of cells known as the ductal plate (DP). The DP first arises from hepatocyte precursors surrounding hilar portal vein vessels, and more peripheral regions of the DP then develop sequentially. During the remainder of gestation, a process of DP remodeling occurs in which small areas of the double layer separate to form tubules, which join to form the intrahepatic biliary tree, while the remaining regions of the DP involute. 4 Hepatic fibropolycystic diseases are thought to originate from failures of this process and are known collectively as DP malformations. Caroli disease is a subcategory of these diseases, characterized by multiple cystic and segmental saccular dilatations of the intrahepatic bile ducts. The term Caroli disease is applied if the hepatic disease is limited to ectasia or segmental dilatation of the larger intrahepatic ducts. This form is much less common than Caroli syndrome, in which malformations of smaller bile ducts and congenital hepatic fibrosis are also present, 4 marked by portal tract enlargement with irregular and broad bands of collagenous tissue, in which variable numbers of abnormally shaped bile ducts are embedded. 1 Caroli syndrome is often associated with autosomal recessive polycystic kidney disease (ARPKD), and both the hepatic and renal processes are thought to reflect a developmental process, albeit in the context of different organs. 1,4
Recently, an animal model for ARPKD (polycystic kidney rat [PCK-rat]) was described. 2, 3 This rat was a spontaneous mutant animal from a colony of Crj : CD rats (Charles River Japan Inc., Sprague Dawley) that were found to show constant renal and hepatic cysts. Sibling mating of the female offspring has led to the establishment of the PCK-rat animal model, in which the lesion is controlled by an autosomal recessive gene. More recently, the PCK-rat was postulated as a useful and promising animal model of Caroli syndrome, on the basis of the observation of constant association in PCK-rats between cystic dilatation of renal tubules and extensive hepatoportal fibrosis with multiple dysplastic and sacculated bile ducts. 5
The finding in our case of hepatorenal lesions is identical to those described previously 2,5 suggesting that in an European standard colony of Sprague-Dawley rats, the same mutation described and breeded by Katsuyama and colleagues 2 has occurred incidentally.